

Abstract
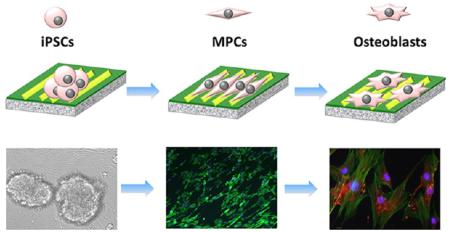
Here we report the design of a unique matrix, assembled from engineered M13 phage bionanofibers with specific cues of nanotopographies and versatile signal peptides to simulate native niche for directing the fate of induced pluripotent stem cells (iPSCs). By independently varying the peptide sequences and nanotopographies, we find that the resident iPSCs on the phage matrix are first differentiated into mesenchymal progenitor cells (MPCs), which are further differentiated into osteoblasts in the absence of osteogenic supplements due to the elongation induced by phage nanofibers. The phage-based matrix represents not only a biomimetic stem cell niche enabling independently varying biochemical and biophysical cues in one system but also a substrate for generating a safe and efficient cell source for tissue engineering.
Keywords: Stem cell niche, bionanofiber, phage display, peptides, viruses, topography
Stem cells have the capacity to self-renew and generate functional differentiated cells that replenish lost cells and hence participate in tissue regeneration, maintenance, and repair throughout the lifetime of an organism.1 Current studies on stem cells from diverse systems have shown that stem cell functions are controlled by their extracellular microenvironment, termed stem cell niche, which provides a location for positioning stem cells and integrates a complex array of molecular signals from both intrinsic and extrinsic cues.2,3 Since the stem cell niche plays a pivotal role in modulating stem cell fate, biomaterials that can potentially provide biomimetic microenvironment are recently developed to design an artificial smart matrix for simulating the native niche.4–6 Nonetheless, the current progress in biomaterial-based niche is still limited due to the lack of excellent biomaterials that can reproduce the biochemical and biophysical stimulation in a native niche. Moreover, it is difficult to independently vary the biochemical and biophysical cues in a material system. Most of the current studies on the biomaterial-based niche focus on an independent factor, either biochemical or biophysical stimulation for dictating stem cell fate.7,8 No reports are available where these two distinct factors are explored independently to understand their effects on directing stem cell fate. Therefore, an ideal biomimetic niche is needed to untangle the effects of biochemical and biophysical cues on stem cell fate.
On the other hand, induced pluripotent stem cells (iPSCs) have recently received great attention for their innovative applications in stem cell therapy.9 With the reprogramming of somatic cells to be pluripotent, it is possible to successfully overcome the serious immune rejection by enabling patient-specific cell therapy and bypass the ethical issue of embryonic stem cells (ESCs).10,11 Although mesenchymal stem cells (MSCs) from autologous bone marrow have been successfully used for bone repair, they are associated with several drawbacks including limited availability of autologous MSCs, low in vitro expandability, and decreased osteogenic differentiation potential with aging of bone marrow.12,13 Additionally, the formation of fully functional bone requires multiple cell types including osteoblasts, neural cells, and vascular cells to act in concert in order to maintain healthy bone structure and function.14,15 Hence, the iPSCs can be considered as a promising alternative cell source for the construction of fully functional bone substitutes because they can give rise to all of these different cell lineages due to their pluripotent differentiation capacity. However, inducing the osteoblastic differentiation of iPSCs by a biomaterial is still a challenge and the iPSCs and their derived cells may risk the formation of teratoma. Hence, there is a pressing need in identifying a material that can induce the differentiation of iPSCs into osteoblasts that can safely be used in bone regeneration.
To tackle the aforementioned challenges, we used a phage display approach to form a biomaterial that mimics the native stem cell niche and to use the resultant biomaterial as a biomimetic niche to induce the directed osteoblastic differentiation of iPSCs. Specifically, we employed a unique bionanofiber, M13 phage, to design a biomimetic matrix with specific biochemical cue (signal peptide) and biophysical cue (nanotopography). Such a matrix allowed us to systematically study the effect of biochemical and biophysical cues on manipulating stem cell fate by varying the two cues independently. M13 phage, a virus that specifically infects bacteria and is harmless to human beings, is indeed a bionanofiber because it measures ~880 nm long and ~6.6 nm wide.16,17 It is made of a protein coat encapsulating a ssDNA. The side wall of the phage is made of ~2700 copies of a major coat protein (termed pVIII), to which a foreign peptide can be fused by genetic insertion of the foreign gene coding for the peptide in the ssDNA (Figure 1). In addition, it can be mass-produced in large quantities of monodisperse nanofibers by infecting host bacteria in an error-free format. Therefore, compared to other nanofibers, M13 phage is unique in that it can be self-assembled into different patterns. Different peptides can be genetically displayed on the side wall, but the peptide display does not influence the assembly of phage nanofibers into a specific pattern (parallel versus random),16,18 enabling us to vary peptides while keeping the phage assembly pattern constant or to vary the phage assembly pattern while keeping the peptide sequences constant. These unique properties of M13 phage make it possible to use phage-assembled matrix for studying the effect on stem cell fate of both nanotopography (biophysical cue) and peptide sequences (biochemical cue) in one system.
Figure 1.
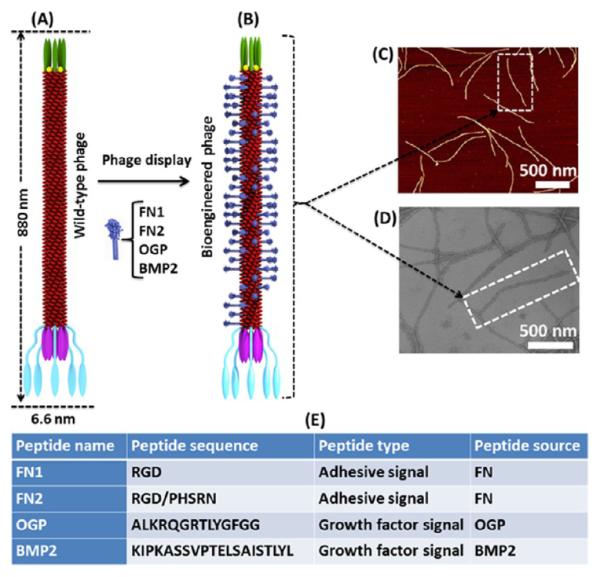
Phage display technique. A foreign peptide was fused to (and thus displayed on) the major coat (side wall) of wide type M13 phage (A) to form a bioengineered phage (B). The displayed peptide was helically assembled along the long axis of the phage nanofiber. M13 phage could be pictured as a bionanofiber with around 880 nm in length and 6.6 nm in width (C, atomic force microscopy (AFM) image; D, transmission electron microscopy (TEM) image). Two adhesive signal peptides and two growth factor signal peptides derived from different functional proteins that are highly associated with bone extracellular matrix (ECM) were separately displayed on the phage (E). FN2 denotes that two peptides (RGD and PHSRN) are double displayed on the major coat of phage.
Recent studies have demonstrated that designed peptide–amphiphile and selected active peptides from growth factors are of paramount importance to direct stem cell fate.6,19 Hence, here we displayed four peptides derived from different functional proteins on the major coat (side wall) of filamentous M13 phage by phage display technique (Figure 1A,B). These four peptides contain two adhesive signal peptides, including RGD and RGD/PHSRN (a combination of RGD and PHSRN) from fibronectin (FN), and two growth factor signal peptides, including ALKRQGRTLYGFGG from osteogenic growth peptide (OGP) and KIPKASSVPTELSAISTLYL from bone morphogenetic protein 2 (BMP2) (Figure 1E). These peptides as biochemical cues have been selected and displayed on the major coat of phage because they are derived from proteins highly associated with bone extracellular matrix (ECM) and bone regeneration. FN is a large adhesive glycoprotein found in the ECM and significantly impacts adhesion, proliferation, and differentiation of many cell lines due to two of its specific peptide domains, RGD and PHSRN, which interact with integrin.20–22 Bone morphogenetic proteins (BMPs) are members of the transforming growth factor-β superfamily and can induce ectopic bone formation in animal models.23 The sequence corresponding to a domain (residues 73–92) of BMP2, KIPKASSVPTELSAISTLYL, can bind to a BMP2-specific receptor and elevate mRNA of both alkaline phosphatase (ALP) and osteocalcin (OCN) in the mouse mesenchymal cell line.24 OGP is a short, naturally occurring 14-mer growth factor peptide (ALKRQGRTLYGFGG) found in serum at a concentration level of μmol/L and regulates proliferation, differentiation, and matrix mineralization in osteoblast lineage cells.25
The bioengineered phages displaying different signal peptides showed a filamentous structure with ~880 nm in length and 6.6 nm in width and consequently could be pictured as a bionanofiber (Figure 1C,D). These engineered bionanofibers were assembled to form phage-based matrix by our reported layer-by-layer self-assembly method (Figure 2A).16,20 The resultant matrix presented an ordered surface nanotopography (Figure 2B–D). Moreover, no matter what peptides were displayed on the constituent phage, we discovered that the phage matrix exhibited a similar unique surface topography where phage bundles as ridges were parallel to each other and separated by grooves. Thus, we termed this nanotopography of phage-assembled matrix as a ridge/groove structure. Such structure was formed because M13 phage was readily assembled into bundles that were further assembled in a parallel format to form a phage-based matrix due to the long-rod structure and monodispersity of phage nanofibers (Supporting Information, Figure S1).26,27 In addition, the electrostatic interaction between negatively charged phage nanofibers and positively charged poly-l-lysine substrates used in the layer-by-layer assembly may provide another driving force to promote the assembly of phage matrix.27 We also fabricated the phage matrix with random nanotopography as a control using a simple spinning-drying method (Supporting Information, Figure S2). These phage-assembled matrices combine specific biochemical factors (phage-displayed peptides) with unique biophysical cue (ordered or random surface nanotopography) in a fabulous biomaterial and can design diverse stem cell niche to evaluate the interaction between stem cells and biochemical and biophysical cues.
Figure 2.

Fabrication of phage-assembled matrix and the interaction between the phage matrix and iPSCs. The phage-assembled matrix with an ordered surface nanotopography was generated by a layer-by-layer self-assembly method, where the glass substrate was alternately immersed into the cationic poly-l-lysine solution and anionic phage nanofiber solution to achieve the electrostatic self-assembly of phage matrix, and phage layer was the last layer deposited (A). The resultant phage matrix showed specific ridge/groove nanotopography (B, bright-field image; C, scanning electron microscopy (SEM) image; D, AFM image). The resident iPSCs-derived EBs showed a quick response to the phage matrix and exhibited a significant morphological alteration over culture time (E, 6 h; F: 24 h; G, 48 h; H-J, 2 weeks). The fibroblast-like cells were first present at the cluster edge (E), and their number increased after 24 h (F). Some fibroblast-like cells were detached from the cluster edge and became slightly aligned along the phage bundles after 48 h (G). We also discovered that these fibroblast-like cells were significantly elongated and aligned along the long axis of the phage bundles constituting the phage matrix (H–J, red arrows showed the direction of cell elongation along phage bundles). Cell nuclei and F-actin were stained by DAPI (blue) and FITC-labeled phalloidin (green), respectively.
We discovered that the shape of resident iPSCs-derived embryonic bodies (EBs) on the phage matrix presented several major changes from nearly spherical, first to bipolar fibroblast-like and then to polygonal during the culture period. With morphological alteration of EBs, more fibroblast-like cells were present from outside to inside of EBs over time (Figure 2F,G). Interestingly, we found that the phage matrix with ordered ridge/groove nanotopography significantly induced the elongation and alignment of these fibroblast-like cells along the phage bundles. These specific regulations on cell elongation and alignment were independent of the peptide sequences displayed on the constituent phage nanofibers and solely relied on the ordered surface topography (Figure 2H–J). Because such elongated and aligned fibroblast-like cells were not detected in both random control (phage matrix substrate with random surface topography) and smooth blank control (poly-l-lysine substrate without phage matrix). In addition, the resident cells were randomly oriented on the two controls due to the lack of contact guidance by the ordered topography of phage matrix (Figure S3). Our findings suggest that the surface nanotopography (ordered, random, and smooth) of culture substrate can significantly influence stem cell behavior by altering cell morphology.
The resident iPSCs-derived EBs seeded on the phage matrix were directed to differentiate into mesenchymal lineage by induction medium prior to studying the osteoblastic differentiation of iPSCs. At the initial time, the seeded EBs showed classic rosette staining with specific pluripotent markers of Oct3/4 and Nanog (Figure S4). After mesenchymal induction for 3 days, the EBs showed a significant morphological alteration and presented a gradual differentiation from outside to inside based on flow cytometry analysis and immunofluorescence staining. We confirmed that the bipolar fibroblast-like cells on the phage matrix expressed varying levels of surface antigen markers including CD90, CD44, and Sca-1 from a mesenchyme of embryonic connective tissue and lacked the hematopoietic marker of CD45 (Figure 3A). Thus, these bipolar cells elongated by the phage matrix can be termed as mesenchymal progenitor cells (MPCs), because they express most of the surface antigens of MSCs, and can be considered as early MSCs (Figure S5). The immunofluorescence staining also showed a consistent outcome, and the fibroblast-like cells located on the EB edge exhibited positive staining of MPCs markers (Figure 3B). Therefore, our current results demonstrate that the resident EBs show a quick response to the phage matrix and can be guided to differentiate into the mesenchymal lineage at the early stage of EB differentiation.
Figure 3.

Differentiation of iPSCs-derived EBs on the phage matrix. The resident iPSCs-derived EBs on the phage matrix with an ordered nanotopography showed two-step differentiation from iPSCs to osteoblasts including mesenchymal differentiation (A, B, after 3 days) and the subsequent osteoblastic differentiation (C–E, after 4 weeks). The cells were initially guided to differentiate into mesenchymal progenitor cells (MPCs) induced by mesenchymal differentiation medium and subsequently differentiated into osteoblasts by the induction of the phage matrix in the absence of any osteogenic supplements. The specific mesenchymal markers of CD44, CD90, and Sca-1 showed positive expression, whereas the CD45, a hemopoietic marker as a negative control, exhibited a significant negative expression based on the analysis of flow cytometry (A). The double staining of both pluripotent marker (Oct3/4, Nanog) and mesenchymal markers (CD44, CD90, and Sca-1) further demonstrated the mesenchymal differentiation (the mesenchymal markers stained by green fluorescence, whereas the pluripotent markers stained by red fluorescence) (B). The ALP activity (C) showed a significant increase in all of the phage matrices compared with the control (**, p < 0.01). Both real-time PCR (D) and immunofluorescence staining (E) confirmed that the successful osteoblastic differentiation of MPCs was induced by the phage matrix according to the formation of specific osteogenic makers of osteocalcin (OCN) and osteopontin (OPN) at both gene and protein level. CON denoted control (poly-l-lysine substrates without phage). WT, FN1, FN2, OGP, and BMP2 denote the phage matrix made of WT-phage, FN1-phage, FN2-phage, OGP-phage, and BMP2-phage, respectively. OCN, OPN, and COL were stained by rhodamine-labeled antibody (red), cell nuclei were stained by DAPI (blue), and F-actin were stained by FITC-labeled phalloidin (green).
We,27 along with others,28,29 have clearly demonstrated that the nanotopography-induced elongation of human and rat MSCs initiated their directed osteoblastic differentiation without any osteogenic differentiation supplements. Furthermore, the peptides displayed on the surface of phage matrix are highly related to bone formation. Consequently, we further evaluate the osteoblastic differentiation of the above bipolar fibroblast-like MPCs induced by the unique phage matrix with ordered nanotopography and controlled biochemical cue in the absence of any osteogenic supplements.
ALP, a specific marker protein of osteoprogenitor, is normally used to verify the osteogenic status at the initial stage. The ALP analysis demonstrated that all of the phage matrix with ordered nanotopography including wild-type (WT) and bioengineered phage matrix showed a significantly high ALP activity compared with the blank control (**, p < 0.01) (Figure 3C). Moreover, among the bioengineered phages, OGP-phage and BMP2-phage showed a higher level of ALP expression than FN1-phage and FN2-phage. However, the phage matrix with random nanotopography (random control) showed a low level of ALP expression and exhibited no significant difference in comparison with the smooth control (Figure S6). These results indicate that the biophysical stimulation of ordered nanotopography plays a crucial role in inducing osteogenic differentiation, and the biochemical cue of signal peptide is dependent on nanotopography.
Immunofluorescence staining, as a qualitative analysis at the protein level, was used to investigate the osteogenic markers (Figure 3E). We found that OCN and osteopontin (OPN), two osteogenesis-specific markers, showed positive staining on all of the phage matrices with an ordered nanotopography. In addition, the OCN and OPN exhibited a higher expression level in the OGP-phage and BMP2-phage than in FN1-phage and FN2-phage. Collagen I (COL) as a positive control is a nonspecific osteogenic marker. It showed a high expression level in all groups, and there was no significant difference between the phage matrix and controls. Nevertheless, both OCN and OPN were not detected on all phage matrices with a random nanotopography. These findings imply that the same bioengineered phage matrix without an ordered nanotopography is unable to induce the osteogenic differentiation of iPSCs, and the ordered nanopattern of the phage matrix is a primary factor for inducing differentiation. Namely, our work demonstrated that the topographical cue is more important than the protein cues in directing the osteoblastic differentiation of iPSCs. It should be noted that, even though the phage bundles are shorter and narrower than the ridges in the microgrooved structures produced by electron beam lithography30,31 and photolithography,32,33 the unique biochemical and biophysical cues carried by phage-based microgrooved matrix could still induce the differentiation of iPSCs.
A real-time polymerase chain reaction (PCR) assay was used to further analyze the relative transcription level of osteogenic genes associated with osteogenic differentiation (Figure 3D). The results demonstrated that both OCN and OPN genes showed a significant up-regulation on the phage matrix with an ordered nanotopography in comparison with the smooth control (*, p < 0.05). The genes of OCN and OPN also showed a significantly higher mRNA level in the groups of OGP-phage and BMP2-phage than in FN1-phage and FN2-phage. Moreover, no significant mRNA level of both OCN and OPN genes was detected in the phage matrices with a random nanotopography. The nonspecific osteogenic marker, COL gene, was detected and showed a higher level in all groups. All of these results are highly consistent with the immunofluorescence assay and ALP analysis.
Overall, by comparing the ordered phage matrices with their corresponding random control, we can conclude that the ordered nanopatterns on the phage matrix can direct the osteoblastic differentiation of iPSCs, which are predifferentiated into MPCs in the conditioned media, in the absence of any osteogenic supplements. A comparison between the ordered bioengineered phage matrix and the ordered WT-phage matrix (no peptide display) shows that the signal peptides can further influence osteoblastic differentiation and the peptides from growth factor proteins of OGP and BMP2 can significantly boost the osteoblastic differentiation in comparison with those from the adhesive factor of FN1 and FN2. However, the signal peptide for modulating osteoblastic differentiation is dependent on how their carriers, the phage nanofibers, are organized but cannot act independently.
One major risk in the clinical use of iPSCs is that they can induce teratoma formation in vivo.34,35 The same issue applies to the specialized mature cells derived from iPSCs. Consequently, we tracked the performance of iPSCs-derived osteoblasts (iPSCs-OBs) in vivo and evaluated their potential risk in inducing teratoma formation. The animal test showed that teratomas were not formed in the group of iPSCs-OBs one month after cells were injected into nude mice (Figure 4B). On the contrary, the iPSCs as a control group generated a large teratoma under the same condition (Figure 4A–C). By histological analysis, we found that the teratoma contained three embryonic germ layers, including endoderm, mesoderm and ectoderm (Figure 4D–I), which were all differentiated from iPSCs. These data confirm that the iPSCs-OBs achieved by our current protocol are safe and unable to induce any teratoma formation and thus could serve as an alternative cell source for bone tissue engineering.
Figure 4.

Teratoma evaluation of iPSCs-derived osteoblasts (iPSCs-OBs). The animal test demonstrated that the iPSCs-Ob did not induce obvious teratoma formation in vivo, whereas pure iPSCs generated a large teratoma (around 3 cm in length) one month after both iPSCs-Ob and iPSCs were separately injected into the nude mice (A, injection of iPSCs into live nude mouse to generate a large visible teratoma; B, comparison of nude mice injected with iPSCs-Ob and iPSCs; C, teratoma excised from the animal). Histological analysis further demonstrated that the formed teratoma contained three embryonic germ layers including ectoderm (D and E), mesoderm (F, G and H) and endoderm (I). D, Immature neural tissue, ectoderm; E, Blood vessel tissue, ectoderm; F, Muscle-like tissue, mesoderm; G, Cartilage tissue, mesoderm; H, Bone tissue, mesoderm, I, Intestinal track tissue, endoderm. The red arrows in (A) and (B) showed the injection site.
In summary, a unique bionanofiber-assembled matrix as a biomimetic stem cell niche with a controlled biochemical cue and ordered nanotopography was precisely designed to direct the iPSCs fate. Our current data demonstrated that both the biochemical cue (i.e., displayed peptides) and ordered nanotopographical cue (i.e., ridge/groove nanotopography) had an effect on the osteoblastic differentiation of the iPSCs. The ordered nanotopographical cue changed the shape of resident cells by inducing cell elongation along the phage nanofibers and eventually stimulated osteoblastic differentiation of iPSCs. The peptides that were highly associated with bone regeneration and displayed on phage nanofibers further enhanced osteogenic differentiation level. The growth factor signals of OGP and BMP2 are more effective in inducing the osteoblastic differentiation of iPSCs than the adhesive signals of FN1 and FN2. We first demonstrated that a cooperative combination of the ordered ridge/groove nanotopography and growth factor signal peptide cues can significantly promote the osteoblastic differentiation of iPSCs in one system. The iPSCs-derived osteoblasts are safe and do not induce teratoma formation in vivo. This work provides a powerful approach to the use of phage display for creating a unique matrix that can direct the osteoblastic differentiation of iPSCs and develops an alternative cell source for bone tissue engineering.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Oklahoma Center for Adult Stem Cell Research (434003). It was also in part supported by National Science Foundation (CMMI-1234957, CBET-0854414, CBET-0854465, and DMR-0847758), National Institutes of Health (1R01DE015633 and 1R21EB015190), Department of Defense Peer Reviewed Medical Research Program (W81XWH-12-1-0384), and Oklahoma Center for the Advancement of Science and Technology (HR14-160). M.Y. also thanks the generous support from National High Technology Research and Development Program 863 (2013AA102507), Zhejiang Provincial Natural Science Foundation of China (LZ12C17001), National Natural Science Foundation of China (20804037 and 21172194), and Silkworm Industry Science and Technology Innovation Team (2011R50028).
Footnotes
Supporting Information Materials and methods, detailed material characterizations, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Morrison SJ, Spradling AC. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Scadden DT. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- (3).Li LH, Xie T. Annu. Rev. Cell Dev. Biol. 2005;21:605–631. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- (4).Dawson E, Mapili G, Erickson K, Taqvi S, Roy K. Adv. Drug Delivery Rev. 2008;60:215–228. doi: 10.1016/j.addr.2007.08.037. [DOI] [PubMed] [Google Scholar]
- (5).Lutolf MP, Gilbert PM, Blau HM. Nature. 2009;462:433–441. doi: 10.1038/nature08602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lutolf MP, Hubbell JA. Nat. Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- (7).Vogel V, Sheetz MP. Curr. Opin. Cell Biol. 2009;21:38–46. doi: 10.1016/j.ceb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gilbert PM, Havenstrite KL, Magnusson KEG, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dravid GG, Crooks GM. Adv. Drug Delivery Rev. 2011;63:331–341. doi: 10.1016/j.addr.2010.12.006. [DOI] [PubMed] [Google Scholar]
- (10).Takahashi K, Yamanaka S. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- (11).Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- (12).Caplan AI. Tissue Eng. 2005;11:1198–1211. doi: 10.1089/ten.2005.11.1198. [DOI] [PubMed] [Google Scholar]
- (13).Colnot C. Tissue Eng., Part B. 2011;17:449–457. doi: 10.1089/ten.teb.2011.0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).de Peppo GM, Marcos-Campos I, Kahler DJ, Alsalman D, Shang L, Vunjak-Novakovic G, Marolt D. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8680–8685. doi: 10.1073/pnas.1301190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marcos-Campos I, Marolt D, Petridis P, Bhumiratana S, Schmidt D, Vunjak-Novakovic G. Biomaterials. 2012;33:8329–8342. doi: 10.1016/j.biomaterials.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhu H, Cao B, Zhen Z, Laxmi AA, Li D, Liu S, Mao CB. Biomaterials. 2011;32:4744–4752. doi: 10.1016/j.biomaterials.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mao CB, Liu A, Cao B. Angew. Chem., Int. Ed. 2009;48:6790–6810. doi: 10.1002/anie.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Merzlyak A, Indrakanti S, Lee S-W. Nano Lett. 2009;9:846–852. doi: 10.1021/nl8036728. [DOI] [PubMed] [Google Scholar]
- (19).Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- (20).Liu A, Abbineni G, Mao CB. Adv. Mater. 2009;21:1001–1005. [Google Scholar]
- (21).Benoit DSW, Anseth KS. Biomaterials. 2005;26:5209–5220. doi: 10.1016/j.biomaterials.2005.01.045. [DOI] [PubMed] [Google Scholar]
- (22).Petrie TA, Raynor JE, Reyes CD, Burns KL, Collard DM, Garcia AJ. Biomaterials. 2008;29:2849–2857. doi: 10.1016/j.biomaterials.2008.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Cell. Signal. 2011;23:609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- (24).Saito A, Suzuki Y, Ogata S, Ohtsuki C, Tanihara M. J. Biomed. Mater. Res., Part A. 2005;72A:77–82. doi: 10.1002/jbm.a.30208. [DOI] [PubMed] [Google Scholar]
- (25).Moore NM, Lin NJ, Gallant ND, Becker ML. Biomaterials. 2010;31:1604–1611. doi: 10.1016/j.biomaterials.2009.11.011. [DOI] [PubMed] [Google Scholar]
- (26).Wang J, Yang M, Zhu Y, Wang L, Tomsia AP, Mao CB. Adv. Mater. 2014;26:4961–4966. doi: 10.1002/adma.201400154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang J, Wang L, Li X, Mao CB. Sci. Rep. 2013;3:1242. doi: 10.1038/srep01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CDW, Oreffo ROC. Nat. Mater. 2007;6:997–1003. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- (29).Oh S, Brammer KS, Li YSJ, Teng D, Engler AJ, Chien S, Jin S. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2130–2135. doi: 10.1073/pnas.0813200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).McMurray RJ, Gadegaard N, Tsimbouri PM, Burgess KV, McNamara LE, Tare R, Murawski K, Kingham E, Oreffo ROC, Dalby MJ. Nat. Mater. 2011;10:637–644. doi: 10.1038/nmat3058. [DOI] [PubMed] [Google Scholar]
- (31).Bettinger CJ, Langer R, Borenstein JT. Angew. Chem., Int. Ed. 2009;48:5406–5415. doi: 10.1002/anie.200805179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee MR, Kwon KW, Jung H, Kim HN, Suh KY, Kim K, Kim KS. Biomaterials. 2010;31:4360–4366. doi: 10.1016/j.biomaterials.2010.02.012. [DOI] [PubMed] [Google Scholar]
- (33).Kilian KA, Bugarija B, Lahn BT, Mrksich M. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4872–4877. doi: 10.1073/pnas.0903269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yamanaka S. Cell. 2009;137:13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- (35).Zhao T, Zhang Z-N, Rong Z, Xu Y. Nature. 2011;474:212–U251. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.