

Abstract
Despite years of research dedicated to preventing the sexual transmission of herpes simplex virus 2 (HSV-2), there is still no protective vaccine or microbicide against one of the most common sexually transmitted infections in the world. Using a phage display library constructed from a llama immunized with recombinant HSV-2 glycoprotein D, we identified a single-domain antibody VHH, R33, which binds to the viral surface glycoprotein D. Although R33 does not demonstrate any HSV-2 neutralization activity in vitro, when expressed with the cytotoxic domain of exotoxin A, the resulting immunotoxin (R33ExoA) specifically and potently kills HSV-2-infected cells, with a 50% neutralizing dilution (IC50) of 6.7 nM. We propose that R33ExoA could be used clinically to prevent transmission of HSV-2 through killing of virus-producing epithelial cells during virus reactivation. R33 could also potentially be used to deliver other cytotoxic effectors to HSV-2-infected cells.
INTRODUCTION
Herpes simplex virus 2 (HSV-2) is one of the most prevalent sexually transmitted infections (STIs) in the world and infects approximately 16% of people ages 15 to 49 (1). While generally not life threatening, HSV-2 can have severe sequelae in immunocompromised individuals and in infants (2). Additionally, HSV-2 infection is associated with a significantly increased risk of acquisition of human immunodeficiency virus type 1 (HIV-1) (3, 4). Complicating the effort to prevent transmission, a large proportion of HSV-2 primary infections and reactivations are subclinical, so asymptomatic individuals may still transmit the virus (5, 6). Vaccine strategies to prevent HSV-2 transmission have not been broadly protective, and condoms are not always effective at preventing transmission (7, 8). These failures have generated renewed interest in alternative prevention strategies.
Members of the camelid family (camels, llamas, and alpacas) can produce antibodies lacking light chains and CH1 domains (9). The variable domains of these heavy-chain-only antibodies, termed VHH, represent the smallest naturally occurring functional domain of the antibody molecule. They demonstrate the same antigen binding capability as full-length antibodies yet are typically approximately 15 kDa in size (9). When cloned and purified as monomeric domains, VHH demonstrate remarkable stability under a wide range of denaturing, temperature, and pH conditions (10). VHH exhibit increased solubility compared to full-length antibodies or other antibody fragments, and very high expression levels have been achieved in Escherichia coli, yeast, and tobacco expression systems (11–13). The human immune system is unlikely to react to VHH due to a high degree of sequence homology among mammalian variable domains, and VHH have been shown not to be immunogenic in mice (14). As a result of their small size, VHH have enhanced tissue penetration (14), and an extended CDR3 loop may allow VHH access to cryptic epitopes in enzymatically active sites that are unavailable for binding by full-length antibodies (15, 16).
We have sought to exploit these properties of VHH to develop a reagent that could be applied intravaginally for prophylaxis or treatment of HSV-2 infection. Several groups have previously demonstrated that antibodies directed against glycoprotein D of HSV-2 (gD2), a glycoprotein expressed on the surface of the virion that is required for entry into cells, can be applied vaginally to protect against genital HSV-2 infection in animal models (17–19). Based on previously published protocols describing the immunization of llamas or alpacas to obtain an antigen-specific VHH (20–22), we immunized a llama with recombinant gD2 and created a phage library displaying the VHH repertoire of the immunized animal. We identified a VHH, termed R33, which specifically binds to gD2 but fails to neutralize the virus. However, fusion of R33 to the cytotoxic domain of Pseudomonas aeruginosa exotoxin A yielded an immunotoxin which, unlike antivirals targeting HSV-2, was highly efficient in killing virus-infected cells. The potential therapeutic implications of these findings are discussed.
MATERIALS AND METHODS
Expression and purification of recombinant gD2 in Pichia pastoris.
Using genomic DNA from HSV-2 (strain 186) as a template, we amplified DNA encoding amino acids 1 to 314 of gD2 (ectodomain) from the viral genome using the primers CCCGAATTCACCATGAAATACGCCTTAGCAGACCCCTCG (forward) and ATTGCGGCCGCGTTAATGGTGATGGTGATGGTGCGGGTTGCTGGGGGC (reverse), which introduced a His tag at the C terminus. The gD2 sequence was cloned into the expression vector pPIC9 and transformed into Pichia pastoris by electroporation. Recombinant gD2 was expressed in P. pastoris using a previously published protocol (23) and purified with Ni-nitrilotriacetic acid (NTA) Superflow resin (Qiagen, Valencia, CA) using the manufacturer's protocol. Recombinant gD2 was eluted from the resin with elution buffer (250 mM imidazole in phosphate-buffered saline [PBS]), filtered through a 0.22-μm filter, and dialyzed overnight against PBS. The protein concentration was measured using a Bradford assay (Bio-Rad, Hercules, CA), and protein samples were separated by SDS-PAGE for visualization with Coomassie brilliant blue staining. The purified protein was detected with gD-specific antibodies by enzyme-linked immunosorbent assay (ELISA) and Western blotting using standard protocols. Antibodies used included R45 (rabbit polyclonal; a gift from R. Eisenberg and G. Cohen, University of Pennsylvania, Philadelphia, PA), HSV8 (human monoclonal; a gift from L. Zeitlin, Mapp BioPharmaceuticals, San Diego, CA), DL6 (mouse monoclonal; Santa Cruz Biotechnology, Dallas, TX), and anti-His (mouse monoclonal; Sigma-Aldrich, St. Louis, MO).
Llama immunizations.
Llama immunizations were performed by Triple J Farms in Bellingham, WA (protocol 110, approved by Triple J Farms IACUC, USDA registration number 91-R-0054) in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The llama Rayo was immunized on days 0, 21, 42, and 63. Each immunization included 0.5 mg of gD2, mixed with complete Freund's adjuvant for the first injection and incomplete Freund's adjuvant for subsequent injections. Prior to the first immunization and following each immunization, ∼20 ml of serum was collected to monitor for the presence of anti-gD2 antibody. After the fourth immunization, 500 ml of blood was taken from the llama and peripheral blood mononuclear cells (PBMCs) were purified using a Ficoll-Paque Plus gradient (GE Healthcare Life Sciences, Piscataway, NJ). PBMCs were aliquoted and frozen at −80C until further use.
Llama serum ELISA.
Nunc MaxiSorp ELISA plates (Thermo Fisher Scientific Inc., Waltham, MA) were coated with 100 μl of gD2 at 10 μg/ml and incubated overnight (ON) at 4°C. The plate was blocked with 2% bovine serum albumin (BSA) in PBS for 30 min at room temperature (RT). Freshly thawed serum samples were diluted in PBS and added in duplicate to wells for 1 h at RT. Wells were washed 5 times with 200 μl PBS-0.05% Tween (PBS-T) per well, horseradish peroxidase (HRP)-conjugated anti-llama secondary antibody (Bethyl Laboratories, Inc.) was diluted 1:10,000 in PBS-T, and 100 μl was added to wells for 1 h at RT. Wells were washed 5 times with 200 μl PBS-T per well and developed with 200 μl 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS) ELISA HRP substrate (KPL, Gaithersburg, MD). The plate was read at 405 nm using a BioTek (Winooski, VT) Synergy HT plate reader.
Amplification of VHH regions and construction of T7 phage display library.
Using PBMCs that were isolated from the llama following the final immunization, RNA was extracted using an RNeasy minikit (Qiagen, Valencia, CA) and reverse transcribed into cDNA (SuperScript II reverse transcriptase; Invitrogen, Carlsbad, CA). Nested PCR was performed to amplify the VHH regions from the cDNA using primers that bind to the conserved regions flanking the VHH genes. The first round of PCR was performed with primers as previously published (24), while the second round of primers introduced the appropriate restriction sites for ligation into the phage genome. The VHH band of ∼450 bp was gel extracted and ligated into predigested T7 phage vector arms as described in the manufacturer's handbook (Novagen Inc., Madison, WI). The ligation reaction mixture was packaged into the phage according to the manufacturer's protocol, and the titer was determined to assess the diversity of the packaged library prior to amplification. After amplification, the library was aliquoted and stored at −80°C until further use. VHH expressed on the phage surface are referred to as VHH-phage.
Biopanning of VHH/T7 library against gD2 and isolation of VHH sequences.
For the first round of biopanning, 109 PFU from the phage library was added to a well coated with 0.5 μg gD2 and incubated at room temperature for 1 h. Wells were then washed 10 times with shaking for 1 min with Tris-buffered saline (TBS) with 0.05% Tween (TBS-T) and 10 times with TBS. Bound phage were eluted using 200 μl of 1% SDS in TBS incubated on wells for 1 h at room temperature. A sample of the eluted phage was used to determine the titer of phage present, and the remaining eluted phage were added to 50 ml of E. coli strain BLT5403 grown in LB-ampicillin at an optical density at 600 nm (OD600) of 0.5 and shaken at 37°C until lysis occurred. The titer of this phage lysate was determined, and it was used as the input for the next round of biopanning, which was carried out using the same procedure. Additional rounds of biopanning were performed against gD2, and individual plaques from the phage elution after the sixth round of biopanning were picked, amplified, and tested in an ELISA to confirm gD2-binding capability. Nunc MaxiSorp ELISA plates (Thermo Fisher Scientific Inc., Waltham, MA) were coated with 0.5 μg gD2 per well and incubated ON at 4°C. The plate was blocked for 1 h with 2% BSA in PBS, and then 109 PFU of each phage clone was added in duplicate and incubated at RT for 1 h. After removal of phage, the plate was washed 5 times with 200 μl PBS-T per well. Anti-T7 tail fiber monoclonal antibody (GE Healthcare Life Sciences, Piscataway, NJ) was diluted to 1:1,000 and added at 100 μl to each well for 1 h at RT. After washing the plate 5 times with 200 μl PBS-T per well, HRP-conjugated anti-mouse IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA) was added at 1:3,000 in 100 μl and incubated at RT for 1 h. After a final wash, 5 times with 200 μl PBS-T per well, 200 μl of ABTS ELISA HRP substrate (KPL, Gaithersburg, MD) was added. The plate was read at 405 nm using a BioTek (Winooski, VT) Synergy HT plate reader.
Cloning and expression of VHH in E. coli.
VHH sequences were amplified from VHH-phage by PCR and cloned into the expression plasmid pET-47b (Novagen Inc., Madison, WI). To create a bivalent VHH construct (bvR33), another primer set was used to amplify a second R33 sequence and incorporate a GS linker between the two R33 sequences. The VHH constructs were transformed into E. coli BL21(DE3) competent cells (New England BioLabs, Ipswich, MA). An osmotic shock protocol was utilized to purify the soluble VHH from the periplasmic space, as described by Graef et al. (25). Briefly, an ON 30-ml midscale culture was diluted in 450 ml Terrific broth and grown at 25°C for 3 h. Cells were induced at 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) (Lab Scientific, Inc., Highlands, NJ) and grown for an additional 3 h at 25°C. After centrifugation, the cell pellet was lysed in Tris-sucrose buffer with lysozyme. Contents of periplasmic space were separated from cellular debris by centrifugation, and Ni-NTA Agarose (Qiagen, Valencia, CA) was added to the supernatant ON with rocking at 4°C. Agarose was collected by centrifugation and washed, and protein was eluted by addition of 3 ml elution buffer. Eluted VHH were dialyzed against PBS with 1 mM dithiothreitol (DTT) with at least 4 buffer changes. VHH were concentrated using an Amicon Ultra-15 centrifugal filter unit (EMD Millipore, Billerica, MA) and centrifuged at 16,000 × g for 10 min to remove precipitated protein, and the protein concentration was measured by Bradford assay (Bio-Rad, Hercules, CA). Protein samples were separated by SDS-PAGE for visualization with Coomassie brilliant blue staining.
Flow cytometry to validate VHH binding to cell surface-expressed gD2.
Z4/6 cells (gift from D. Johnson, Oregon Health and Science University) are a derivative of L cells that stably express gD2 at the cell surface (26). Nearly confluent z4/6 cells were trypsinized, washed once with PBS, and resuspended at 0.5 × 106 cells/ml. Five hundred microliters of cells were aliquoted, centrifuged at 500 × g for 5 min, resuspended with 1 ml 1% BSA-PBS, and incubated at 37°C for 30 min for blocking. Samples were centrifuged at 500 × g for 5 min, resuspended in VHH or DL6 antibody (Santa Cruz Biotechnology, Dallas, TX) diluted in 1% BSA-PBS, and incubated for 1 h at 4°C. Cells were washed twice with 2 ml PBS and resuspended in a fluorescein isothiocyanate (FITC)-conjugated anti-His antibody for VHH detection (Abcam, Cambridge, MA) or FITC-conjugated anti-mouse secondary antibody for DL6 detection (Jackson ImmunoResearch, West Grove, PA) diluted in 1% BSA-PBS for 30 min at 4°C, followed by a final wash with 2 ml PBS. Samples were run on a Becton-Dickinson FACSCalibur cytometer, and data were analyzed using the software program FlowJo (Tree Star Inc., Ashland, OR).
HSV-2 neutralization assay with VHH.
Vero cells (ATCC CCL-81; ATCC, Manassas, VA) were plated in Falcon 12-well trays (Thermo Fisher Scientific Inc., Waltham, MA) at 4 × 106 cells per tray and incubated ON at 37°C. VHH samples were serially diluted in Dulbecco's modified Eagle medium (DMEM)-2% FBS with HSV-2 G (ATCC VR-734; ATCC, Manassas, VA) at 5 × 103 PFU/ml, and all dilutions were incubated at 37°C for 1 h. Medium was removed from the Vero cells, and 100 μl of VHH dilutions with virus were added in duplicate to cells for 1 h at 37°C, with gentle shaking every 10 min to distribute liquid over cells. Cells were overlaid with 2 ml of 2% methylcellulose overlay–5% FBS in DMEM (Cellgro, Manassas, VA). Trays were incubated for 3 days at 37°C and stained with crystal violet, and plaques were counted.
Expression, purification, and refolding of immunotoxins.
The previously published exotoxin A sequence HA22 (27) with point mutations to remove immunogenic human B-cell epitopes (28) was synthesized (GenScript, Inc., Piscataway, NJ) and cloned in frame to the C terminus of the VHH (R33 and P10), already present in the pET-47b vector, to create R33ExoA and P10ExoA. Expression, purification, and refolding of immunotoxin proteins were performed based on a previously published protocol by Buchner et al. (29). Briefly, large-scale cultures (800 ml) of transformed BL21(DE3) cells (New England BioLabs, Ipswich, MA) were grown to an OD600 of 0.6 and induced with 1 mM with IPTG (Lab Scientific, Inc., Highlands, NJ) for 3 h at 37°C. After cells were harvested and lysed under denaturing conditions (6 M guanidine hydrochloride [GuHCl]), Ni-NTA agarose (Qiagen, Valencia, CA) was added to the clarified lysate to purify the His-tagged immunotoxin. After washing, immunotoxin protein was eluted (8 M urea, 250 mM imidazole, 50 mM NaH2PO4, 500 mM NaCl, 300 mM DTT), diluted 1:100 in refolding buffer (100 mM Tris, 500 mM l-arginine, 8 mM oxidized glutathione, and 2 mM EDTA), and incubated at 10°C overnight. After completion of the refolding reaction, the refolded immunotoxin was concentrated using an Amicon Ultra-15 centrifugal filter unit (EMD Millipore, Billerica, MA), and buffer exchange was performed by repeatedly bringing up the volume of the concentrated protein with PBS. The final volume of the protein was brought to ∼1 ml, and it was aliquoted and frozen at −80°C until use. The protein concentration was measured using a Bradford assay (Bio-Rad, Hercules, CA), and protein samples were separated by SDS-PAGE for visualization with Coomassie brilliant blue staining.
ELISA: binding of VHH and immunotoxins to gD2.
ELISAs were performed to determine if the purified VHH and immunotoxins were capable of binding to gD2. Nunc ELISA plates were coated with dilutions of VHH and immunotoxins, and after a blocking step, dilutions of purified gD2 were added to wells in duplicate. After a washing step of 5 times with PBS-T wash buffer, the anti-gD antibody DL6 (Santa Cruz Biotechnology, Dallas, TX) was added at 1:1,000 for 1 h. Wells were washed again, and HRP-conjugated anti-mouse (Sigma-Aldrich, St. Louis, MO) was added at 1:3,000 for 1 h. A final wash step was performed, and the plate was developed by adding 100 μl/well ABTS ELISA HRP substrate (KPL, Gaithersburg, MD). The plate was read at 405 nm using a BioTek (Winooski, VT) Synergy HT plate reader.
Toxicity assay.
The CellTiter 96 AQueous One solution cell proliferation assay was used to determine toxicity of VHH-ExoA for cell lines, and the assay was carried out using the protocol recommended by the manufacturer (Promega, Madison, WI). Z4/6 cells (26), expressing gD2 at the cell surface, and Vero cells were plated in 96-well trays at 3 × 105 cells/well overnight. The following day, dilutions of the VHH-ExoA proteins were added to wells and incubated overnight. About 16 h after the addition of protein, 20 μl of the CellTiter 96 AQueous One solution reagent was added to each well and incubated for 4 h at 37°C. The plate was read at 490 nm using a BioTek (Winooski, VT) Synergy HT plate reader. Higher OD values indicate greater cell viability and therefore less toxicity.
In vitro infectious center assay (ICA).
Vero cells were plated in 12-well trays at 3 × 105 cells/well and after 24 h were infected with HSV-2 G (ATCC VR-734; ATCC, Manassas, VA) at 500 PFU/well. Following the 1-h adsorption time, dilutions of the VHH or VHH immunotoxins were added to wells in duplicate, and complete medium (5% FBS-DMEM; CellGro, Manassas, VA) was added to bring the volume up to 700 μl per well. About 16 h later, supernatant was removed, and cells were trypsinized briefly with 250 μl trypsin-EDTA (CellGro, Manassas, VA) before adding an equal volume of complete medium. Cells were centrifuged at 500 × g for 5 min to pellet cells and then resuspended in 200 μl of complete medium. A 10-fold dilution series from 1:10 to 1:1,000 of the infected Vero cells was made in uninfected Vero cells harvested the same day (resuspended at 6 × 105 cells/ml), and 0.5 ml of the dilution series was added in duplicate to wells of a 12-well tray. Cells were overlaid with 0.5% methylcellulose-5% FBS to bring the volume to 1 ml. After 2 days, cells were stained with crystal violet and plaques were counted.
Statistical analysis.
For the neutralization assays, the significance of the difference in plaque numbers was calculated using an analysis of variance (ANOVA) test, with a Bonferroni correction (STATA Corp., College Station, TX). Prism software (Graphpad) was used to calculate a variable-slope sigmoidal dose-response curve to determine the 50% neutralizing dilution (IC50) and the 95% confidence interval for the antibodies tested in the neutralization assays and ICAs.
RESULTS
Construction of VHH-phage library and biopanning against gD2.
Recombinant gD2 was amplified from the viral genome (Fig. 1A), expressed in P. pastoris, and purified as previously published (23). The size and purity of purified gD2 were verified by separation with SDS-PAGE and Coomassie staining (Fig. 1B) and by Western blotting using R45, a polyclonal anti-gD antibody (Fig. 1C). A band of approximately 48 kDa was detected by both methods, somewhat smeared due to the variable glycosylation pattern from P. pastoris, as has been reported previously (23). The antigenicity of the purified protein was verified by ELISA using a panel of conformational and nonconformational anti-gD2 antibodies (Fig. 1D).
FIG 1.

Production and purification of gD2. (A) DNA encoding gD2 was amplified by PCR from the HSV-2 strain186 genome. (B and C) Purified gD2 from P. pastoris was separated by SDS-PAGE and stained with Coomassie brilliant blue (B) or detected with a polyclonal anti-gD2 antibody (R45) by Western blotting (C). The left lanes of all three gels are molecular markers, while right lanes show a single amplified PCR product (A), purified gD2 (B), or immunoreactive gD2 (C). (D) In an ELISA, gD2 was recognized by a panel of anti-gD2 antibodies: R45 (1:5,000), HSV8 (1:5,000), DL6 (1:1,000), and anti-His (1:1,000). As controls, wells were coated with gD2, and only HRP-conjugated secondary antibody (anti-rabbit for R45, anti-human for HSV8, and anti-mouse for DL6 and anti-His) was added. Error bars represent maximum and minimum values.
A llama was immunized four times with gD2, and the induction of anti-gD2 antibodies was monitored by ELISA. As shown in Fig. 2, the llama serum demonstrated notable reactivity to gD2 following the second immunization compared to results for serum collected prior to immunization (naive), with the highest reactivity occurring after the fourth immunization. The llama serum was tested in an HSV-2 neutralization assay and failed to neutralize the virus (data not shown). PBMCs were separated from whole blood following the final immunization, and RNA purified from these cells was used as the template for cDNA. VHH genes were amplified from the cDNA using primers published by Arbabi Ghahroudi et al. (24) and ligated into the T7 phage vector to generate a library with an initial diversity of 3.9 × 107 PFU. Using protocols recommended by the manufacturer, the VHH-phage library was panned against gD2 immobilized in wells of a 96-well plate. After the first round of biopanning, the titer of eluted phage increased stepwise following each round of biopanning, indicating a gradual enrichment for VHH-phage binding to gD2 (data not shown). Individual plaques from the phage eluted after the sixth round of biopanning were picked and amplified for analysis. Sixty VHH-phage clones were amplified and tested in an ELISA to determine reactivity to gD2. Of the 60 VHH-phage tested, 56 reacted to gD2 by ELISA (data not shown), and it was determined by sequencing that 91% of these sequences were identical, a VHH clone we termed R33. The VHH-phage clone P10 was amplified from a VHH phage library derived from a different llama prior to any biopanning for use as a negative-control VHH that did not bind to gD2. The specificity of P10 is undetermined. The VHH sequences are shown in Fig. 3C.
FIG 2.
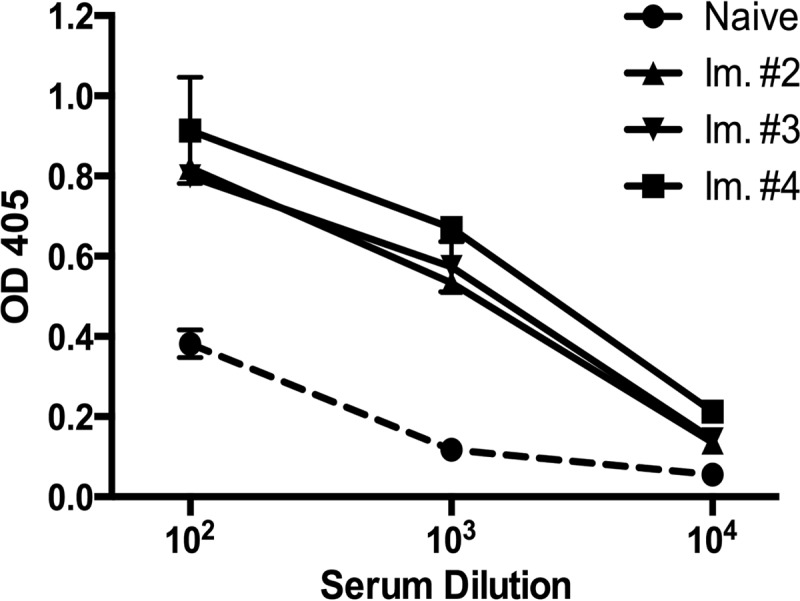
Reactivity of sera from llamas immunized with gD. A 1:100 dilution of sera collected before the initiation of immunization (naive) and after the second through fourth immunizations (Im. #2 to #4) was serially diluted 10-fold and tested in an ELISA for reactivity to gD2. The llama sera from Immunizations 2 to 4 demonstrate higher reactivity to gD2 than naive sera, demonstrating that the llama mounted an antibody response against the gD2 immunizations. Data represent the averages from three wells, and error bars are standard deviations.
FIG 3.

Purification of VHH and immunotoxins and comparison of sequences from VHH of different specificities. (A and B) Representative gels that demonstrated the size and purity of purified R33 and bvR33(A) or R33ExoA and P10ExoA (B). (C) R33 and P10 amino acid sequences. VHH genes, originally amplified from the variable region of heavy-chain-only antibodies, were sequenced from VHH-phage clones and aligned to determine unique VHH sequences identified from the gD2 biopanning process. R33 is able to bind gD2, while P10 is a negative-control VHH that does not bind gD2.
VHH binding to gD2 and HSV-2 neutralization assay.
To further validate the VHH sequences, they were recloned, expressed, and purified from E. coli using an osmotic shock procedure described by others (25). The purified VHH proteins were separated by SDS-PAGE for Coomassie staining to determine size and purity (Fig. 3A). In an ELISA with recombinant gD2 protein-coated plates, R33 but not P10 was able to bind to gD2 (see Fig. 7A). Using a cell line that expresses gD2 at the cell surface, z4/6 cells (26), flow cytometry was used to confirm that R33 and bvR33 could recognize gD2 that is expressed at the surface of the cell, as it would be in an infected cell (Fig. 4). R33 and bvR33 did not demonstrate any reactivity to the parental L cell line, which does not express gD2 (data not shown). While a known neutralizing antibody, HSV8 (17), demonstrated potent neutralization activity (IC50, 1.5 nM; 95% confidence interval [CI], 0.8 to 2.5 nM) against HSV-2 in a neutralization assay, R33 and bivalent R33 were unable to neutralize the virus at any of the concentrations tested, and an IC50 could not be calculated (Fig. 5A and B).
FIG 7.

(A) R33 and R33ExoA bind to recombinant gD2. An ELISA in which wells were coated with the indicated VHH or VHH immunotoxin, and gD2 was added to assay for their ability to bind gD2, was performed. While R33 and R33ExoA are able to bind to recombinant gD2, neither P10 nor P10ExoA demonstrates any reactivity. The graph is representative of three separate experiments. Each dilution was assayed in duplicate, and error bars represent maximum and minimum values. (B) R33ExoA antiviral activity in infectious center assay (ICA). An ICA with dilutions of R33ExoA and P10Exo shows that only R33ExoA has antiviral activity, with an IC50 of 6.7 nM. This is a representative graph from six independent experiments. Error bars represent standard errors of the means.
FIG 4.

Binding of R33 and bvR33 VHH to surface-expressed gD2 on z4/6 cells. z4/6 cells (surface expression of gD2) were stained with various VHH (R33, P10, and bvR33) and detected using fluorescence-activated cell sorting (FACS) by a FITC-conjugated anti-His secondary antibody, demonstrating that R33 and bvR33 but not P10 can bind to gD2. DL6 was used as a positive control to verify that gD2 was expressed, and a secondary antibody control (anti-His) with no VHH or primary antibody was also used as a negative control.
FIG 5.

HSV-2 neutralization assay. Virus was incubated with dilutions of R33 and HSV8 (known neutralizing antibody) (A) or R33, bvR33, and P10 (B) and then plated on Vero cells to assay for HSV-2 neutralizing activity. While HSV8 is capable of neutralizing HSV-2 (IC50 of 1.5 nM), R33 and bvR33 do not differ from P10 in terms of inhibiting viral infection. Each dilution was assayed in duplicate, and error bars represent maximum and minimum plaque numbers. These graphs are representative of at least three independent experiments, and results are expressed as percent inhibition compared to plaque numbers from untreated virus. Statistical significance compared to results for untreated virus was calculated by ANOVA and is indicated by asterisks (P < 0.05).
Specific toxicity of R33ExoA for gD2-expressing cells.
We next tested the efficacy and specificity of the immunotoxin. To do this, we used a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay with z4/6 cells that express gD2, as well as Vero cells, which do not express gD2. It is known that expression of gD2 by Z4/6 cells is not uniform (26), as staining with the anti-gD antibody DL6 shows (Fig. 4). The levels of gD2 expression, however, should be sufficient to determine if R33ExoA can exert a cytotoxic effect on cells expressing gD2. Figure 6 demonstrates that cytotoxic activity of R33ExoA affects only z4/6 cells and not the parental cell line. Non-gD2-binding P10ExoA has no cytotoxic effect on either cell line compared to results with cells treated with medium alone.
FIG 6.

Specific toxicity of R33ExoA for gD2-expressing cells. Dilutions of immunotoxins were added to Vero cells (A) or z4/6 cells (B), and their cytotoxicity was measured using an MTS assay. While R33ExoA demonstrates cytotoxic activity against gD2-expressing cell lines (IC50 = 0.5 nM) (95% CI, 0.1810 to 1.403), P10ExoA does not. Neither immunotoxin demonstrates activity against Vero cells, which do not express gD2. Dilutions of each protein were added to wells in triplicate, and error bars represent standard deviations.
In vitro activity of anti-gD2 immunotoxin.
Although monovalent and bivalent versions of R33 were ineffective at neutralizing HSV-2, we next sought to examine whether the anti-gD2 VHH, when fused to the cytotoxic domain of P. aeruginosa exotoxin A, might still be effective in targeting HSV-2-infected cells. The immunotoxins were purified and refolded based on protocols published by Buchner et al. (29). R33ExoA was still able to bind gD2 at levels comparable to those with R33 alone, while P10 and P10ExoA had no gD2 binding activity (Fig. 7A). To test the cytotoxic effect of R33ExoA on HSV-2-infected cells, an infectious center assay (ICA) was performed. After a 1-h virus adsorption on Vero cells, dilutions of R33, R33ExoA, and P10ExoA were added, and the infection was allowed to proceed for roughly 16 h. Cells were harvested at this time, mixed with uninfected Vero cells, and then diluted and plated so that the number of infectious centers could be quantified. R33ExoA consistently demonstrated potent antiviral activity compared to those of R33 and P10ExoA, which had no antiviral activity (Fig. 7B). Multiple repetitions of the ICA revealed that the IC50 of R33ExoA is approximately 6.7 nM (95% CI, 4.8 to 9.4 nM), while the IC50 of P10ExoA could not be calculated.
DISCUSSION
After immunizing a llama with recombinant gD2 and creating a T7 phage library displaying the VHH antibody repertoire of the immunized llama, a VHH that binds to gD2 was identified after multiple rounds of biopanning. This VHH, termed R33, was able to bind gD2 when expressed and purified from E. coli but was not able to neutralize HSV-2 in vitro. Bivalent R33 was also unable to neutralize the virus. Creation of a single domain antibody immunotoxin by expression of R33 with the cytotoxic domain of exotoxin A from Pseudomonas aeruginosa resulted in specific and potent killing of HSV-2-infected cells in vitro with an IC50 of 6.7 nM (95% CI, 4.8 to 9.4 nM). R33 has no activity in the infectious center assay, indicating that the antiviral activity exhibited by R33ExoA is due to the immunotoxin portion of the molecule rather than the antibody portion.
The majority of previous immunotoxin research has focused on targeting blood cancers, in which case the immunotoxin treatment is administered intravenously for systemic distribution to reach the circulating cancer cells (30–33). Additionally, the candidate immunotoxins that have been published directed against viruses such as HIV-1 (34, 35), Kaposi's sarcoma-associated herpesvirus (KSHV) (36, 37), rabies virus (38), or human cytomegalovirus (39) would also require systemic treatment. To our knowledge, the immunotoxin described here represents the first immunotoxin designed for targeting a pathogen at a mucosal site of infection. Therefore, there are many unknown variables that could affect the efficacy of the R33ExoA immunotoxin when it is administered within the genital tract. The mouse model, while an important tool in evaluating HSV-2 microbicide or antiviral candidates, may not be representative of humans, particularly due to the differences in pH in the mouse and human vagina (40). As a result, further in vitro studies evaluating the activity of R33ExoA under the lower-pH conditions found in the human vagina, for example, should be undertaken. In this regard, having a VHH as the antibody portion of the immunotoxin may be advantageous due to its stability at a wide range of temperatures and pHs (41).
One of the potential disadvantages that has emerged during systemic administration of immunotoxin for treatment of cancer is the development of an antibody response against the immunotoxin, which reduces its therapeutic efficacy (42–44). Several different approaches have been tried to reduce the immunogenicity of the exotoxin component while maintaining its efficacy (28, 30, 45–47). The construct used in these studies introduced amino acid substitutions into the immunotoxin which were designed to eliminate potential B cell epitopes (28) and which did not alter the ability of the immunotoxin to kill infected cells. Given the generally attenuated immune response in the female genital tract due to the need for tolerance of the major histocompatibility complex (MHC)-incompatible fetus or semen (48), it is possible that within this setting the immunotoxin would not elicit the problematic immune response that limits other immunotoxin therapies.
Therapeutic and prophylactic intervention in the HSV-2 infection cycle has primarily targeted the infectious virus or its intracellular replication cycle. The use of a toxin that interrupts protein synthesis within infected eukaryotic cells represents a novel approach to blocking the infection by targeting the sites from which virus disseminates upon initial exposure and, potentially, the reservoirs from which virus reactivates. The expression of gD2 on the surface of epithelial cells upon infection will make these cells susceptible to the gD2-binding R33ExoA immunotoxin. Given its efficacy at very low concentrations, application prior to intercourse or through continuous secretion from genetically engineered commensal genital flora could provide sufficient levels of the immunotoxin to achieve a level of protection not observed with the use of antivirals alone (49). Similar delivery methods for other antivirals have been used successfully to prevent STI transmission in various model systems (50, 51).
In a vaginal HSV-2 infection, virus is shed essentially continuously (52) and from multiple locations throughout the genital tract (53). During reactivation, the virus is released from infected neurons, leading to infection of the adjacent epithelial cells (54). The majority of viral shedding originates with these infected epithelial cells (54), which, as in the case of new infection, could be targeted by R33ExoA through the cells' surface gD2 expression. As a result, a potential use of this immunotoxin is in the context of an HSV-2-discordant couple hoping to prevent transmission to the uninfected partner. Because shedding episodes are frequent and subclinical shedding accounts for 80% of shedding, it is unrealistic to rely on avoidance of sexual contact during times of shedding as a means to prevent transmission (55). While oral antiviral drugs such as valacyclovir decrease viral shedding, they do not completely eliminate viral shedding or prevent transmission (49). Daily application of this immunotoxin formulated as a microbicide could help suppress the amount of virus shed by the infected partner and therefore decrease the chance of transmission to the uninfected partner. Immunotoxins targeting KSHV through binding of surface glycoproteins were tested in combination with nucleoside analog antivirals and demonstrated greater efficacy with combination treatment than with immunotoxin treatment alone (36, 37). R33ExoA treatment in combination with oral antiviral drugs could potentially maintain the amount of virus shed below the level estimated to be needed for transmission (55). In a newly acquired HSV-2 infection, R33ExoA could also be used to limit the number of latently infected neurons, thereby reducing future shedding and occurrence of ulcers following reactivation.
The frequency with which genital herpes infections have been attributed to HSV-1 has been increasing in recent years (56). While this immunotoxin was not tested for antiviral activity against HSV-1, recent studies indicate a high degree of cross-immunoreactivity between gD from the two viruses (57), consistent with the >88% sequence identity of the gD ectodomains from HSV-1 and HSV-2 (58, 59).
Conceptually, it is also possible that this immunotoxin could be used to kill infected neurons in the genital tract, thereby eliminating the latent viral reservoir. While the penetrative capability of R33ExoA through the vaginal epithelium is unknown, R33ExoA could potentially target infected neurons which express gD2 upon reactivation of the virus, neurons which, based on in vitro studies (60), are already dysfunctional.
While it is significant that we have demonstrated that an anti-gD2 VHH can be used as the antibody portion of an immunotoxin to specifically target HSV-2-infected cells, there are several caveats that must be addressed before its clinical application as a microbicide or antiviral treatment is undertaken. Although the apoptosis that results from internalization of exotoxin A is a noninflammatory process (61), the effect of vaginal treatment with R33ExoA must be further studied to determine if it could possibly lead to inflammation or the infiltration of immune cells, which could facilitate the transmission of other STIs, particularly HIV-1 (62). This risk would have to be weighed against the demonstrated ability of HSV-2 to promote transmission of other STIs, including HIV-1 (63). Also, since infected cells, such as dendritic cells, involved in the generation of the adaptive immune response might be targeted by this immunotoxin, the possibility that this treatment might prevent development of a protective adaptive immune response must be considered. However, recent studies have indicated that once infected, dendritic cells, as is the case for neurons, become dysfunctional. It is therefore unclear if further immunologic compromise would result from elimination of gD-expressing antigen-presenting cells (64).
Given the progress that cancer immunotoxins have made in terms of efficacy and safety, there is reason for optimism that a similar strategy could be used to prevent or treat HVS-2 infections. E. coli and yeast expression systems have been used to generate standard antibody-based expression systems (65, 66) with production of sufficient material for use in clinical trials (67). Further, reports of efficient production of VHH in yeast and insect cell expression systems support the potential commercial development of this type of antibody for therapeutic or prophylactic indications, obviating the concerns associated with bacterial and mammalian expression systems (68, 69).
Beyond the described potential applications, this study demonstrates that R33 is able to specifically deliver an effector molecule to HSV-2-infected cells. This VHH could be expressed with a myriad of other effectors for their delivery to HSV-2-infected cells, potentially greatly increasing the repertoire of tools to treat or prevent HSV-2 infection.
ACKNOWLEDGMENTS
We thank Janet Guedon, Harold Marcotte, Lennart Hammarstrom, Richard Cone, Russell Graef, and David FitzGerald for helpful advice and discussion. We also thank R. Eisenberg and G. Cohen for generously sharing purified protein and antibodies with us.
E.M.G. was supported by a Sommer Scholarship from the Johns Hopkins Bloomberg School of Public Health, and the research was funded by NIH grant AI079794. This research was also supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
E.M.G. and R.B.M. are coinventors on a provisional U.S. patent application for R33ExoA. P.J.D., A.B., and H.Z. declare no competing interest.
REFERENCES
- 1.Looker K. 2008. An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. Bull World Health Organ 86:805–812. doi: 10.2471/BLT.07.046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta R, Warren T, Wald A. 2007. Genital herpes. Lancet 370:2127–2137. doi: 10.1016/S0140-6736(07)61908-4. [DOI] [PubMed] [Google Scholar]
- 3.Holmberg SD, Stewart JA, Gerber AR, Byers RH, Lee FK, O'Malley PM, Nahmias AJ. 1988. Prior herpes simplex virus type 2 infection as a risk factor for HIV infection. JAMA 259:1048–1050. doi: 10.1001/jama.1988.03720070048033. [DOI] [PubMed] [Google Scholar]
- 4.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. 2006. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83. doi: 10.1097/01.aids.0000198081.09337.a7. [DOI] [PubMed] [Google Scholar]
- 5.Langenberg AGM, Corey L, Ashley RL, Leong WP, Straus SE. 1999. A prospective study of new infections with herpes simplex virus type 1 and type 2. N Engl J Med 341:1432–1438. doi: 10.1056/NEJM199911043411904. [DOI] [PubMed] [Google Scholar]
- 6.Fleming DT, McQuillan GM, Johnson RE, Nahmias AJ, Aral SO, Lee FK, St Louis ME. 1997. Herpes simplex virus type 2 in the United States, 1976 to 1994. N Engl J Med 337:1105–1111. doi: 10.1056/NEJM199710163371601. [DOI] [PubMed] [Google Scholar]
- 7.Johnston C, Koelle DM, Wald A. 2011. HSV-2: in pursuit of a vaccine. J Clin Invest 121:4600–4609. doi: 10.1172/JCI57148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth K, Ferreira VH, Kaushic C. 2013. HSV-2 vaccine: Current state and insights into development of a vaccine that targets genital mucosal protection. Microb Pathog 58:45–54. doi: 10.1016/j.micpath.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. 1993. Naturally occurring antibodies devoid of light chains. Nature 363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 10.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LG, Muyldermans S, Wyns L, Matagne A. 2002. Single-domain antibody fragments with high conformational stability. Protein Sci 11:500–515. doi: 10.1110/ps.34602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajabi-Memari H, Jalali-Javaran M, Rasaee MJ, Rahbarizadeh F, Forouzandeh-Moghadam M, Esmaili A. 2006. Expression and characterization of a recombinant single-domain monoclonal antibody against MUC1 mucin in tobacco plants. Hybridoma 25:209–215. doi: 10.1089/hyb.2006.25.209. [DOI] [PubMed] [Google Scholar]
- 12.Smolarek D, Bertrand O, Czerwinski M. 2012. Variable fragments of heavy chain antibodies (VHHs): a new magic bullet molecule of medicine? Postepy Hig Med Dosw 66:348–358. doi: 10.5604/17322693.1000334. [DOI] [PubMed] [Google Scholar]
- 13.Omidfar K, Rasaee MJ, Kashanian S, Paknejad M, Bathaie Z. 2007. Studies of thermostability in Camelus bactrianus (Bactrian camel) single-domain antibody specific for the mutant epidermal-growth-factor receptor expressed by Pichia. Biotechnol Appl Biochem 46:41–49. doi: 10.1042/BA20060104. [DOI] [PubMed] [Google Scholar]
- 14.Cortez-Retamozo V, Lauwereys M, Hassanzadeh Gh G, Gobert M, Conrath K, Muyldermans S, De Baetselier P, Revets H. 2002. Efficient tumor targeting by single-domain antibody fragments of camels. Int J Cancer 98:456–462. doi: 10.1002/ijc.10212. [DOI] [PubMed] [Google Scholar]
- 15.Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Hölzer W, De Genst E, Wyns L, Muyldermans S. 1998. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J 17:3512–3520. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrath KE, Lauwereys M, Galleni M, Matagne A, Frère JM, Kinne J, Wyns L, Muyldermans S. 2001. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother 45:2807–2812. doi: 10.1128/AAC.45.10.2807-2812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeitlin L, Whaley KJ, Sanna PP, Moench TR, Bastidas R, De Logu A, Williamson RA, Burton DR, Cone RA. 1996. Topically applied human recombinant monoclonal IgG1 antibody and its Fab and F(ab′)2 fragments protect mice from vaginal transmission of HSV-2. Virology 225:213–215. doi: 10.1006/viro.1996.0589. [DOI] [PubMed] [Google Scholar]
- 18.Sanna PP, De Logu A, Williamson RA, Hom YL, Straus SE, Bloom FE, Burton DR. 1996. Protection of nude mice by passive immunization with a type-common human recombinant monoclonal antibody against HSV. Virology 215:101–106. doi: 10.1006/viro.1996.0011. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Davé SK, Simmons A. 2004. Prevention of genital herpes in a guinea pig model using a glycoprotein D-specific single chain antibody as a microbicide. Virol J 1:11. doi: 10.1186/1743-422X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maass DR, Sepulveda J, Pernthaner A, Shoemaker CB. 2007. Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs). J Immunol Methods 324:13–25. doi: 10.1016/j.jim.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Linden R, de Geus B, Stok W, Bos W, van Wassenaar D, Verrips T, Frenken L. 2000. Induction of immune responses and molecular cloning of the heavy chain antibody repertoire of Lama glama. J Immunol Methods 240:185–195. doi: 10.1016/S0022-1759(00)00188-5. [DOI] [PubMed] [Google Scholar]
- 22.Tanha J, Dubuc G, Hirama T, Narang SA, MacKenzie CR. 2002. Selection by phage display of llama conventional V(H) fragments with heavy chain antibody V(H)H properties. J Immunol Methods 263:97–109. doi: 10.1016/S0022-1759(02)00027-3. [DOI] [PubMed] [Google Scholar]
- 23.van Kooij A, Middel J, Jakab F, Elfferich P, Koedijk D, Feijlbrief M, Scheffer A, Degener J, The T, Scheek R. 2002. High level expression and secretion of truncated forms of herpes simplex virus type 1 and type 2 glycoprotein D by the methylotrophic yeast Pichia pastoris. Protein Expr Purif 25:400–408. doi: 10.1016/S1046-5928(02)00034-7. [DOI] [PubMed] [Google Scholar]
- 24.Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. 1997. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett 414:521–526. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- 25.Graef RR, Anderson GP, Doyle KA, Zabetakis D, Sutton FN, Liu JL, Serrano-González J, Goldman ER, Cooper LA. 2011. Isolation of a highly thermal stable lama single domain antibody specific for Staphylococcus aureus enterotoxin B. BMC Biotechnol 11:86. doi: 10.1186/1472-6750-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson DC, Smiley JR. 1985. Intracellular transport of herpes simplex virus gD occurs more rapidly in uninfected cells than in infected cells. J Virol 54:682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker T, Oelsner M, Kreitman RJ, Salvatore G, Wang QC, Pastan I, Peschel C, Licht T. 2004. Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood 103:2718–2726. doi: 10.1182/blood-2003-04-1317. [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L, Pastan I. 2012. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci U S A 109:11782–11787. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchner J, Pastan I, Brinkmann U. 1992. A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal Biochem 205:263–270. doi: 10.1016/0003-2697(92)90433-8. [DOI] [PubMed] [Google Scholar]
- 30.Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, Pastan I. 2011. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci U S A 108:5742–5747. doi: 10.1073/pnas.1102746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastan I, Beers R, Bera TK. 2004. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol 248:503–518. doi: 10.1385/1-59259-666-5:503. [DOI] [PubMed] [Google Scholar]
- 32.Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. 2002. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res 8:995–1002. [PubMed] [Google Scholar]
- 33.Sharkey RM, Goldenberg DM. 2008. Use of antibodies and immunoconjugates for the therapy of more accessible cancers. Adv Drug Deliv Rev 60:1407–1420. doi: 10.1016/j.addr.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bera TK, Kennedy PE, Berger EA, Barbas CF, Pastan I. 1998. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med 4:384–391. [PMC free article] [PubMed] [Google Scholar]
- 35.Till MA, Zolla-Pazner S, Gorny MK, Patton JS, Uhr JW, Vitetta ES. 1989. Human immunodeficiency virus-infected T cells and monocytes are killed by monoclonal human anti-gp41 antibodies coupled to ricin A chain. Proc Natl Acad Sci U S A 86:1987–1991. doi: 10.1073/pnas.86.6.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Y, Berger EA. 2011. An immunotoxin targeting the gH glycoprotein of KSHV for selective killing of cells in the lytic phase of infection. Antiviral Res 90:143–150. doi: 10.1016/j.antiviral.2011.03.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee D, Chandran B, Berger EA. 2012. Selective killing of Kaposi's sarcoma-associated herpesvirus lytically infected cells with a recombinant immunotoxin targeting the viral gpK8.1A envelope glycoprotein. MAbs 4:233–242. doi: 10.4161/mabs.4.2.19262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mareeva T, Wanjalla C, Schnell MJ, Sykulev Y. 2010. A novel composite immunotoxin that suppresses rabies virus production by the infected cells. J Immunol Methods 353:78–86. doi: 10.1016/j.jim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnett BB, Smee DF, Malek SM, Sidwell RW. 1996. Selective cytotoxicity of ricin A chain immunotoxins towards murine cytomegalovirus-infected cells. Antimicrob Agents Chemother 40:470–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castle PE, Karp DA, Zeitlin L, García-Moreno EB, Moench TR, Whaley KJ, Cone RA. 2002. Human monoclonal antibody stability and activity at vaginal pH. J Reprod Immunol 56:61–76. doi: 10.1016/S0165-0378(02)00013-X. [DOI] [PubMed] [Google Scholar]
- 41.Ewert S, Cambillau C, Conrath K, Plückthun A. 2002. Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry 41:3628–3636. doi: 10.1021/bi011239a. [DOI] [PubMed] [Google Scholar]
- 42.Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, Tsang KY, Benhar I, Pastan I. 2012. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci U S A 109:E3597–E3603. doi: 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. 2007. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res 13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 44.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. 2009. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res 15:5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weldon JE, Xiang L, Zhang J, Beers R, Walker DA, Onda M, Hassan R, Pastan I. 2013. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol Cancer Ther 12:48–57. doi: 10.1158/1535-7163.MCT-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen JK, Weldon JE, Xiang L, Beers R, Onda M, Pastan I. 2010. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother 33:297–304. doi: 10.1097/CJI.0b013e3181cd1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, Pastan I. 2009. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 113:3792–3800. doi: 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark GF, Schust DJ. 2013. Manifestations of immune tolerance in the human female reproductive tract. Front Immunol 4:26. doi: 10.3389/fimmu.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corey L, Wald A, Patel R, Sacks SL, Tyring SK, Warren T, Douglas J, John M, Paavonen J, Morrow RA, Beutner KR, Stratchounsky LS, Mertz G, Keene ON, Watson HA, Tait D, Vargas-Cortes M. 2004. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med 350:11–20. doi: 10.1056/NEJMoa035144. [DOI] [PubMed] [Google Scholar]
- 50.Lagenaur LA, Sanders-Beer BE, Brichacek B, Pal R, Liu X, Liu Y, Yu R, Venzon D, Lee PP, Hamer DH. 2011. Prevention of vaginal SHIV transmission in macaques by a live recombinant Lactobacillus. Mucosal Immunol 4:648–657. doi: 10.1038/mi.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Lagenaur L, Lee P, Xu Q. 2008. Engineering of a human vaginal Lactobacillus strain for surface expression of two-domain CD4 molecules. Appl Environ Microbiol 74:4626–4635. doi: 10.1128/AEM.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiffer JT, Abu-Raddad L, Mark KE, Zhu J, Selke S, Magaret A, Wald A, Corey L. 2009. Frequent release of low amounts of herpes simplex virus from neurons: results of a mathematical model. Sci Transl Med 1:7ra16. doi: 10.1126/scitranslmed.3000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tata S, Johnston C, Huang ML, Selke S, Magaret A, Corey L, Wald A. 2010. Overlapping reactivations of herpes simplex virus type 2 in the genital and perianal mucosa. J Infect Dis 201:499–504. doi: 10.1086/650302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schiffer JT, Swan D, Al Sallaq R, Magaret A, Johnston C, Mark KE, Selke S, Ocbamichael N, Kuntz S, Zhu J, Robinson B, Huang ML, Jerome KR, Wald A, Corey L. 2013. Rapid localized spread and immunologic containment define Herpes simplex virus-2 reactivation in the human genital tract. eLife 2:e00288. doi: 10.7554/eLife.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schiffer JT, Mayer BT, Fong Y, Swan DA, Wald A. 2014. Herpes simplex virus-2 transmission probability estimates based on quantity of viral shedding. J R Soc Interface 11:20140160. doi: 10.1098/rsif.2014.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieuwenhuis RF, van Doornum GJ, Mulder PG, Neumann HA, van der Meijden WI. 2006. Importance of herpes simplex virus type-1 (HSV-1) in primary genital herpes. Acta Derm Venereol 86:129–134. doi: 10.2340/00015555-0029. [DOI] [PubMed] [Google Scholar]
- 57.Awasthi S, Belshe RB, Friedman HM. 2014. Better neutralization of herpes simplex virus type 1 (HSV-1) than HSV-2 by antibody from recipients of GlaxoSmithKline HSV-2 glycoprotein D2 subunit vaccine. J Infect Dis 210:571–575. doi: 10.1093/infdis/jiu177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zago A, Spear PG. 2003. Differences in the N termini of herpes simplex virus type 1 and 2 gDs that influence functional interactions with the human entry receptor Nectin-2 and an entry receptor expressed in Chinese hamster ovary cells. J Virol 77:9695–9699. doi: 10.1128/JVI.77.17.9695-9699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carfi A, Gong H, Lou H, Willis SH, Cohen GH, Eisenberg RJ, Wiley DC. 2002. Crystallization and preliminary diffraction studies of the ectodomain of the envelope glycoprotein D from herpes simplex virus 1 alone and in complex with the ectodomain of the human receptor HveA. Acta Crystallogr D Biol Crystallogr 58:836–838. doi: 10.1107/S0907444902001270. [DOI] [PubMed] [Google Scholar]
- 60.Fukuda J, Kurata T. 1981. Loss of membrane excitability after herpes simplex virus infection in tissue-cultured nerve cells from adult mammals. Brain Res 211:235–241. doi: 10.1016/0006-8993(81)90090-1. [DOI] [PubMed] [Google Scholar]
- 61.Keppler-Hafkemeyer A, Kreitman RJ, Pastan I. 2000. Apoptosis induced by immunotoxins used in the treatment of hematologic malignancies. Int J Cancer 87:86–94. doi:. [DOI] [PubMed] [Google Scholar]
- 62.Mayer KH, Venkatesh KK. 2011. Interactions of HIV, other sexually transmitted diseases, and genital tract inflammation facilitating local pathogen transmission and acquisition. Am J Reprod Immunol 65:308–316. doi: 10.1111/j.1600-0897.2010.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barnabas RV, Wasserheit JN, Huang Y, Janes H, Morrow R, Fuchs J, Mark KE, Casapia M, Mehrotra DV, Buchbinder SP, Corey L. 2011. Impact of herpes simplex virus type 2 on HIV-1 acquisition and progression in an HIV vaccine trial (the Step study). J Acquir Immune Defic Syndr 57:238–244. doi: 10.1097/QAI.0b013e31821acb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stefanidou M, Ramos I, Mas Casullo V, Trepanier JB, Rosenbaum S, Fernandez-Sesma A, Herold BC. 2013. Herpes simplex virus 2 (HSV-2) prevents dendritic cell maturation, induces apoptosis, and triggers release of proinflammatory cytokines: potential links to HSV-HIV synergy. J Virol 87:1443–1453. doi: 10.1128/JVI.01302-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gurkan C, Ellar DJ. 2005. Recombinant production of bacterial toxins and their derivatives in the methylotrophic yeast Pichia pastoris. Microb Cell Fact 4:33. doi: 10.1186/1475-2859-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsai A, Gallo M, Petterson T, Shiloach J. 1995. Large-scale production and purification of clinical grade pseudomonas aeruginosa exotoxin A from E. coli. Bioprocess Eng 12:115–118. doi: 10.1007/BF00369587. [DOI] [Google Scholar]
- 67.Weldon JE, Pastan I. 2011. A guide to taming a toxin—recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J 278:4683–4700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez-Sebastian S, Nunez MC, Garaicoechea L, Alvarado C, Mozgovoj M, Lasa R, Kahl A, Wigdorovitz A, Parreno V, Escribano JM. 2012. Rotavirus A-specific single-domain antibodies produced in baculovirus-infected insect larvae are protective in vivo. BMC Biotechnol 12:59. doi: 10.1186/1472-6750-12-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gorlani A, de Haard H, Verrips T. 2012. Expression of VHHs in Saccharomyces cerevisiae. Methods Mol Biol 911:277–286. doi: 10.1007/978-1-61779-968-6_17. [DOI] [PubMed] [Google Scholar]