

Abstract
Atovaquone is a component of Malarone, a widely prescribed antimalarial combination, that targets malaria respiration. Here we show that parasites with high-level resistance to an inhibitor of dihydroorotate dehydrogenase demonstrate unexpected atovaquone tolerance. Fortunately, the tolerance is diminished with proguanil, the second partner in Malarone. It is important to understand such “genetic cross talk” between respiration and pyrimidine biosynthesis since many antimalarial drug development programs target these two seemingly independent pathways.
TEXT
Malarone is an antimalarial drug combination, often prescribed for travelers as a prophylactic. but its high cost so far has limited its general use in countries where malaria is endemic. The patent for this drug expired in 2013, and its global usage could soon surge. Atovaquone and proguanil are the synergistic pair of antimalarials that make up this effective, well-tolerated cocktail. While the mechanism of action for proguanil remains uncertain (1, 2), it is well know that atovaquone targets the cytochrome bc1 complex of the Plasmodium falciparum respiratory chain (3, 4). One function of this mitochondrial pathway is to generate a proton gradient that is required for maintaining the membrane potential for important processes such as protein synthesis, heme biogenesis, and the citric acid cycle (reviewed in reference 2). Importantly, the P. falciparum cytochrome bc1 complex has been implicated in the direct regeneration of oxidized ubiquinone for dihydroorotate dehydrogenase (DHODH), a key enzyme in the biosynthesis of pyrimidine nucleotides (5–7). Thus, DHODH is physically and functionally tied to the respiratory chain.
Recently, P. falciparum parasites resistant to DSM1, a potent inhibitor of DHODH (8, 9), were generated at two levels (333 nM for round 1 and 3 or 10 μM for round 2) (10). Amplification of the genome segments that encompassed the DHODH gene were responsible for the observed phenotype. Whole-genome sequence methods ruled out resistance-conferring mutations elsewhere, and no alterations in the sensitivities to several unrelated antimalarials were detected. However, here we report the unexpected development of tolerance to atovaquone in parasites resistant to higher levels of the DHODH inhibitor DSM1 (round 2).
Following DSM1 selection and subcloning (10), atovaquone sensitivity was tested. Round 1 parasites (∼3-fold DSM1 resistance) exhibited wild-type sensitivity to atovaquone in a 72-h assay (Fig. 1A and Table 1). Long-term exposure to low levels of atovaquone confirmed the sensitivity of these parasites: 10 nM atovaquone completely prevented the proliferation of parental Dd2, as well as round 1 parasites within 2 days, and continued to do so over an extended period of 8 days (Fig. 1C and D, red lines). We further assessed the survival of Dd2 and round 1 clone C by exposing small clonal populations of parasites to atovaquone before extensive washing and replating in the absence of drug and following the number of viable colonies over 60 days (11). In this clonal viability assay, with the exception of a slightly increased survival of clone C at the lowest exposure level (24 h, 10 nM), we observed the full cidal activity of atovaquone on these parasite clones (average survival of 0% compared with that of untreated controls) (Table 2).
FIG 1.
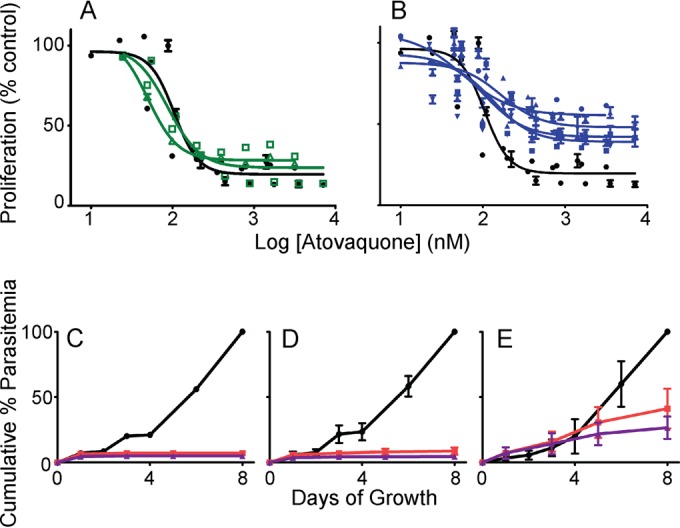
Atovaquone tolerance in high-level DSM1-resistant parasites. (A and B) SYBR Green-based dose responses of various P. falciparum clones to atovaquone are shown in the absence of DSM1 (combined from 3 independent experiments). (A) DSM1-sensitive clonal Dd2 (black filled circles) compared with partial DSM1-resistant round 1 clones (C, green open squares; and D, green open triangles) (10). (B) DSM1-sensitive clonal Dd2 (black filled circle) compared with high-level DSM1-resistant round 2 clones (C53-1, blue squares; C710-1b, blue circles; C710-2a, blue inverted triangles; and D73-1, blue triangles) (10). The decrease in proliferation in the presence of atovaquone is calculated as a percentage of activity from dimethyl sulfoxide (DMSO) controls. The lines on the plots show the nonlinear curve fits of data points from individual clones. EC50s, where measurable, are listed in Table 1. (C to E) Growth of different P. falciparum clones in the presence of continuous levels of 0 (black), 10 (red), or 100 nM (purple) atovaquone over 8 days (see Table 1 for the respective DSM1 concentrations). (C) Clonal Dd2 (single values). (D) Partially DSM1-resistant round 1 clones (means of clone C and clone D with standard deviations). (E) Highly DSM1-resistant round 2 clones (means of C53-1, C710-1b, C710-2a, and D73-1 clones with standard deviations). Cumulative % parasitemia was calculated by normalizing parasitemia values (measured using SYBR Green-based flow cytometry method) to the starting parasitemia and calculating the percentage of the maximum value achieved over 8 days.
TABLE 1.
EC50s of DSM1-resistant clones for various antimalarialsa
| Antimalarial(s) | Method | Dd2 | EC50s (± confidence interval) for: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Round 1 |
Round 2 |
||||||||||
| C | D | C53-1 | D53-1 | D73-1 | C710-1a | C710-1b | C710-2a | C710-2b | |||
| DSM1 selection (μM) | 0 | 0.3 | 0.3 | 3 | 3 | 3 | 10 | 10 | 10 | 10 | |
| Atovaquone (nM) | SYBR | 1.2 (0.2) | 1.0 (0.4) | 0.5 (0.4) | Tolb | —c | Tol | — | Tol | Tol | — |
| Atovaquone (nM) + 1 μM proguanile | SYBR | 0.3 (0.1) | — | — | — | — | 0.6 (0.5) | — | 0.2 (NDd) | 0.4 (0) | — |
| Myxothiazol (μM)f | Hypo | 0.3 (0.1) | 0.2 (0.1) | 0.5 (0.3) | 1.5 (1.1) | — | 0.4 (0.6) | — | 1.1 (0.7) | 0.7 (0.7) | — |
| Antimycin A (μM)f | Hypo | 0.4 (0.07) | 0.2 (0.1) | 0.5 (0.4) | 1.1 (1.4) | — | 0.3 (0.6) | — | 0.9 (0.4) | 0.6 (0.7) | — |
| DSM1 (μM)g | SYBR | 0.1 (0.02) | 0.4 (0.04) | 0.3 (0.04) | — | — | 11.1 (6.2) | — | 4.7 (2.3) | 5.8 (1.6) | |
| Atovaquone (nM) | Hypo | 5.9 (0.3) | 8.9 (1.1) | 4.6 (0.3) | Tol | Tol | — | Tol | — | — | Tol |
| Proguanil (μM) | Hypo | 14.1 (3.6) | 14.8 (3.6) | 14.8 (3.4) | 10.3 (4.4) | 7.0 (2.1) | — | 10.3 (2.7) | — | — | 17.6 (3.9) |
| Artemisinin (nM) | SYBR | 12.5 (ND) | 8.3 (0.8) | 7.4 (1.0) | 12.5 (ND) | — | 4.7 (0.5) | — | 5.0 (ND) | 5.4 (0.4) | — |
| Artemisinin (nM) | Hypo | 5.2 (0.6) | 5.3 (1.3) | 5.2 (1.3) | 7.5 (2.1) | — | 6.6 (3.2) | — | 6.4 (0.8) | 6.2 (6.1) | — |
The development of infected erythrocytes exposed to antimalarials was measured in triplicate by flow cytometry using SYBR green fluorescence (SYBR) or by hypoxanthine uptake assays (Hypo). EC50s were calculated as previously described (10). Values listed are from a single representative experiment.
Tolerance (Tol) is assigned when the EC50 could not be determined due to incomplete inhibition, but parasites grew at attenuated rates (see Fig. 1B and E).
—, experiment not performed.
ND, could not be determined.
We have also performed this experiment with 100 and 500 nM proguanil. Similar to what we observed for 1 μM proguanil (included in the table), the lower nM proguanil concentrations shift the EC50s of atovaquone to Dd2 levels (our unpublished observation).
Dose responses of various P. falciparum clones to these two compounds are presented in Fig. 2.
DSM1 EC50s were also measured by the hypoxanthine uptake assay and published in reference 10.
TABLE 2.
Survival of DSM1-resistant cell lines after exposure to lethal atovaquone concentrationsa
| Condition | Average % survivalb (range of days to detectionc) of clonal population: |
|||
|---|---|---|---|---|
| Dd2 | C12 | C53-1 | C710-2a | |
| 1-day exposure, 10 nM ATOd | 0 (NAe) | 14 (16–18) | 86 (9–13) | 88 (11–13) |
| 5-day exposure, 10 nM ATO | 0 (NA) | 0 | 29 (21) | 63 (16–18) |
| 1-day exposure, 100 nM ATO | 0 (NA) | 0 | 57 (25–28) | 38 (23–25) |
| 5-day exposure, 100 nM ATO | 0 (NA) | 0 | 14 (34) | 25 (30–37) |
This method is derived from our earlier work (11). Starting from freshly thawed parasites of low parasitemia, serial dilutions were performed to isolate clonal populations. Triplicate T25 flasks were seeded with 10 asynchronous infected erythrocytes and incubated for 1 or 5 days with solvent only (control) or 10 nM or 100 nM atovaquone (tests). Each culture was treated at approximately the same time, and at the end of incubation, the erythrocyte pellets were washed three times with complete medium and transferred to 24 wells of a flat-bottom 96-well plate. The plated cultures were grown in inhibitor-free medium under standard culture conditions with three medium changes per week out to 60 days. Viable populations were identified using flow cytometry-detected SYBR green fluorescence after each medium change.
Average % survival = 100 × (average positive wells [test] ÷ average positive wells [control]).
Recovered parasites were observable around the same time as the solvent-only controls (our unpublished data).
ATO, atovaquone.
NA, not applicable.
In contrast, 3 or 10 μM level resistance to DSM1 (round 2, ∼20- to 50-fold resistance) (Table 1) conferred atovaquone “tolerance” (Fig. 1B and Table 1). This phenotype is not like the full resistance seen in traditional studies (12–14) because proliferation stalls at ∼50%, and parasites persist at a low level even at atovaquone concentrations as high as 10 μM in a 72-h study (as measured by SYBR Green DNA labeling or by hypoxanthine uptake assays) (Table 1 and Fig. 1B). During longer exposures, unlike parental Dd2 clones and round 1 low-level DSM1-resistant clones, these round 2 parasites were not inhibited and continued to proliferate in the presence of 10 nM or 100 nM atovaquone. After 5 days, parasite growth slowed, but viable parasites continued to persist in culture even longer (Fig. 1E). When the relative growth is quantified from the later part of this experiment (from 3 to 8 days), round 2 DSM1-resistant clones continued to increase by ∼40% in the presence of 10 nM atovaquone, while parental Dd2 and round 1 clones did not proliferate at all. Further supporting the atovaquone tolerance phenotype in round 2 clones, clonal viability assays on two round 2 clones displayed marked increases in survival under all conditions (average survival of up to ∼90% compared with that of untreated controls) (Table 2).
Atovaquone tolerance in round 2 DSM1-resistant parasites is diminished following the addition of the synergistic partner drug proguanil: the 50% effective concentration (EC50) of round 2 clones shifted to Dd2 levels in a 72-h assay (Table 1). In this way, the tolerance phenotype is similar to that in previous reports of atovaquone resistance (5, 13, 15). Occasionally, proliferation of round 2 clones did not disappear completely in these assays, indicating that viable parasites may still be present (our unpublished observation). Interestingly, tolerance was also observed when round 2 parasites were exposed to other cytochrome bc1 complex inhibitors such as myxothiazol and antimycin A (Fig. 2 and Table 1). These two inhibitors target different sites of the enzyme, and, therefore, cross-tolerance indicates that there may be a global perturbation of the complex that alters the binding of both inhibitors (3, 16).
FIG 2.
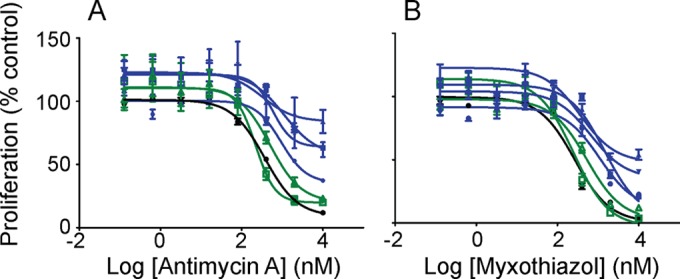
Tolerance to alternative cytochrome bc1 inhibitors in high-level DSM1-resistant parasites. Hypoxanthine uptake dose response of various P. falciparum clones to antimycin A (A) or myxothiazol (B) in the absence of DSM1. DSM1-sensitive clonal Dd2 (black filled circle) compared to partial DSM1-resistant round 1 clones (C, green open square; and D, green open triangle) and high-level DSM1-resistant round 2 clones (C53-1, blue square; C710-1b, blue circle; C710-2a, blue inverted triangle; and D73-1, blue triangle). Proliferation is calculated as a percentage of activity from dimethyl sulfoxide (DMSO) controls. Lines on the plots show the nonlinear curve fits of the data points from individual clones. EC50s are listed in Table 1.
At first it is puzzling to understand how high-level resistance to a DHODH inhibitor (DSM1) confers tolerance to inhibitors of the cytochrome bc1 complex. The present phenomenon is clearly different from traditional atovaquone resistance. While mutations in the mitochondrially encoded cytochrome b gene are commonly observed in parasites that are resistant to antimalarials that target the cytochrome bc1 complex (3, 17–22), such parasites remain susceptible to DHODH inhibitors (23). Furthermore, whole-genome sequencing of round 1 and round 2 DSM1-resistant parasites failed to show causal mutations in the cytochrome bc1 complex or anywhere in the nuclear or mitochondrial genome (10). This was also the case in a previous study where parasites developed resistance against a broad range of cytochrome bc1 complex inhibitors; the genetic cause of this resistance was not identified (13). Since it had been proposed that the sole function of the respiratory chain was to regenerate ubiquinone for DHODH (5), the authors had speculated that DHODH had been uncoupled from the electron transport chain through an alternate source of ubiquinone oxidation. Despite the potential differences between atovaquone resistance and tolerance, a cytochrome bc1“bypass” could also explain the current observations made in DSM1-resistant clones. Fumarate reductase activity, for example, had previously been proposed as an alternate way to regenerate ubiquinone for the use of DHODH, but this has not been biochemically investigated (24).
An alternative theory to explain atovaquone tolerance might involve some contribution of the neighboring upregulated genes besides DHODH. In the DSM1-selected parasites, round 2 resistance always involved high-level amplification of at least 35 kb of sequence surrounding the DHODH gene on chromosome 6. Intriguingly, the smallest conserved amplicon (8 genes) includes PlasmoDB identifier no. PF3D7_0603200, which is annotated as a putative mitochondrial chaperone BCS-1. In both bacteria and mitochondria, this gene product is required for the translocation of an essential subunit of the cytochrome bc1 complex from the mitochondrial matrix to the intermembrane space (25). This protein has not yet been investigated in P. falciparum, but it is possible that amplification of the BSC-1 gene, along with DHODH, might confer enhanced functions to the complex and somehow increase atovaquone tolerance.
There are several antimalarials in the drug development pipeline that target pyrimidine biosynthesis (23, 26), and ones that target the respiratory chain (17, 27–32). Some of the new cytochrome bc1 inhibitors are effective against atovaquone-resistant parasites (33), but it would be of interest to determine if they also participate in the cross talk described here. Since we routinely observe cross-resistance between DHODH inhibitors (our unpublished observations), the present studies raise important questions about the mechanisms of action and of cross-resistance directed at mitochondrial functions among antimalarials. This alert must be tempered by the fact that atovaquone tolerance is only seen at very high-level amplifications of the DHODH locus for cell lines that had undergone two sequential rounds of selection and that this tolerance is lessened with the addition of proguanil.
ACKNOWLEDGMENTS
We thank Ali Guler for help with statistical analysis and Sreekanth Kokonda for DSM1 synthesis.
P.K.R. acknowledges support from the U.S. NIH NIAID for the study of mutagenesis in malaria parasites (grants AI089688 and AI099280). The development of DHODH inhibitors is supported by NIH grant AI103947 (to M.A.P. and P.K.R.).
REFERENCES
- 1.Srivastava IK, Vaidya AB. 1999. A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob Agents Chemother 43:1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaidya AB, Mather MW. 2009. Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol 63:249–267. doi: 10.1146/annurev.micro.091208.073424. [DOI] [PubMed] [Google Scholar]
- 3.Fisher N, Abd Majid R, Antoine T, Al-Helal M, Warman AJ, Johnson DJ, Lawrenson AS, Ranson H, O'Neill PM, Ward SA, Biagini GA. 2012. Cytochrome b mutation Y268S conferring atovaquone resistance phenotype in malaria parasite results in reduced parasite bc1 catalytic turnover and protein expression. J Biol Chem 287:9731–9741. doi: 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fry M, Pudney M. 1992. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566c80). Biochem Pharmacol 43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 5.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. 2007. Specific role of mitochondrial electron transport in blood-stage plasmodium falciparum. Nature 446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 6.Rawls J, Knecht W, Diekert K, Lill R, Loffler M. 2000. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur J Biochem 267:2079–2087. doi: 10.1046/j.1432-1327.2000.01213.x. [DOI] [PubMed] [Google Scholar]
- 7.Malmquist NA, Gujjar R, Rathod PK, Phillips MA. 2008. Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry 47:2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. 2008. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J Med Chem 51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA. 2009. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem 284:26999–27009. doi: 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guler JL, Freeman DL, Ahyong V, Patrapuvich R, White J, Gujjar R, Phillips MA, DeRisi J, Rathod PK. 2013. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog 9:e1003375. doi: 10.1371/journal.ppat.1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young RD, Rathod PK. 1993. Clonal viability measurements on Plasmodium falciparum to assess in vitro schizonticidal activity of leupeptin, chloroquine, and 5-fluoroorotate. Antimicrob Agents Chemother 37:1102–1107. doi: 10.1128/AAC.37.5.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gassis S, Rathod P. 1996. Frequency of drug resistance in Plasmodium falciparum: a nonsynergistic combination of 5-fluoroorotate and atovaquone suppresses in vitro resistance. Antimicrob Agents Chemother 40:914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smilkstein MJ, Forquer I, Kanazawa A, Kelly JX, Winter RW, Hinrichs DJ, Kramer DM, Riscoe MK. 2008. A drug-selected Plasmodium falciparum lacking the need for conventional electron transport. Mol Biochem Parasitol 159:64–68. doi: 10.1016/j.molbiopara.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke H, Morrisey JM, Ganesan SM, Painter HJ, Mather MW, Vaidya AB. 2011. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot Cell 10:1053–1061. doi: 10.1128/EC.05049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srivastava IK, Rottenberg H, Vaidya AB. 1997. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272:3961–3966. doi: 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- 16.Covian R, Trumpower BL. 2006. Regulatory interactions between ubiquinol oxidation and ubiquinone reduction sites in the dimeric cytochrome bc1 complex. J Biol Chem 281:30925–30932. doi: 10.1074/jbc.M604694200. [DOI] [PubMed] [Google Scholar]
- 17.Nam TG, McNamara CW, Bopp S, Dharia NV, Meister S, Bonamy GM, Plouffe DM, Kato N, McCormack S, Bursulaya B, Ke H, Vaidya AB, Schultz PG, Winzeler EA. 2011. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol 6:1214–1222. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musset L, Bouchaud O, Matheron S, Massias L, Le Bras J. 2006. Clinical atovaquone-proguanil resistance of Plasmodium falciparum associated with cytochrome b codon 268 mutations. Microbes Infect 8:2599–2604. doi: 10.1016/j.micinf.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Berry A, Senescau A, Lelievre J, Benoit-Vical F, Fabre R, Marchou B, Magnaval JF. 2006. Prevalence of Plasmodium falciparum cytochrome b gene mutations in isolates imported from Africa, and implications for atovaquone resistance. Trans R Soc Trop Med Hyg 100:986–988. doi: 10.1016/j.trstmh.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Fivelman QL, Butcher GA, Adagu IS, Warhurst DC, Pasvol G. 2002. Malarone treatment failure and in vitro confirmation of resistance of Plasmodium falciparum isolate from Lagos, Nigeria. Malar J 1:1. doi: 10.1186/1475-2875-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz E, Bujanover S, Kain KC. 2003. Genetic confirmation of atovaquone-proguanil-resistant Plasmodium falciparum malaria acquired by a nonimmune traveler to East Africa. Clin Infect Dis 37:450–451. doi: 10.1086/375599. [DOI] [PubMed] [Google Scholar]
- 23.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. 2011. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem 54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher N, Bray PG, Ward SA, Biagini GA. 2008. Malaria-parasite mitochondrial dehydrogenases as drug targets: too early to write the obituary. Trends Parasitol 24:9–10. doi: 10.1016/j.pt.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Wagener N, Ackermann M, Funes S, Neupert W. 2011. A pathway of protein translocation in mitochondria mediated by the AAA-ATPase Bcs1. Mol Cell 44:191–202. doi: 10.1016/j.molcel.2011.07.036. [DOI] [PubMed] [Google Scholar]
- 26.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. 2011. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem 54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bueno JM, Herreros E, Angulo-Barturen I, Ferrer S, Fiandor JM, Gamo FJ, Gargallo-Viola D, Derimanov G. 2012. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med Chem 4:2311–2323. doi: 10.4155/fmc.12.177. [DOI] [PubMed] [Google Scholar]
- 28.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Bagby GC, Rathbun RK, Levin JI, Hinrichs D, Riscoe MK. 2006. Evaluation and lead optimization of anti-malarial acridones. Exp Parasitol 114:47–56. doi: 10.1016/j.exppara.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Cross RM, Maignan JR, Mutka TS, Luong L, Sargent J, Kyle DE, Manetsch R. 2011. Optimization of 1,2,3,4-tetrahydroacridin-9(10H)-ones as antimalarials utilizing structure-activity and structure-property relationships. J Med Chem 54:4399–4426. doi: 10.1021/jm200015a. [DOI] [PubMed] [Google Scholar]
- 30.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, Morrisey JM, Steuten J, Mutka T, Li Y, Wirjanata G, Ryan E, Duffy S, Kelly JX, Sebayang BF, Zeeman AM, Noviyanti R, Sinden RE, Kocken CH, Price RN, Avery VM, Angulo-Barturen I, Jimenez-Diaz MB, Ferrer S, Herreros E, Sanz LM, Gamo FJ, Bathurst I, Burrows JN, Siegl P, Guy RK, Winter RW, Vaidya AB, Charman SA, Kyle DE, Manetsch R, Riscoe MK. 2013. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 5:177ra37. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuck DC, Ferreira SB, Cruz LN, da Rocha DR, Moraes M, Nakabashi M, Rosenthal PJ, Ferreira VF, Garcia CR. 2013. Biological evaluation of hydroxynaphthoquinones as anti-malarials. Malar J 12:234. doi: 10.1186/1475-2875-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biagini GA, Fisher N, Shone AE, Mubaraki MA, Srivastava A, Hill A, Antoine T, Warman AJ, Davies J, Pidathala C, Amewu RK, Leung SC, Sharma R, Gibbons P, Hong DW, Pacorel B, Lawrenson AS, Charoensutthivarakul S, Taylor L, Berger O, Mbekeani A, Stocks PA, Nixon GL, Chadwick J, Hemingway J, Delves MJ, Sinden RE, Zeeman AM, Kocken CH, Berry NG, O'Neill PM, Ward SA. 2012. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc Natl Acad Sci U S A 109:8298–8303. doi: 10.1073/pnas.1205651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barton V, Fisher N, Biagini GA, Ward SA, O'Neill PM. 2010. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol 14:440–446. doi: 10.1016/j.cbpa.2010.05.005. [DOI] [PubMed] [Google Scholar]