

Abstract
The human pathogen Streptococcus pneumoniae has been treated for decades with β-lactam antibiotics. Its resistance is now widespread, mediated by the expression of mosaic variants of the target enzymes, the penicillin-binding proteins (PBPs). Understanding the mode of action of β-lactams, not only in molecular detail but also in their physiological consequences, will be crucial to improving these drugs and any counterresistances. In this work, we investigate the piperacillin paradox, by which this β-lactam selects primarily variants of PBP2b, whereas its most reactive target is PBP2x. These PBPs are both essential monofunctional transpeptidases involved in peptidoglycan assembly. PBP2x participates in septal synthesis, while PBP2b functions in peripheral elongation. The formation of the “lemon”-shaped cells induced by piperacillin treatment is consistent with the inhibition of PBP2x. Following the examination of treated and untreated cells by electron microscopy, the localization of the PBPs by epifluorescence microscopy, and the determination of the inhibition time course of the different PBPs, we propose a model of peptidoglycan assembly that accounts for the piperacillin paradox.
INTRODUCTION
Streptococcus pneumoniae is a facultative bacterial pathogen that belongs to the human nasopharyngeal microbiota (1). When a pneumococcus invades other areas, it can cause mild diseases, such as otitis media, sinusitis, and bronchitis, or life-threatening pneumonia, meningitis, or septicemia. Following the introduction of β-lactams to treat bacterial infections, the first pneumococcal penicillin-resistant strain was reported in 1967 (2), and extensively multidrug-resistant pneumococci have now emerged (3, 4). Understanding the molecular mechanisms of antibiotic resistance in pneumococci is therefore of major importance.
β-Lactam antibiotics interfere with the assembly of peptidoglycan, the main constituent of the bacterial cell wall. The peptidoglycan is a network of glycan chains reticulated by peptide links, constituting a single macromolecule that encases the cell. This essential layer protects bacteria from turgor pressure and provides a scaffold to anchor other surface molecules (5). Fine-tuning the dynamics of peptidoglycan assembly is essential for proper cell division and shape determination. In ovoid bacteria, the synthesis of peptidoglycan is thought to involve two machineries that allow bacteria to elongate and divide (6, 7). A set of six penicillin-binding proteins (PBPs) catalyzes the last step of peptidoglycan assembly in S. pneumoniae. The three class A PBPs (PBP1a, PBP1b, and PBP2a) are bifunctional enzymes that polymerize glycan chains (glycosyltransferase activity) and cross-link them through peptide bonds (transpeptidase activity). The two class B PBPs (PBP2x and PBP2b) are monofunctional transpeptidases. PBP3 is a class C PBP with d,d-carboxypeptidase activity, which is involved in the maturation of peptidoglycan (8). While the class A and C PBPs are not essential in pneumococci (9, 10), both class B PBPs are necessary, with PBP2b being essential for elongation and PBP2x for division (11–14). In pneumococci, β-lactam-resistant strains harbor PBP variants with reduced reactivities toward the drugs (15, 16).
An early study intended to correlate the specificities of 18 β-lactams for different PBPs and the morphological defects they caused in S. pneumoniae (17). A very interesting phenotype of pneumococcal cells was observed in the presence of piperacillin, an extended-spectrum β-lactam antibiotic used to treat polymicrobial infections (often in combination with tazobactam, a β-lactamase inhibitor) (18). The cells treated with 0.5× MIC of piperacillin for 4 h acquired a characteristic lemon shape, with a central bulge and pointy ends (17). In addition, it was reported that piperacillin used at the MIC inhibits about 50% of PBP2b and PBP3 and 27% of PBP2a. However, since PBP2x was not known at that time because it comigrated with PBP2a in the experimental setup, the reported PBP2a inhibition corresponds to that of both PBP2x and PBP2a. Accordingly, PBP2x was not considered in the conclusions drawn at that time. Meanwhile, it is apparent that there is a complex redundancy of the different PBPs, and the morphological, physiological, and biochemical effects induced by one particular PBP remain largely open today.
Further studies investigated the resistance of pneumococci to piperacillin in the laboratory (19–21). Pneumococci were gradually selected in increasing concentrations of piperacillin, and the PBP sequences of the resulting strains were determined. Interestingly, in the three selected piperacillin-resistant lineages, PBP2b variants appeared first, and PBP2x variants were found only in strains subsequently selected for a higher resistance level (21). In subsequent experiments in which pbp genes were randomly mutagenized, PBP2b variants were again selected by piperacillin exposure (20). The T446A substitution in PBP2b, which is commonly found in resistant clinical isolates, increased the MIC for piperacillin 2-fold. The same piperacillin MIC was determined for a strain having incorporated the full PBP2b sequence of a clinical isolate.
These selection studies strongly suggest that the primary essential target of piperacillin is PBP2b. However, the morphological defects induced by piperacillin challenge (i.e., the lemon shape) resemble those induced by the depletion of PBP2x (11, 13). Moreover, of the PBPs, PBP2x was found to be the most reactive with piperacillin (12, 19). In this work, we revisited the effect of piperacillin treatment on the morphology and localization of both class B PBPs, as well as the specificities for the different PBPs. We confirm here that the primary target of piperacillin is PBP2x, causing morphological defects that are consistent with PBP2x inhibition, despite the fact that it is the lesser inhibition of PBP2b that prevents growth, a behavior that we dub the piperacillin paradox. The results are discussed in light of the recent knowledge on the respective roles of PBP2x and PBP2b, as well as peptidoglycan composition modifications that play a role in resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains (Table 1) were grown on Columbia blood agar plates supplemented with defibrinated horse blood to 4% (CB agar) or in liquid C+Y medium (22) at 37°C in a 5% CO2 atmosphere. A stock of each strain was prepared from an exponentially growing culture (optical density at 600 nm [OD600], between 0.2 and 0.4) and stored at −80°C in 20% glycerol.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| Strains | ||
| R6 | Unencapsulated laboratory strain | 42 |
| R6 ΔlytA | lytA::CAT; Cmr | 43 |
| R6 ΔlytA gfp-ftsZ | lytA::CAT bga::tetM-PczcD-gfp+-ftsZ; Cmr Tetr | This study |
| DKL031 | bga::tetM-PczcD-gfp+-pbp2x pbp2x::aad9; Tetr Spcr | 13 |
| R6 ΔlytA gfp-pbp2x Δpbp2x | lytA::kanR bga::tetM-PczcD-gfp+-pbp2x pbp2x::aad9; Kanr Tetr Spcr | This study |
| sspCM54 | R6; bga::tetM-PczcD-gfp+-pbp2b; Tetr | This study |
| sspCM99 | R6; bga::tetM-PczcD-gfp+-pbp2b pbp2b::CAT; Tetr Cmr | This study |
| R6 ΔlytA gfp-pbp2b Δpbp2b | lytA::kanR bga::tetM-PczcD:gfp+-pbp2b pbp2b::CAT; Kanr Tetr Cmr | This study |
| R6 ΔlytA pbp2bR | ΔlytA::CAT pbp2bT446A; Cmr | This study |
| R6 ΔlytA pbp2b5 | ΔlytA::CAT pbp2b5204; Cmr | This study |
| R6 ΔlytA pbp2xR | ΔlytA::CAT pbp2xT338A/M339F; Cmr | This study |
| R6 ΔlytA pbp2x5 | ΔlytA::CAT pbp2x5204; Cmr | This study |
| Plasmids | ||
| pNM15 | PCR-Script::pbp2b; Cmr | This study |
| pCM25 | pJWV25 bga::tetM-PczcD-gfp+-pbp2b; Tetr | This study, 44 |
| pCM57 | PCR-Script::ftsZ; Cmr | This study |
| pCM49e | pJWV25 bga::tetM-PczcD-gfp+-ftsZ; Tetr | This study, 44 |
| pET28-his-lytA | pET28a::his-lytA | This study |
| pET30-2bT446A | pET30 pbp2bT446A; Kanr | 30 |
| pGEX-2b5204 | pGEX pbp2b5204; Ampr | 30 |
| pGEX-2xT338A,M339F | pGEX pbp2xT338A,M339F; Ampr | 33 |
| pGEX-2x5204 | pGEX pbp2x5204; Ampr | 33 |
Cm, chloramphenicol; Tet, tetracycline; Spc, spectinomycin; Kan, kanamycin.
The S. pneumoniae R6 ΔlytA gfp-ftsZ strain and the R6 ΔlytA strains with low-susceptibility PBPs (Table 1; see also Fig. S1 in the supplemental material) were constructed by transformation of the appropriate plasmid in the R6 ΔlytA and homologous recombination at the bgaA locus. The clones of R6 ΔlytA gfp-ftsZ were selected on CB agar containing 2.5 μg/ml tetracycline. The clones of R6 ΔlytA pbp2xT338A/M339F (pbp2x encoding the T338A/M339F substitutions) (R6 ΔlytA pbp2xR) and R6 ΔlytA pbp2x5204 (R6 ΔlytA pbp2x5) (pbp2x5204 is the pbp2x allele from S. pneumoniae strain 5204) were selected on CB agar containing 0.1 μg/ml cefotaxime. The clones of R6 ΔlytA pbp2bT446A (R6 ΔlytA pbp2bR) (pbp2bT446A is pbp2b encoding the T446A substitution) and R6 ΔlytA pbp2b5204 (R6 ΔlytA pbp2b5) were selected on CB agar containing 0.03 μg/ml piperacillin.
The R6 ΔlytA gfp-pbp2x Δpbp2x and ΔlytA gfp-pbp2b Δpbp2b strains were obtained by transformation of their lytA+ parent, with a PCR fragment consisting of a kanamycin resistance cassette flanked by 1,000-bp-long fragments homologous to the upstream and downstream regions of the lytA gene, as described by Fadda et al. (23).
Transformations of pneumococci with PCR products or plasmids were performed using the competence-stimulating peptide 1 (CSP1) for convenience (24). The region surrounding the modified or introduced gene was systematically sequenced in the clones used for further experiments. In the case of R6 ΔlytA pbp2b5, only a partial fragment of pbp2b5204 missing the 3′ region of the gene was found to have recombined in the genome (see Results and Fig. S1 in the supplemental material). The growth curves were measured and compared to that of the R6 ΔlytA strain (see protocol below). All strains grew identically (doubling time, 34 ± 2 min), except the strains grown in the presence of 150 μM ZnCl2, which had a longer (1.3-fold) generation time.
Growth curves.
Volumes of 2.5 ml of C+Y medium were inoculated with 250 μl of preculture at an OD600 of 0.3 in 24-well plates, sealed, and grown at 37°C without added CO2 in a FLUOstar plate reader (BMG Labtech) equipped with a 595-nm filter. The OD595 was recorded every 20 min after shaking. Each strain was grown in triplicate in parallel with the R6 ΔlytA control strain, in two independent experiments. The strains containing green fluorescent protein (GFP) fusions under the control of the PczcD zinc-inducible promoter were grown with 150 μM ZnCl2.
MICs.
The MICs were determined in liquid cultures to determine the piperacillin concentrations to be used in the various experiments. Piperacillin was obtained from Sigma (P8396). Glycerol stocks were diluted 100-fold in 30 ml of C+Y medium. Nine hundred fifty microliters of this dilution was added to 50 μl of a 20× piperacillin solution in 96-deep-well plates. The final piperacillin concentrations were 0, 0.02, 0.03, 0.04, 0.05, 0.06, 0.08, and 0.1 μg/ml. The cultures were incubated for 6 h at 37°C before 200 μl of culture was transferred to a 96-well microplate to record the OD595 in a FLUOstar plate reader (BMG Labtech). The MIC was defined as the lowest piperacillin concentration that did not permit growth. MIC determination was always carried out in triplicate, in two independent experiments, including the R6 ΔlytA control strain.
Piperacillin treatment for optical microscopy analysis.
Glycerol stocks were diluted 100-fold in 5 ml of C+Y medium and grown to an OD600 of 0.3. Nine hundred fifty microliters of this culture was added to 50 μl of a 20× piperacillin solution to obtain the desired concentrations and incubated further for 2 h. The samples were centrifuged at 5,000 × g for 3 min and resuspended in 50 μl of supernatant. Four microliters of concentrated bacteria was transferred to microscope slides. Images were acquired with an Olympus BX61 microscope equipped with a UPFLN 100× O-2PH/1.3 objective and processed using the Volocity software package (luminosity and contrast adjustment). The cell dimensions were determined using the ObjectJ plugin written by Norbert Vischer (University of Amsterdam [http://simon.bio.uva.nl/objectj/]), which runs with the ImageJ software. Global representations of length and diameter were generated using ObjectJ.
Time-lapse microscopy.
A glycerol stock of R6 ΔlytA gfp-ftsZ was diluted 107 times in 15 ml of C+Y medium and grown overnight to an OD600 of 0.1. An agarose pad containing C+Y medium, 1.5% low-melting agarose, and 150 μM ZnCl2 was prepared on a microscopy slide, as described by de Jong et al. (25). To avoid antibiotic degradation, the medium was incubated at 60°C until complete melting of the agarose, and piperacillin was added just prior to casting on the slide. A volume of 2.5 μl of culture was inoculated on the agarose pad. When the droplet was completely absorbed in the pad, a coverslip was mounted and the samples were observed using an inverted Olympus IX81 microscope equipped with a Plan Apo 60×/1.42 objective and a 37°C incubation chamber. Videos were acquired and processed using the Volocity software.
Electron microscopy.
Glycerol stocks were diluted 109 times in 50 ml of C+Y medium and incubated overnight. At an OD600 of 0.3, piperacillin was added at a concentration of 0.06 μg/ml, and incubation was pursued for 2 h. The bacteria were then centrifuged at 3,220 × g for 10 min. A pellet volume of 1.4 μl was dispensed on the 200-μm side of a type A 3-mm gold platelet (Leica Microsystems), covered with the flat side of a type B 3-mm aluminum platelet (Leica Microsystems), and was vitrified by high-pressure freezing using an HPM100 system (Leica Microsystems). Next, the samples were freeze substituted at −90°C for 80 h in acetone supplemented with 2% OsO4 and warmed up slowly (1°C/h) to −60°C in an automated freeze substitution device (AFS2; Leica Microsystems). After 8 to 12 h, the temperature was raised (1°C/h) to −30°C, and the samples were kept at this temperature for another 8 to 12 h before being rinsed several times in acetone. The samples were then infiltrated with gradually increasing concentrations of Epon in acetone (1:2, 1:1, 2:1 [vol/vol], and pure) for 2 to 3 h while raising the temperature. Pure Epon was added at room temperature. After polymerization at 60°C, 80-nm-thin sections were obtained using an ultramicrotome UC7 (Leica Microsystems) and were collected on Formvar-carbon-coated 100-mesh copper grids. The thin sections were poststained for 5 min with 5% aqueous uranyl acetate, rinsed, and incubated for 2 min with lead citrate. Note that no negative staining was performed with the samples presented in Fig. S2 in the supplemental material. The samples were observed using a CM12 (Philips) or Tecnai 12 (FEI) microscope operating at 120 kV with an Orius charge-coupled-device (CCD) camera (Gatan) at 5,000 to 45,000× magnifications.
Recombinant protein purification.
Recombinant His-tagged LytA was purified from Escherichia coli Rosetta cells transformed with the pET28a-his-lytA plasmid. After induction with 500 μM isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.6 and overnight expression at 25°C, the cell pellet from 2 liters of culture was resuspended in 50 ml of 50 mM Tris-HCl (pH 8), 500 mM NaCl, 25 mM imidazole, and 10% glycerol, and it was frozen at −80°C. Once thawed, the cells were lysed using a Microfluidizer M-110P (Microfluidics), and the lysate was centrifuged at 40,000 × g for 30 min at 4°C. The supernatant was applied onto a 10-ml Ni-nitrilotriacetic acid (NTA) column (Qiagen) in the same buffer. The protein was eluted with a 25 mM to 500 mM imidazole gradient. Pooled fractions were concentrated and further purified by size exclusion chromatography using a Superdex S200 10/300 GL column (GE Healthcare) in 25 mM Tris-HCl (pH 8) and 150 mM NaCl. Ten milliliters of purified His6-LytA was obtained at a concentration of 500 μg/ml.
Recombinant PBP2b from R6 and from the R6 ΔlytA pbp2b5 strain was purified as described before with a C-terminal Strep tag (26). The pbp2b5 gene was PCR amplified from the genomic DNA of the R6 ΔlytA pbp2b5 strain. PBP2b5 was expressed from a pET30-derived plasmid after cloning, as described previously (26).
PBP inhibition profiles.
PBP profiles were determined by a procedure derived from those previously reported (11, 12). Glycerol stocks were diluted 100-fold in 30 ml of C+Y medium and grown to an OD600 of 0.3. Piperacillin was added at various concentrations. The cultures were further incubated for 10 min at 37°C and centrifuged at 3,220 × g at room temperature for 10 min. The pellets were resuspended in 150 μl of phosphate-buffered saline (PBS) containing 25 μg/ml purified LytA. After complete lysis, typically obtained after 1 h at 37°C (determined visually as the samples become transparent), membrane proteomes were isolated by centrifugation at 20,000 × g for 20 min at 4°C. The pellets were resuspended in 150 μl of fresh PBS and stored at −20°C. The total protein concentration of each sample was estimated by the absorbance at 280 nm determined with a NanoVue Plus spectrophotometer (GE Healthcare). The sample protein concentrations were adjusted to 1 mg/ml, and 10 μl of each sample was supplemented with 0.5 μl of Bocillin FL (Life Technologies) at 300 μM (final concentration, 15 μM) and incubated for 10 min at room temperature. After boiling for 10 min in Laemmli buffer, the proteins were separated by SDS-PAGE at 4°C using 4 to 12% acrylamide gradient gels (Criterion; Bio-Rad) with the XT MOPS running buffer (Bio-Rad). The PBP profiles were revealed under UV illumination in a ChemiDoc imager (Bio-Rad). Densitometry of the band pattern was performed using the ImageJ freeware.
In vitro peptidoglycan synthesis and PBP2b inhibition.
The reactions were performed and analyzed as described previously (26). Briefly, unlabeled isoglutamine-containing lipid II and dansylated lipid II were mixed in a 10:1 ratio and dried prior to dissolution to final concentrations of 50 and 5 μM, respectively, in a reaction mixture containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 25% (vol/vol) dimethyl sulfoxide (DMSO), 0.02% (wt/vol) Triton X-100, 1 μM PBP2a with the S410A substitution (PBP2aS410A), and 0.5 μM PBP2b. Aliquots were withdrawn after various times of incubation at 30°C, and the reaction was stopped by adding penicillin G (1 mM) and moenomycin (0.5 mM). The samples were analyzed by SDS-PAGE and visualized with UV transillumination.
The reactivities of R6 PBP2b and PBP2b5 with piperacillin were evaluated by incubation of 0.5 or 0.25 μM enzyme, respectively, for 10 min at room temperature in their purification buffer with serial dilutions of the drug. Bocillin FL was then added to a concentration of 100 μM for a further 10 min prior to SDS-PAGE analysis and imaging under UV transillumination and Coomassie blue staining.
RESULTS
The primary target of piperacillin is PBP2x.
As piperacillin primarily selects substitutions in PBP2b of pneumococci in the laboratory, this β-lactam is expected to specifically inhibit PBP2b. However, the most reactive PBP with piperacillin was reported to be PBP2x (12, 19). To investigate this paradoxical behavior, we set out to characterize the effect of piperacillin on pneumococci.
The MIC of piperacillin on the R6 strain of S. pneumoniae was determined (Table 2). The effect of piperacillin on the morphology of pneumococci was observed 2 h after its addition at twice the MIC (0.06 μg/ml) during exponential growth (OD600 of 0.3). The cells were lemon shaped or bulging and elongated, as observed by phase-contrast and differential interference contrast microscopy. However, a large proportion of cells had also lysed at this stage. To facilitate observation, we then used an R6 strain depleted of the major autolysin LytA by substituting the coding gene with an antibiotic resistance cassette. Autolysis was nearly nonexistent in this ΔlytA strain and allowed the observation of morphological defects due to the toxicity of the antibiotic. The piperacillin MIC was not changed (Table 2), showing that the absence of LytA does not modify susceptibility to piperacillin. Unless otherwise mentioned, all the experiments described below were performed in a ΔlytA background.
TABLE 2.
MICs of piperacillin
| Strain | MIC (μg/ml) |
|---|---|
| R6 | 0.03 |
| R6 ΔlytA | 0.03 |
| R6 ΔlytA gfp-ftsZ | 0.03 |
| R6 ΔlytA gfp-pbp2x Δpbp2x | 0.03 |
| R6 ΔlytA gfp-pbp2b Δpbp2b | 0.03 |
| R6 ΔlytA pbp2b5 | 0.06 |
| R6 ΔlytA pbp2x5 | 0.04 |
| R6 ΔlytA pbp2bR | 0.06 |
| R6 ΔlytA pbp2xR | 0.04 |
Piperacillin treatment of the ΔlytA strain also resulted in the accumulation of lemon-shaped cells with a characteristic bulge at mid-cell, as observed by phase-contrast microscopy (Fig. 1A). In the absence of lysis, nearly all cells adopted abnormal shapes. Most cells were in the shape of a citrus fruit (81% of 155 analyzed misshapen cells and top two cells in Fig. 1A), i.e., with pointy poles bracketing a bulky mid-cell. A number of cells (19% of 155 analyzed misshapen cells and the bottom cell in Fig. 1A) were longer and trilobed, the middle lobe having the wider diameter (Fig. 1A). The ratio between the cell length and the larger diameter was greater in the presence of piperacillin (Fig. 1B). Note that by cell length, we mean the longitudinal dimension of entities that constitute a continuum of forms from the single cell to diplococci, including trilobed elongated cells. Figure 1C represents the cells sorted by length, and the brightness of each vertical line is proportional to the width of a particular cell along its long axis. While the sorting of the control cells shows a dark zone corresponding to the constricted region at mid-cell, the sorting of cells exposed to piperacillin shows a bright central region, revealing the absence of mid-cell constriction. All together, these data indicate that piperacillin treatment results in the enlargement of pneumococcal cells at their middle, regardless of their length. Strikingly, the morphology induced by piperacillin resembles that resulting from the depletion of PBP2x (11, 13) (Fig. 1A).
FIG 1.
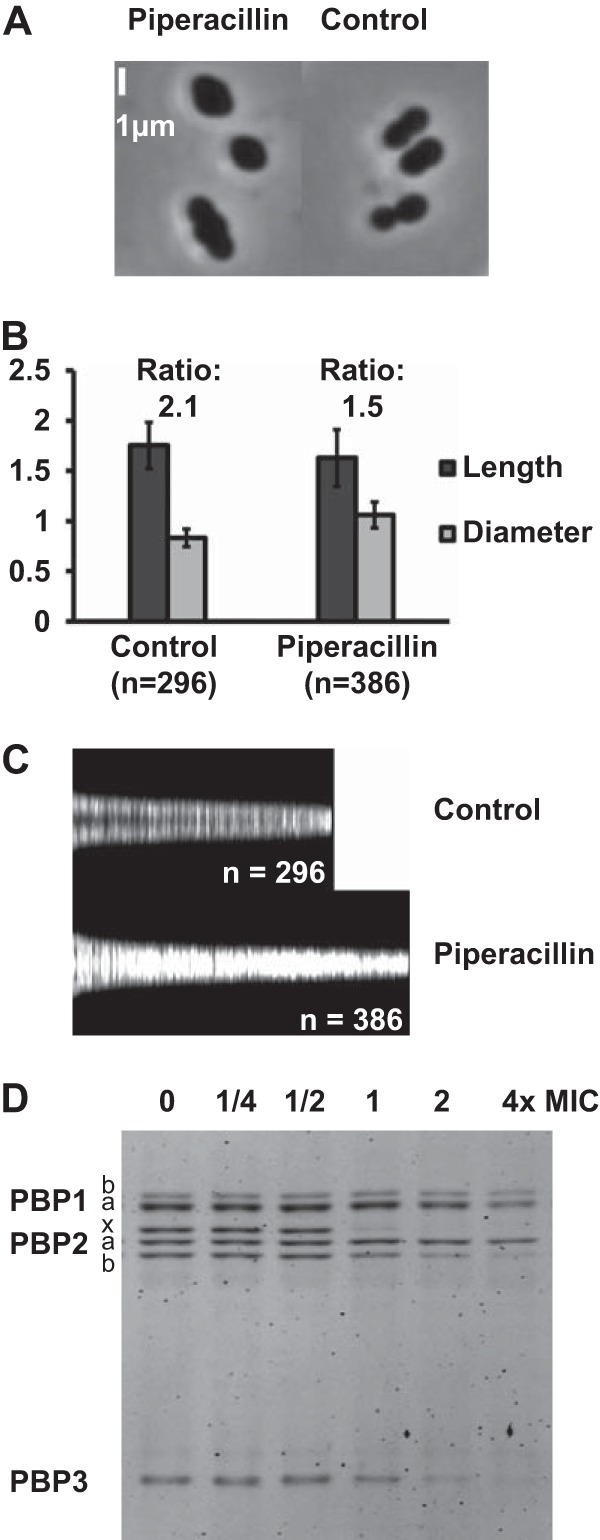
Morphological consequences of piperacillin treatment and PBP inhibition profile. (A to C) R6 ΔlytA cells were observed following 2 h of incubation at twice the piperacillin MIC (0.06 μg/ml). (A) Phase-contrast microscopy. (B) Mean apparent lengths and diameters (with standard deviations). (C) Global representation of cell lengths and diameters. The vertical light bars represent individual cells sorted by length. The intensity of a bar is proportional to the cell width (perpendicular to the long axis). (D) Fluorogram showing the in vivo PBP inhibition profile by piperacillin. The cells were incubated for 10 min with the drug prior to washing, membrane isolation, and PBP labeling with Bocillin FL.
To further investigate this observation, we determined which PBPs are targeted by piperacillin. Exponentially growing cultures were incubated with different concentrations of piperacillin prior to lysis and incubation with a large excess of Bocillin FL. The membrane proteins were finally separated by SDS-PAGE, and Bocillin-labeled proteins were visualized by UV illumination. The PBP inhibition profile of the R6 strain was identical to that of its ΔlytA derivative, indicating that the absence of LytA did not influence the accessibilities or reactivities of the PBPs to piperacillin (Fig. 2A and B). The band corresponding to PBP2x was the first to disappear from the fluorogram with increasing piperacillin concentration, followed by PBP3 and PBP2b (Fig. 1D and 4B). PBP2x has the strongest affinity for piperacillin, consistent with the observation that morphological defects caused by piperacillin resemble the morphological defects caused by the depletion of PBP2x (11, 13).
FIG 2.

In vivo PBP inhibition profile with different concentrations of piperacillin. The cells were incubated for 10 min with the drug prior to washing, membrane isolation, and PBP labeling with Bocillin FL. A representative fluorogram is shown for each strain. The intensity of the bands was quantified by densitometry, and the results are presented for each PBP as the fraction remaining reactive (r.f.) with Bocillin FL after piperacillin treatment (means from three independent experiments, with the error bars as the standard deviations). The vertical dashed lines represent the MICs.
FIG 4.

Electron micrographs of negatively stained cryo-thin sections of R6 ΔlytA cells treated for 2 h with 0.06 μg/ml piperacillin (2× the MIC). The scale bars are 200 nm (A and B) and 50 nm (C to E). Bulging cells with complete (A and C), absent (B and D), or partial septa (B and E) are shown. The white arrows point to the two leaflets of the plasma membrane. The black arrows point to the cell wall.
Inhibition by piperacillin of PBP2b and PBP2x does not affect their localization, nor that of FtsZ.
To probe the mechanism underlying the morphological defects induced by piperacillin, we investigated whether the drug affects the localization of FtsZ and the class B PBPs. FtsZ is the tubulin-like protein that polymerizes on the cytoplasmic face of the membrane at the division site and recruits the other proteins involved in the division process (15, 27). Three different strains expressing FtsZ, PBP2x, or PBP2b fused to the green fluorescent protein (GFP) from an ectopic site under the control of a Zn-inducible promoter were created in the R6 ΔlytA background. In the cases of GFP-PBP2x and GFP-PBP2b, the fluorescence signal observed in the initial merodiploid strains was unsatisfactory. The fluorescence signal was improved by deleting the endogenous copy of pbp2x or pbp2b. The GFP-PBP fusions were functional, as they complemented the absence of the corresponding native proteins. The essentiality of pbp2x and pbp2b was confirmed, as the expression of the GFP-fused version was required, and the cells could not grow in the absence of zinc. GFP-PBP2x and GFP-PBP2b were localized at mid-cell and at the equators, as expected from previous immunofluorescence and GFP fusion studies (13, 14, 28, 29) (Fig. 3A, control [Ctl]). The GFP fusions were overexpressed compared to the native proteins (Fig. 3B). Note that an additional Bocillin FL-reactive species appeared at the size of PBP2b. This product might result from the cleavage of the GFP fusion protein, but the corresponding GFP fragment was not detected by immunoblotting (Fig. S3 in the supplemental material). Some synthesis of PBP2b may have been initiated from an alternative start codon. Also, a faint band is visible at the size of PBP2x in the Δpbp2x strain. This protein is not a cleavage product of GFP-PBP2x, as it does not disappear in the absence of zinc and is not detected by immunoblotting with an anti-PBP2x serum (see Fig. S3). It may correspond to a fragment of PBP1a or PBP1b.
FIG 3.

Localization of FtsZ and class B PBPs. (A) Localization of GFP-FtsZ, GFP-PBP2b, and GFP-PBP2x after 2 h of treatment with (Pip) or without (Ctl) piperacillin at 2× the MIC. (B) Bocillin FL fluorogram of R6 ΔlytA, R6 ΔlytA gfp-pbp2x Δpbp2x, and R6 ΔlytA gfp-pbp2b Δpbp2b strains in the presence (Zn) or absence (−) of inducer (Zn2+). Depletion was achieved by growing the cells to an OD600 of 0.3 with zinc, diluting the culture 16× (Δpbp2x) or 256× (Δpbp2b) in fresh medium without zinc and further incubation to an OD600 of 0.3. (C) Time-lapse localization of GFP-FtsZ in the presence (Pip) or absence (Ctl) of piperacillin at 2× the MIC. The arrows point to bulging cells. The scale bars represent 1 μm. DIC, differential interference contrast.
Also, the zinc-inducible GFP-PBPs retained their reactivities with Bocillin FL (Fig. 3B). The determination of the MICs of the three strains showed that the induction of expression with zinc did not affect their viabilities or susceptibilities to piperacillin (Table 2).
To localize the GFP fusions, the appropriate strains were grown with zinc to an OD600 of 0.3, and piperacillin was added or not to the culture at twice the MIC (0.06 μg/ml). Unexpectedly, piperacillin did not cause delocalization of the FtsZ ring, in the sense that FtsZ was observed at mid-cell and at the equators, even when the mid-cell was grossly enlarged (Fig. 3A). Similarly, the equatorial and mid-cell localizations of both PBP2b and PBP2x were not affected by the action of piperacillin (Fig. 3A).
In order to gain insight into the dynamics of formation of the mid-cell bulge, we imaged the growth of the GFP-FtsZ-expressing strain using time-lapse microscopy (Fig. 3C). Pneumococci were grown to an OD600 of 0.3 and transferred to an agarose pad of culture medium supplemented or not with 0.06 μg/ml piperacillin. The cells were then observed over time at 37°C. The strain expressing GFP-FtsZ in the presence of zinc is shown in Fig. 3C. In the absence of piperacillin, the cells divided normally in 20 to 30 min. In contrast, the presence of piperacillin inhibited proper cell separation, and a mid-cell bulge appeared after 30 min. In some cases, the cells continued to inflate until lysis after >1 h of piperacillin treatment. The FtsZ ring was found to enlarge with the bulge formation at mid-cell. Remarkably, bulging occurred equally in cells that were at different stages of the cell cycle at the time of piperacillin addition. Indeed, in Fig. 3C at time 0′, the cell on the left was at an advanced stage of the cell cycle (elongated diplococcus), whereas the cell on the right was at an early stage. In both cases, the FtsZ ring enlarged during bulge formation.
We did not obtain good time-lapse fluorescence microscopy data with strains expressing GFP-PBPs, as the illumination intensity required to obtain a good fluorescence signal caused phototoxicity.
Morphological details of piperacillin-treated pneumococci.
To shed light on the bulge formation, piperacillin-treated cells were observed by electron microscopy. R6 ΔlytA cells were grown to exponential phase and treated with piperacillin as for optical microscopy. Next, the cells were flash-frozen under high pressure to keep the cellular structures intact. The cells were then slowly brought back to room temperature while being included in Epon resin. Thin sections of resin-embedded pneumococci were then negatively stained with uranyl-acetate to enhance the contrast of lipid membranes and peptide-containing peptidoglycan.
Interestingly, three distinct types of abnormal bulging cells were observed (Fig. 4; see also a colorized version in Fig. S4 in the supplemental material highlighting the features of the cell envelope). First, some bulging cells were in fact constituted of two cells adjoined by a continuous peptidoglycan septum, at least in the plane of the thin section, which apparently failed to split (Fig. 4A and C). Second, some lemons were single cells, as the membrane underlying their cell wall was clearly continuous along the bulge (Fig. 4B and D). Finally and surprisingly, some bulging shapes were formed by two distinct cells separated by their respective plasma membranes but without a normal continuous peptidoglycan septum (Fig. 4C and E). In those cases, a nascent cross-wall is present close to the periphery but does not extend inward. However, we cannot exclude that a thinner incomplete peptidoglycan layer is present and undetectable. To our knowledge, this is the first observation of this phenomenon in ovoid bacteria. The three types of bulging shapes may result from cells that were at different stages of the cell cycle at the time of piperacillin addition, as proposed in the Discussion.
To determine whether the absence of LytA-induced lysis plays a role in the observed morphological defects, these experiments were repeated with the R6 parental strain. In this case, cells were observed after both 1 and 2 h of piperacillin treatment. Most cells were lysed 2 h after the addition of piperacillin. However, after only 1 h, the bulging morphology was observed, but none of the 20 cells observed had a peptidoglycan or membrane septum, complete or partial (see Fig. S2 in the supplemental material). This observation suggests that upon piperacillin inhibition, LytA may be involved in the specific lysis of pneumococcal cells with a septum through the targeted degradation of septal peptidoglycan.
PBP2b variants confer piperacillin resistance, in contrast to PBP2x variants.
We showed that the primary target of piperacillin is PBP2x (Fig. 1D). PBP2b and PBP3 are less reactive to this β-lactam. In the late 1980s, piperacillin-resistant pneumococci were selected in the laboratory by successive plating on medium with increasing piperacillin concentrations (20, 21). Interestingly, the first level of piperacillin resistance was conferred by low-affinity PBP2b variants. The PBP2x variants did not provide pneumococci with this first level of resistance (20). Low-affinity PBP2x variants were selected in lineages in which a low-affinity PBP2b was already present, conferring a second higher level of resistance.
To further document this paradox, we constructed four strains in the R6 ΔlytA background. Two strains had a variant pbp2b allele introduced by selection on piperacillin. One allele codes for the T446A PBP2b point mutant (20, 30) (pbp2bR), while the other encodes a PBP2b derivative from the clinical resistant strain S. pneumoniae 5204 (31, 32) (pbp2b5). PBP2b5 has 23 substitutions, including the critical T446A, but compared to PBP2b from strain 5204, it lacks 21 substitutions following residue 520. This impossibility of introducing the complete pbp2b sequence from strain 5204 into an R6 background was reported previously (30).
Similarly, two strains had their pbp2x alleles replaced with alleles coding for low-affinity variants, which were introduced by selection with cefotaxime. The first allele coded for PBP2x with the T338A-M339F mutations (herein referred to as pbp2xR), known to reduce its affinity to β-lactams (33). The second incorporated the full-length pbp2x allele of strain 5204 (herein referred to as pbp2x5) that encodes a variant with 80 amino acid substitutions, including 41 in the transpeptidase domain (31, 33, 34).
The MICs of piperacillin were determined for the four strains (Table 2). The low-affinity PBP2b variants increased the MIC 2-fold. In contrast, low-affinity PBP2x variants conferred only a modest increase in the piperacillin MIC. This modest effect on the MIC was not sufficient to select the introduction of pbp2xR and pbp2x5 alleles by piperacillin, in contrast to the pbp2bR and pbp2b5 alleles.
Profiles of PBP inhibition by piperacillin in strains with low-affinity class B PBPs.
Given the apparent conflict between the morphological observations, which point to a predominant role of PBP2x inhibition in the action of piperacillin, and the selection experiments that indicate the importance of the inhibition of PBP2b, we determined the profile of PBP inhibition of our four resistant strains compared to that of their parental R6 ΔlytA and R6 strains (Fig. 2). After 10 min of incubation with various concentrations of piperacillin below and above the MIC, the fraction of free PBP remaining reactive was determined by a reaction with an excess of Bocillin FL and SDS-PAGE separation. All the strains were challenged in three independent experiments. For each PBP, the data were normalized against the intensity of the Bocillin FL labeling without piperacillin treatment (Fig. 2).
The PBP inhibition profiles with piperacillin were similar for the R6 strain and its ΔlytA derivative (Fig. 2A and B). At the piperacillin MIC (0.03 μg/ml), <20% of PBP2x remained reactive, whereas about 50% of PBP2b and PBP3 were so. The class A PBPs were nearly not impaired (>80% still reactive). The two strains with low-affinity PBP2b variants had similar profiles, regardless of their pbp2b allele (Fig. 2C and D). Both retained >75% of their PBP2b with a free active site after incubation with 0.12 μg/ml piperacillin, and PBP2b was nearly not affected at 0.03 and 0.06 μg/ml piperacillin, which are the MICs of the parental and transformed strains, respectively. In those strains, PBP2x showed the same inhibition profile as those of the parental strains. At 0.03 μg/ml piperacillin, i.e., below the MIC, most of PBP2x (>80%) was inhibited. Thus, <20% of the PBP2x present in the cell is sufficient to sustain growth and cell division. In these strains, the class A and C PBP inhibition profiles showed no variation compared to those of the parental strains.
Both strains with variants of PBP2x showed a lower affinity of PBP2x for piperacillin than those containing the R6 pbp2x allele (Fig. 2E and F). However, whereas PBP2x5 was nearly not inhibited even at the highest piperacillin concentration, the double mutation in PBP2xR caused only a modest decrease in the reactivity for piperacillin. In the case of PBP2xR, its inhibition profiles are very similar to those of PBP2b. Note that despite their differing inhibition profiles, the strains carrying pbp2xR and pbp2x5 have the same MIC for piperacillin (Table 2).
In the case of the strain carrying pbp2b5, the low affinity of PBP2b5 impaired Bocillin FL. Thus, we had to increase the concentration of Bocillin FL to allow a reaction with PBP2b5 during the preparation of the membrane samples. With this higher concentration of Bocillin FL, the SDS-PAGE profile gave a saturated fluorescent smear that was not interpretable. To avoid this, Bocillin FL-treated membranes of this strain were pelleted again and resuspended in fresh PBS, removing the free Bocillin FL responsible for the smear. However, some material was probably lost during this additional step, which explains why the PBP profile of this strain is less intense and the standard deviations are larger.
Kinetics of in vivo inhibition of PBPs by piperacillin.
A major caveat of the experiments presented above is that they monitor different parameters at different times. The profiles of PBP inhibition were recorded after 10 min of incubation with piperacillin, whereas the morphological consequences were observed after 1 or 2 h. MICs, on the other hand, were determined after inoculation in fresh medium containing the antibiotic. To verify that the observations reported above are significant, we performed time course experiments in which the growth, viability, morphology, and PBP inhibition profiles were determined at various times following the addition of piperacillin at different concentrations. The R6 ΔlytA and R6 ΔlytA pbp2x5 strains were examined, and the mean values from three independent experiments are shown in Fig. 5.
FIG 5.

Consequences of the exposure of strain R6 ΔlytA (A to E) and strain R6 ΔlytA pbp2x5 (F to J) to piperacillin (pip) at 0 (green), 1/2 (blue), 1 (red), 2 (purple), and 4 (orange) times the MIC. The means from three independent experiments are shown with standard deviations. (A and F) Turbidity measured after addition of piperacillin. (B and G) Viability after addition of piperacillin. (C and H) Morphology observed by phase-contrast microscopy after 1 h of further incubation without piperacillin. (D and I) Morphology after 1 h of incubation with piperacillin at the MIC. The white arrows point to lemon-shaped (D) or lentil-shaped (I) cells. (E and J) Quantification of PBP profiles. The bars represent the amount of a particular PBP that has reacted with Bocillin FL after incubation with piperacillin. The amounts are given relative to that of PBP2x (or PBP2x5) at time zero (set arbitrarily to 100). a.u., arbitrary units.
After 10 min at their respective piperacillin MICs, both strains continued to gain mass, as observed by the optical density of the culture (Fig. 5A and F), but they stopped multiplying (Fig. 5B and G). This result is consistent with the way the MIC was determined and its definition. At the lowest concentrations of piperacillin, PBP2x is the most inhibited PBP in the R6 ΔlytA strain, with PBP2b and PBP3 being impacted to a lesser extent (Fig. 5E). In the R6 ΔlytA pbp2x5 strain, PBP2x is not affected by piperacillin, whereas PBP2b and PBP3 remain inhibited (Fig. 5J). The very limited impact of the noninhibition of PBP2x5 on the MIC indicates that it is determined by the inhibition of PBP2b, which is consistent with the selection of PBP2b variants by piperacillin.
The viability of the R6 ΔlytA strain decreases after 1 h of incubation with piperacillin above the MIC (Fig. 5B). This loss of viability likely correlates with the morphological aberrations (lemon shape) arising from the inhibition of PBP2x (Fig. 5D). Indeed, in the R6 ΔlytA pbp2x5 strain, the viability decreases less sharply in the presence of piperacillin (Fig. 5G). Interestingly, with this strain, the morphological changes observed after 1 h at the MIC of piperacillin resemble those (chained “lentils”) brought by the depletion of PBP2b (Fig. 5I) (11), which is consistent with the inhibition of PBP2b and the noninhibition of PBP2x5.
On the other hand, when the R6 ΔlytA pbp2b5 strain was observed after 2 h of incubation with piperacillin at the MIC (0.6 μg/ml), cells were found in a variety of shapes, including a majority of lemon-shaped cells, as well as elongated and trilobed cells, resembling the effect of piperacillin on the susceptible R6 ΔlytA strain (see Fig. S5 in the supplemental material). In the absence of drug, both R6 ΔlytA pbp2x5 and R6 ΔlytA pbp2b5 showed normal morphologies.
It is remarkable that the amount of Bocillin FL-reactive PBPs decreases with time, with the exception of PBP2x, even in the absence of piperacillin. This is particularly the case for the class A PBP1a. It is possible that the amount of PBP enzymes is downregulated upon entry into the stationary phase, when the optical density at 600 nm is approaching 1.
In vitro activity of PBP2b5.
To check if the activity of PBP2b in strain R6 ΔlytA pbp2b5 was altered compared to that in the R6 PBP2b, the pbp2b5 gene encoding the full-length protein was cloned in a vector for expression with a C-terminal Strep tag, as described by Zapun et al. (26). PBP2b5 in R6 differs from PBP2b in strain 5204, which served as the sequence donor for the transformation, for two reasons. First, the gene fragment used lacked the sequence for the cytoplasmic and transmembrane segments. Second, despite the addition of a 3′ extension, homologous recombination can occur within the pbp2b gene. Thus, PBP2b5 differs from the R6 PBP2b at 29 positions, including the important T446A substitution (30). PBP2b5 also differs from the 5204 PBP2b at 27 positions in the C-terminal region. The difficulty or impossibility of incorporating the 3′ sequence of pbp2b from clinical strains in the R6 background has already been reported (30).
Recombinant PBP2b5 and R6 PBP2b were purified in parallel in the same way, as described before (26). An activity time course was performed using PBP2aS410A to provide the glycosyltransferase activity, with a mixture of 90% amidated lipid II and 10% nonamidated dansylated lipid II, and it was analyzed by SDS-PAGE. No difference in activity was detected in this manner (Fig. 6A). Note that the polymerization of the glycan chains by PBP2aS410A may have been limiting.
FIG 6.

Transpeptidase activity and reactivity of PBP2b with piperacillin in vitro. (A) Time course of peptidoglycan synthesis by R6 PBP2b or PBP2b5 with PBP2aS410A to provide glycosyltransferase activity. A mixture of 50 μM amidated lipid II and 5 μM nonamidated dansylated lipid II was incubated with 0.5 μM PBP2b and 1 μM PBP2aS410A at 30°C. Aliquots were withdrawn after various time intervals, and the reaction was stopped by adding moenomycin and penicillin G. The samples were analyzed by SDS-PAGE, and the dansyl fluorescence was imaged by UV transillumination. (B) Concentrations of 0.5 μM R6 PBP2b or 0.25 μM PBP2b5 were incubated with various concentrations of piperacillin for 10 min at room temperature prior to a reaction with 100 μM Bocillin FL. The samples were analyzed by SDS-PAGE fluorescence imaging and Coomassie blue staining.
As expected from the observation in vivo, PBP2b5 was less reactive with piperacillin than was the R6 PBP2b (Fig. 6B). After 10 min of incubation at room temperature, the R6 PBP2b was quantitatively titrated by piperacillin, whereas PBP2b5 was not, even with a 4-fold excess of the drug. Further doubling the concentration of piperacillin, however, resulted in the complete inhibition of PBP2b5.
When in vitro peptidoglycan synthesis reactions were set up to test the inhibition by or resistance to piperacillin, no difference was found between the R6 PBP2b and PBP2b5, with both enzymes being inhibited by piperacillin (not shown). The inhibition of the transpeptidase activity was observed even when the drug and protein concentrations were the same as those used to demonstrate that PBP2b5 has a lower reactivity toward piperacillin (0.25 μM PBP2b5 and 0.5 μM piperacillin; Fig. 6B). The reason for this discrepancy is likely due to the rates of the reactions. In vitro assembly of the peptidoglycan is very slow and inefficient with PBP2b under the experimental conditions (requiring typically an overnight incubation). It is likely that even at the very low near-stoichiometric concentration of piperacillin, PBP2b5 has time to be fully inhibited before significant transpeptidation has occurred.
DISCUSSION
PBP2x is inhibited by piperacillin to a greater extent than PBP2b, yet this β-lactam selects low-affinity PBP2b variants. The following explanation can be proposed to solve this paradox. Even a partial inhibition of PBP2b involved in elongation signals the arrest of growth (understood as the completion of cell cycles increasing the number of cells). In the meantime, the inhibition of PBP2x, which is more extensive, leads to the abortion of septation associated with the observed morphological aberrations resulting from piperacillin exposure. Note that there must be a range of low piperacillin concentrations in which the partial inhibition of PBP2b stops multiplication, whereas the near-complete inhibition of PBP2x is not sufficient to completely prevent cell division. Under such fine-line conditions, the incorporation of a low-affinity pbp2b allele is sufficient to maintain cell multiplication, which allowed us to select the strains harboring the PBP2bR or PBP2b5 variants. In support of this interpretation, the piperacillin MIC was nearly not affected by the presence of PBP2x variants that were not inhibited. At the MIC, this strain did not display the morphological defects associated with an inhibition of the septation, and yet the cells failed to multiply.
Note that PBP3, the d,d-carboxypeptidase PBP from S. pneumoniae, is inhibited by piperacillin to the same extent as PBP2b. As PBP3 is not essential and variants do not arise by β-lactam selection, we chose to ignore PBP3 in our simple interpretation. However, the deletion of pbp3 (dacA) causes morphological aberration and growth defects (35, 36). We cannot therefore discount a more complex explanation in which the effect of piperacillin on pneumococcal growth results from the combined synergistic inhibition of PBP2x, PBP2b, and PBP3. The primary selection of pbp2b variants might then be due to it being more prone to mutations or recombination.
Likewise, our explanation ignores a possible role of the class A PBPs. The absence of transpeptidase activity of PBP2x inhibited by piperacillin may be compensated by that of one of the class A PBPs. The activity of PBP2b may more specific and therefore cannot be rescued by a class A PBP.
It is somewhat surprising that even a partial inhibition of PBP2b by piperacillin stops multiplication, as it was shown that the depletion of PBP2b must be very extensive in order to achieve the same arrest (reference 11 and our unpublished data). Note that both types of experiments (β-lactam challenge and genetic depletion) were performed with R6 ΔlytA strains, so that cell lysis does not contribute to the different behavior. We propose here two mutually nonexclusive hypotheses to account for the large impact of PBP2b inhibition.
At the beginning of the cell cycle, peripheral peptidoglycan is inserted, with the participation of the transpeptidase activity of PBP2b. Septal synthesis is initiated using the transpeptidase activity of PBP2x. When splitting of the septal disc occurs, the peripheral machinery, which includes PBP2b, follows the inward moving circular junction between the peripheral and the septal wall. In this configuration, PBP2b and the peripheral machinery insert material into or onto fresh peptidoglycan assembled by the septal machinery and PBP2x. This model (Fig. 7) is consistent with the old measurements of cell wall dimensions (37) and the most recent data about the localization of the synthetic machineries and their activities (12, 14, 38). Upon mild piperacillin challenge, the transpeptidase activity of PBP2x is mostly inhibited, but septal material could still be assembled by class A PBPs, although with a lower degree of cross-linking. The low PBP2x activity could be tolerated, as long as sufficient PBP2b activity in the peripheral machinery consolidates the peptidoglycan at the splitting site to produce a viable cell wall. Thus, PBP2b would be under greater selection pressure by piperacillin than PBP2x, despite being less reactive with this drug.
FIG 7.
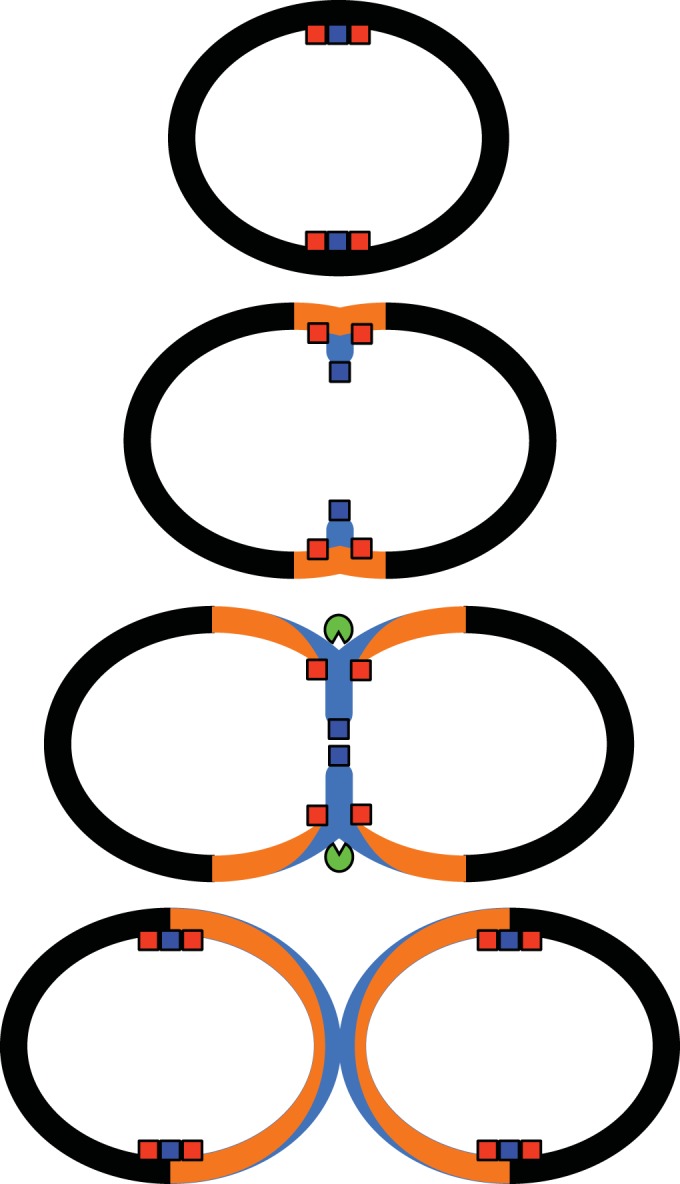
Model for the assembly of peptidoglycan in the pneumococcal cell wall. Peptidoglycan resulting from the septal machinery, which includes PBP2x (blue box), is shown in blue. Peptidoglycan resulting from the peripheral machinery, including PBP2b (red box), is in orange. The septum-splitting hydrolase activity is the green symbol. This model of peptidoglycan deposition can account for the greater required proportion of active PBP2b than PBP2x, since synthesis by the peripheral machinery can reinforce the material laid down by diminished septal machinery.
A second possible explanation may be related to the proportion of branched muropeptides incorporated in the peptidoglycan. In pneumococcal peptidoglycan, peptide stems can be linked either directly, with the third residue l-Lys connected to the fourth residue d-Ala of another peptide, or indirectly, with an Ala-Ala or Ser-Ala intervening dipeptide. The dipeptides are added in the cytoplasm on the precursor by the MurM and MurN enzymes (39). Upon the depletion of PBP2b, the fraction of cross-linked peptides containing a Ser-Ala dipeptide increases, and the deletion of murM increases the amount of necessary PBP2b (11). It is therefore possible that during the depletion of PBP2b by dilution in medium without an inducer, which requires several cell cycles, cells have time to adapt and build up a sufficient amount of branched muropeptides, whereas the effect of PBP2b inhibition by piperacillin is much more rapid and does not allow metabolic adaptation.
Some murM alleles are known to increase the resistance conferred by low-affinity PBPs and increase the proportion of branched muropeptides in the peptidoglycan (40). This appears to be due to the greater catalytic efficiency of the variant MurM (39). A prediction from the above hypothesis is that a murM allele that increases the pool of branched precursors would decrease the susceptibility to piperacillin. We attempted to transform several of our strains with the murM allele from the clinical strain 5204 but failed to select transformants.
β-Lactams that inhibit PBP2b, such as piperacillin, are known to trigger lysis, whereas those that do not react with PBP2b, such as cephalosporins, are nonlytic (41). Could this observation be related to the piperacillin paradox? Now that the functions of PBP2x and PBP2b in the septal and peripheral cell wall assembly, respectively, are firmly established (11, 12, 14), the different lytic responses can be rationalized. In the septal disc, new peptidoglycan can be added directly at the leading inner edge. In contrast, it is a topological necessity that insertion at any other site in order to produce an enlargement of the peptidoglycan sacculus requires cleavage of the preexisting peptidoglycan. Without the associated hydrolytic activity, the addition of new material would only result in thickening of the peptidoglycan layer. Thus, PBP2x can function without an associated hydrolase, whereas PBP2b works necessarily in concert with an unidentified hydrolase. When PBP2b is inhibited, the associated hydrolytic activity would not be compensated and lysis would ensue. Note, however, that lysis cannot be a direct effect, since the absence of the major autolysin LytA prevents lysis when PBP2b is inhibited. Instead, the action of the PBP2b-associated hydrolytic activity may be to sensitize the cell wall to the activity of LytA. Also, piperacillin selection of low-affinity PBP2b variants in a ΔlytA background demonstrates that it is not the susceptibility to lysis that limits cell multiplication. Instead, a change in the peptidoglycan properties (e.g., rigidity, elasticity, and shape) brought about by the imbalance between PBP2b and its cognate hydrolase may constitute the signal that stops growth.
As the morphological modifications resulting from piperacillin exposure resemble those occurring during PBP2x depletion, we conclude that the inhibition of PBP2x is driving these changes in shape. Cells adopted lemon or elongated bulging shapes, with the elongated shapes appearing in phase-contrast or differential interference microscopy as the juxtaposition of three beads, with the middle one often being the largest. Electron microscopy of negatively stained cryo-thin sections showed that the elongated bulging pneumococci can have three distinct anatomies: a single elongated cell with a swollen mid-cell, two distinct cells with an abnormally extended peptidoglycan septal cross-wall, or two cells separated by their respective plasma membranes but without a continuous peptidoglycan septum. We propose that these different morphologies arise from the arrest of septation by piperacillin at different stages of the cell cycle, while some peripheral cell wall building continues (Fig. 8). To explain the absence of cells with complete or partial septa in the presence of LytA, we propose that cells that were actively inserting peptidoglycan at the septum when piperacillin was added were lysed by the septal action of LytA. The PBPs responsible for the ongoing peripheral synthesis are unknown but might involve any of the three class A PBPs that are not affected by piperacillin and the residual activity of PBP2b, which is less affected than PBP2x.
FIG 8.

Model for the generation of abnormal morphologies following exposure to piperacillin. Depending on the stage of the cell cycle at the time of piperacillin inhibition (top row), the ongoing insertion of new cell wall material (light gray) generates the abnormal bulging shapes shown on the lower row. The plasma membrane is shown in dark gray.
The observation of pneumococcal cells separated by continuous membranes without a cross-wall suggests that constriction of the FtsZ ring and the membrane invagination and fission can proceed without simultaneous peptidoglycan septal growth. Note that a delay between the constriction and relocalization of FtsZ and that of PBP2x has been observed during the normal cell cycle of S. pneumoniae (14, 28). The piperacillin inhibition of PBP2x highlights this decoupling.
The examination of the localization of GFP-coupled FtsZ, PBP2x, and PBP2b shows that the inhibition of PBPs by piperacillin does not affect the localization of the morphogenetic machineries. It is particularly notable that FtsZ remains localized at the mid-cell of bulging cells. Time-lapse microscopy showed that the FtsZ ring enlarges with the bulge. This observation supports the idea that the localization of the FtsZ ring in ovococci occurs at the position with the largest diameter. Although no time-lapse data were obtained with GFP-fused PBPs, the localization of GFP-PBP2x and GFP-PBP2b indicates that the PBPs also localize at the largest cell diameter.
The observations in this report reveal the complexity of the response to β-lactam challenge. Most importantly, the PBP most inhibited by a particular β-lactam, even though the resulting morphology is consistent with the inactivation of this PBP, may not be the essential enzyme that determines growth arrest.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Breukink for the generous gift of lipid II, F. Couppey for initiating the work, and G. Cerardi for technical support in the construction of strain sspCM99. We thank R.-L. Revel-Goyet, F. Lacroix, and J.-P. Kleman (Institut de Biologie Structurale, Grenoble, France) for support and access to the microscopy platform for time-lapse acquisitions and C. Morisco and G. Schoehn for support and access to the electron microscopy platform.
J. Philippe was supported by a grant from la Région Rhône-Alpes. The work of R. Hakenbeck and D. Denapaite was supported by a grant from the Deutsche Forschungsgemeinschaft, Ha 1011/11-1. This work was partly funded by the COOPOL Innovation France-Chinese program and by the ANR (grant ANR-2011-BSV5-012-01 NOBLEACH), and it used platforms of the Grenoble Instruct Centre (ISBG; grant UMS3518 CNRS-CEA-UJF-EMBL) with support from the FRISBI (grant ANR-10-INSB-05-02) and GRAL (grant ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04283-14.
REFERENCES
- 1.Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, Bruin J, Montijn R, Bonten M, Sanders E. 2011. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 6:e17035. doi: 10.1371/journal.pone.0017035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansman D, Bullen MM. 1967. A resistant pneumococcus. Lancet 290:264–265. [DOI] [PubMed] [Google Scholar]
- 3.Kang CI, Baek JY, Jeon K, Kim SH, Chung DR, Peck KR, Lee NY, Song JH. 2012. Bacteremic pneumonia caused by extensively drug-resistant Streptococcus pneumoniae. J Clin Microbiol 50:4175–4177. doi: 10.1128/JCM.01642-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henriques-Normak B. 2007. Molecular epidemiology and mechanisms for antibiotic resistance in Streptococcus pneumoniae, p 269–290. In Hakenbeck R, Chhatwal S (ed), Molecular biology of streptococci. Horizon Scientific Press, Wymondham, United Kingdom. [Google Scholar]
- 5.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 6.Massidda O, Nováková L, Vollmer W. 2013. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol 15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- 7.Philippe J, Vernet T, Zapun A. 2014. The elongation of ovococci. Microb Drug Resist 20:215–221. doi: 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 9.Hoskins J, Matsushima P, Mullen DL, Tang J, Zhao G, Meier TI, Nicas TI, Jaskunas SR. 1999. Gene disruption studies of penicillin-binding proteins 1a, 1b, and 2a in Streptococcus pneumoniae. J Bacteriol 181:6552–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Severin A, Schuster C, Hakenbeck R, Tomasz A. 1992. Altered murein composition in a dd-carboxypeptidase mutant of Streptococcus pneumoniae. J Bacteriol 174:5152–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berg KH, Stamsås GA, Straume D, Håvarstein LS. 2013. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol 195:4342–4354. doi: 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. 2013. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol 90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. 2014. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Mol Microbiol 92:733–755. doi: 10.1111/mmi.12588. [DOI] [PubMed] [Google Scholar]
- 14.Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. 2014. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Mol Microbiol 94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 16.Hakenbeck R, Brückner R, Denapaite D, Maurer P. 2012. Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol 7:395–410. doi: 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- 17.Williamson R, Hakenbeck R, Tomasz A. 1980. In vivo interaction of beta-lactam antibiotics with the penicillin-binding proteins of Streptococcus pneumoniae. Antimicrob Agents Chemother 18:629–637. doi: 10.1128/AAC.18.4.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson DM, Biedenbach DJ, Jones RN. 2002. Potency and antimicrobial spectrum update for piperacillin/tazobactam (2000): emphasis on its activity against resistant organism populations and generally untested species causing community-acquired respiratory tract infections. Diagn Microbiol Infect Dis 43:49–60. doi: 10.1016/S0732-8893(02)00358-9. [DOI] [PubMed] [Google Scholar]
- 19.Laible G, Hakenbeck R. 1987. Penicillin-binding proteins in beta-lactam-resistant laboratory mutants of Streptococcus pneumoniae. Mol Microbiol 1:355–363. doi: 10.1111/j.1365-2958.1987.tb01942.x. [DOI] [PubMed] [Google Scholar]
- 20.Grebe T, Hakenbeck R. 1996. Penicillin-binding proteins 2b and 2x of Streptococcus pneumoniae are primary resistance determinants for different classes of beta-lactam antibiotics. Antimicrob Agents Chemother 40:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hakenbeck R, Martin C, Dowson C, Grebe T. 1994. Penicillin-binding protein 2b of Streptococcus pneumoniae in piperacillin-resistant laboratory mutants. J Bacteriol 176:5574–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacks S, Hotchkiss RD. 1960. A study of the genetic material determining an enzyme in pneumococcus. Biochim Biophys Acta 39:508–518. doi: 10.1016/0006-3002(60)90205-5. [DOI] [PubMed] [Google Scholar]
- 23.Fadda D, Pischedda C, Caldara F, Whalen MB, Anderluzzi D, Domenici E, Massidda O. 2003. Characterization of divIVA and other genes located in the chromosomal region downstream of the dcw cluster in Streptococcus pneumoniae. J Bacteriol 185:6209–6214. doi: 10.1128/JB.185.20.6209-6214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison DA. 1997. Streptococcal competence for genetic transformation: regulation by peptide pheromones. Microb Drug Resist 3:27–37. doi: 10.1089/mdr.1997.3.27. [DOI] [PubMed] [Google Scholar]
- 25.de Jong IG, Beilharz K, Kuipers OP, Veening JW. 2011. Live cell imaging of Bacillus subtilis and Streptococcus pneumoniae using automated time-lapse microscopy. J Vis Exp 2011(53):e3145. doi: 10.3791/3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zapun A, Philippe J, Abrahams KA, Signor L, Roper DI, Breukink E, Vernet T. 2013. In vitro reconstitution of peptidoglycan assembly from the Gram-positive pathogen Streptococcus pneumoniae. ACS Chem Biol 8:2688–2696. doi: 10.1021/cb400575t. [DOI] [PubMed] [Google Scholar]
- 27.Lutkenhaus J, Pichoff S, Du S. 2012. Bacterial cytokinesis: from Z ring to divisome. Cytoskeleton (Hoboken) 69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morlot C, Zapun A, Dideberg O, Vernet T. 2003. Growth and division of Streptococcus pneumoniae: localization of the high molecular weight penicillin-binding proteins during the cell cycle. Mol Microbiol 50:845–855. doi: 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- 29.Zapun A, Vernet T, Pinho MG. 2008. The different shapes of cocci. FEMS Microbiol Rev 32:345–360. doi: 10.1111/j.1574-6976.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- 30.Pagliero E, Chesnel L, Hopkins J, Croize J, Dideberg O, Vernet T, Di Guilmi AM. 2004. Biochemical characterization of Streptococcus pneumoniae penicillin-binding protein 2b and its implication in beta-lactam resistance. Antimicrob Agents Chemother 48:1848–1855. doi: 10.1128/AAC.48.5.1848-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chesnel L, Carapito R, Croizé J, Dideberg O, Vernet T, Zapun A. 2005. Identical penicillin-binding domains in penicillin-binding proteins of Streptococcus pneumoniae clinical isolates with different levels of beta-lactam resistance. Antimicrob Agents Chemother 49:2895–2902. doi: 10.1128/AAC.49.7.2895-2902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Contreras-Martel C, Dahout-Gonzalez C, Martins Ados S, Kotnik M, Dessen A. 2009. PBP active site flexibility as the key mechanism for beta-lactam resistance in pneumococci. J Mol Biol 387:899–909. doi: 10.1016/j.jmb.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 33.Chesnel L, Pernot L, Lemaire D, Champelovier D, Croizé J, Dideberg O, Vernet T, Zapun A. 2003. The structural modifications induced by the M339F substitution in PBP2x from Streptococcus pneumoniae further decreases the susceptibility to beta-lactams of resistant strains. J Biol Chem 278:44448–44456. doi: 10.1074/jbc.M305948200. [DOI] [PubMed] [Google Scholar]
- 34.Carapito R, Chesnel L, Vernet T, Zapun A. 2006. Pneumococcal beta-lactam resistance due to a conformational change in penicillin-binding protein 2x. J Biol Chem 281:1771–1777. doi: 10.1074/jbc.M511506200. [DOI] [PubMed] [Google Scholar]
- 35.Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol 191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuster C, Dobrinski B, Hakenbeck R. 1990. Unusual septum formation in Streptococcus pneumoniae mutants with an alteration in the d,d-carboxypeptidase penicillin-binding protein 3. J Bacteriol 172:6499–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higgins ML, Shockman GD. 1976. Study of cycle of cell wall assembly in Streptococcus faecalis by three-dimensional reconstructions of thin sections of cells. J Bacteriol 127:1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wheeler R, Mesnage S, Boneca IG, Hobbs JK, Foster SJ. 2011. Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Mol Microbiol 82:1096–1109. doi: 10.1111/j.1365-2958.2011.07871.x. [DOI] [PubMed] [Google Scholar]
- 39.Lloyd AJ, Gilbey AM, Blewett AM, De Pascale G, El Zoeiby A, Levesque RC, Catherwood AC, Tomasz A, Bugg TD, Roper DI, Dowson CG. 2008. Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J Biol Chem 283:6402–6417. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]
- 40.Filipe SR, Tomasz A. 2000. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc Natl Acad Sci U S A 97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hakenbeck R, Tornette S, Adkinson NF. 1987. Interaction of non-lytic beta-lactams with penicillin-binding proteins in Streptococcus pneumoniae. J Gen Microbiol 133:755–760. [DOI] [PubMed] [Google Scholar]
- 42.Hoskins J, Alborn WE Jr, Arnold J, Blaszczak LC, Burgett S, DeHoff BS, Estrem ST, Fritz L, Fu DJ, Fuller W, Geringer C, Gilmour R, Glass JS, Khoja H, Kraft AR, Lagace RE, LeBlanc DJ, Lee LN, Lefkowitz EJ, Lu J, Matsushima P, McAhren SM, McHenney M, McLeaster K, Mundy CW, Nicas TI, Norris FH, O'Gara M, Peery RB, Robertson GT, Rockey P, Sun PM, Winkler ME, Yang Y, Young-Bellido M, Zhao G, Zook CA, Baltz RH, Jaskunas SR, Rosteck PR Jr, Skatrud PL, Glass JI. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J Bacteriol 183:5709–5717. doi: 10.1128/JB.183.19.5709-5717.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pagliero E, Dublet B, Frehel C, Dideberg O, Venet T, Am DG. 2008. The inactivation of a new peptidoglycan hydrolase Pmp23 leads to abnormal septum formation in Streptococcus pneumoniae. Open Microbiol J 2:107–114. doi: 10.2174/1874285800802010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eberhardt A, Wu LJ, Errington J, Vollmer W, Veening JW. 2009. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol Microbiol 74:395–408. doi: 10.1111/j.1365-2958.2009.06872.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.