

Abstract
Glioblastomas are among the most aggressive human cancers, and prognosis remains poor despite presently available therapies. Angiogenesis is a hallmark of glioblastoma, and the resultant vascularity is associated with poor prognosis. The proteins that mediate angiogenesis, including vascular endothelial growth factor (VEGF) signaling proteins, have emerged as attractive targets for therapeutic development. Since VEGF receptor-2 (VEG-FR-2) is thought to be the primary receptor mediating angiogenesis, direct inhibition of this receptor may produce an ideal therapeutic effect. In this context, we tested the therapeutic effect of CT322, a selective inhibitor of VEGFR-2. Using an intracranial murine xenograft model (U87-EGFRvIII-luciferase), we demonstrate that CT322 inhibited glioblastoma growth in vivo and prolonged survival. Of note, the anti-neoplastic effect of CT322 is augmented by the incorporation of temozolomide or temozolomide with radiation therapy. Immunohistochemical analysis of CT322 treated tumors revealed decreased CD31 staining, suggesting that the tumoricidal effect is mediated by inhibition of angiogenesis. These pre-clinical results provide the foundation to further understand long term response and tumor escape mechanisms to anti-angiogenic treatments on EGFR over-expressing glioblastomas.
Keywords: Glioblastoma, CT322, Vascular endothelial growth factor, Xenograft, Temozolomide, Adnectin™
Introduction
The cure for glioblastoma (GBM) has evaded generations of scientists. It is the most common form of primary brain malignancy and one of the most aggressive forms of human cancer. Without treatment, the median survival is approximately 14 weeks [1]. The current standard treatment includes maximal surgical resection followed by concurrent temozolomide and radiation therapy [2, 3]. This regimen extends the median survival to approximately 14 months [2]. For most patients, however, the disease remains fatal.
Histologically, glioblastoma is characterized by increased vascularity. This vascularity is achieved, in large part, by tumoral secretion of a soluble pro-angiogenic factor termed vascular endothelial growth factor (VEGF) [4–7]. Of note, the extent of vascularity and VEGF expression correlates with poor clinical outcomes for glioblastoma patients [8, 9]. In this context, vascular endothelial growth factor signaling has emerged as a key therapeutic target [10, 11].
Within the VEGF signaling family, there are several ligands (VEGF-A, B, C, D, and placental growth factor) and associated receptors (VEGFR-1, -2, and -3) [12]. While all VEGF family ligands can mediate angiogenesis, VEGFR-2 is the primary receptor that mediates angiogenesis. Thus, the generally accepted principal signal for angiogenesis involves the interaction between the various VEGF isoforms and the receptor VEGFR-2 [12, 13]. Pertinent to our study, human VEGFs have been shown to bind and activate mouse VEGF-receptors to trigger angiogenesis. [14–17]. For instance, Oka et al. demonstrated that injection of VEGF-expressing cancer stem cell into the brains of NOD-SCID mice formed vascular tumors that were negative for human CD31 and positive for mouse CD31 [17]. VEGF-A secretion is regulated by several oncogenes, including epidermal growth factor receptor (EGFR) [7]. One well-described EGFR mutant found in glioblastoma, EGFRvIII, results in constitutively active EGFR signaling [18]. In this way, activating mutations of EGFR can simultaneously trigger oncogenesis, as well as promote angiogenesis [7].
In this context, we tested the efficacy of CT322, a novel therapeutic agent that specifically and effectively blocks VEGFR-2, in an EGFRvIII-expressing glioblastoma model [19]. CT322 is a prototypical Adnectin™, a novel type of targeted biologic derived from human fibronectin, an abundant extracellular protein that naturally binds to other proteins [19]. Adnectins are molecules based upon the 10th type III domain of fibronectin. Variable regions within the stable tertiary structure of this fibronectin domain can be engineered to facilitate binding to specific targets. The variable regions of CT322 have been engineered for specific binding to human and mouse VEGFR-2 [20]. A Phase I clinical trial of CT322 in solid cancers suggests that the drug is acceptably tolerated [21]. CT322 is able to bind and effectively inhibit human and mouse VEGFR-2 and prevent the proliferation effect of VEGF-A on human embryonic vascular endothelial cells. Pertinent to neuro-oncology, CT322 inhibits tumor growth in a mouse subcutaneous xenograft glioblastoma model [19]. Here,we extend this previous work by testing the effect of CT322 alone and in combination with currently used therapeutics in vivo using an intracranial mouse EGFRvIII-expressing glioblastoma model.
Materials and methods
Cell lines and reagents
The U87-EGFRvIII cell line was generously provided by Dr. Xandra Breakefield (Massachusetts General Hospital, Boston MA). The U87-EGFRvIII-luciferase cell line was generated by transducing U87-EGFRvIII with replication-defective lentivirus encoding firefly luciferase as previously described [22]. Cells were maintained in the DMEM with 10 % FBS (Invitrogen), 2 mM Glutamine, and 1 % Penstrep (Invitrogen) at 37 °C with 5 % CO2. Supernatants from the U87-EGFRvIII cells were analyzed for presence of VEGF. An in vitro cell viability assay was performed for the U87-EGFRvIII with CT322 treatment.
Tumor implantation
9 week-old male NOD-SCID mice (NOD-CB17-Prkdcscid/lcrCrl; Charles River Laboratories, Wilmington, MA, USA) were anesthetized by 2 % isoflurane. 5 × 104 U87 EGFRvIII-luciferase cells were implanted intracranially using the following stereotactic coordinates: 0.5 mm posterior, 2.5 mm lateral, and 3.5 mm deep. Treatment groups within each experiment ranged in size from 4 to 7 mice each (indicated in the results section and in each figure legend). Pilot experiments suggested statistically significant differences in treatment outcomes could be demonstrated with groups this size. The weight of the mice was monitored at regular intervals and with each imaging session to assure animal health and lack of treatment toxicity. Animal care was handled and maintained in strict accordance to established protocols from the Institutional Animal Care and Use Committee of the Massachusetts General Hospital.
Chemotherapeutic and radiation administration
Beginning on post-transplant day 4, CT322 was administered at a dose of 60 mg/kg intraperitoneally (IP) 3-times weekly for 5 doses. For negative control, phosphate buffered saline (PBS) was administered in an equal volume. Temozolomide was administered at 34 mg/kg orally daily for the first 5 days of the treatment period. In the absence of radiation therapy, temozolomide administration was concurrent with the combination therapy beginning at day 4 after tumor transplantation. Because experiments involving radiation therapy were conducted subsequent to the non-radiation groups, an independent PBS control group was maintained concurrent to the cohorts receiving radiation plus temozolomide with and without CT322. With radiation therapy, temozolomide was administered concurrent with the start of radiation therapy at post-transplant day 8. Mice randomized to receive radiation therapy were irradiated to a total dose of 30 Gray (Gy) in three daily 10 Gy fractions. Prior to each treatment, mice were anesthetized (ketamine 118 mg/kg IP + xylazine 11.8 mg/kg IP) and shielded in a custom-designed block with an aperture of 0.8 cm.
Bioluminescence imaging (BLI)
In vivo longitudinal monitoring was accomplished using bioluminescence detection. Mice were anesthetized using 1–2 % isoflurane before intraperitoneal injection of d-luciferin salt (150 mg/kg in 200 μl PBS; Caliper Life Sciences, Hopkinton, MA). BLI was performed in a high sensitivity, cooled CCD camera (IVIS®, Xenogen) at five-minute intervals until peak values were recorded for all mice. Living image® software (Xenogen) was used for imaging and signal quantification. Imaging schedules are described in the treatment groups.
Magnetic resonance imaging (MRI)
Two animals per treatment group were subjected to MRI. MRI was performed to obtain serial images with a 4.7 tesla Bruker imaging system (Pharmascan, Karlruhe, Germany). Animals were imaged at baseline and weekly until termination of study. Animals were anesthetized during imaging with 1–1.5 % inhaled isoflurane and monitored during imaging with respiratory monitoring. Imaging protocols included a tri-plane and axial rapid acquisition with relaxation enhancement (RARE) localizer. Multi-slice multi-echo T2-weighted imaging was performed prior to and following intravenous injection of magnetic nano-particle (10 mg/kg Fe). The following parameters were utilized: Flip angle = 900; Matrix size (128 × 64); TR = 2500 ms; TE = 16 equally spaced echoes at 8.6 ms intervals ranging from 8.6 ms to 137 ms; field of view (FOV) = 4.24 × 2.12 cm, slight thickness = 1 mm. T1-weighted imaging was performed following the administration of Gd–DTPA utilizing the following parameters: Flip angle 900; Matrix size (256 × 256); TR = 700 ms; TE = 14 ms; field of view (FOV) = 4.24 × 2.12 cm, slice thickness = 1 mm. OsiriX DICOM software (www.osirix-viewer.com) was used for image analysis. Tumors were outlined in T1 post-contrast scans using all available images containing tumor. Volumes were calculated using the “compute volume” function.
Histology
A set of mice separate from the survival and bioluminescence studies was designated for histologic analysis and comparison, and tumors from these mice were collected at day 14. These mice received the various treatments in parallel with the mice in the survival studies: PBS, CT322 alone, temozolomide alone, CT322 with temozolomide, radiation therapy with temozolomide, and CT322 with radiation and temozolomide. Mice were deeply anesthetized and perfused with chilled PBS, followed by 4 % paraformaldehyde (PF). Brains were removed and placed in 4 % PF for 12 h before being paraffin embedded. Immunohistochemical staining was performed on paraffin wax embedded mouse brain tissue sections using a polymer peroxidase system in accordance to the manufacturer's instruction (Biocare Medical, Concord, CA). Antibodies used include: anti-Ki67 (1:100; Biocare Medical #CRM 325), anti-caspase 3 (1:100; Biocare Medical # CP 229), and anti-CD31 (1:50; Biocare Medical # CM 347). All antibodies were used in accordance to the manufacturer's instruction. Subsequently, sections were incubated with Biocare's Rabbit on Rodent HRP-Polymer or Rat on mouse HRP-Polymer (Biocare Medical) for 10 or 30 min, respectively. Thereafter, tissue sections were incubated with DAB chromogen substrate (Biocare Medical) for 5 min in room temperature. Finally, sections were lightly counter-stained with Mayer's hematoxylin. For negative control, the primary antibody was omitted during the incubation step.
Cell counts of positively staining cells were performed with high power (×200) digital images for each marker (CD31, Ki67 and cleaved-Caspase 3). The average number of marker-positive cells per 200× field was determined for the entire area of each tumor. Depending on the tumor size, the entire area of the tumor ranged from 2 to 6 fields at 200× power. No tumor had reached the size of ten 200× fields.The tumors for histologic analysis were taken from 2 mice in the CT322 monotherapy treatment group, 2 mice in the CT322 plus temozolomide treatment group, 3 mice in the temozolomide monotherapy treatment group, and 3 mice from the control group for this cohort. Additionally, tumors for histologic analysis were taken from 4 mice in the temozolomide plus radiation therapy treatment group, 4 mice from the CT322 plus temozolomide and radiation therapy group, and 4 mice from the control group for this cohort.
Statistical analyses
Survival analyses were made with Kaplan–Meier curves and Gehan's generalized Wilcoxon test. Statistical comparisons between control and experimental groups were carried out with the Mann–Whitney test, and comparisons between control and treated groups were established using the Kruskal–Wallis test. All statistical analysis was made using Prism 5.0 software (Graphpad).
Results
Treatment with CT322 delayed the growth of intracranial U87-EGFRvIII xenografts. We tested CT322 using the U87-EGFRvIII Luciferase because EGFRvIII expression has been associated with increased VEGF secretion and angiogenesis [7]. Production of VEGF was confirmed on supernatant analysis from these U87-EGFRvIII cells. In vitro treatments with CT322 did not affect viability of the cells, suggesting the tumor cell viability was not VEGFR-2 dependent. In this model, CT322 treatment resulted in reduced peak bioluminescence compared to administration of PBS (Fig. 1a), suggesting a delay in growth of the U87-EGFRvIII Luciferase xenograft. By day 13, the mean bioluminescence in the control group (n = 4) was 4,380 photons/s × 106. In comparison, the mean bioluminescence in the CT322 treated group (n = 5) showed roughly a sevenfold decrease to 610 photons/s × 106 (p = 0.08 at day 13 for PBS vs CT322 (unpaired t test)). MRI images from a representative subset of mice further confirmed reduced tumor size with CT322 treatment (Fig. 1b, c). CT322 treatment also resulted in a statistically significant survival advantage relative to the untreated control mice (p = 0.0336, Gehan-Breslow-Wilcoxon Test) (Fig. 1d). The median survival within the control group of mice was 19 days (standard deviation (sd) = 2 days) and 29 days (sd = 6 days) in the CT322 treatment group.
Fig. 1.
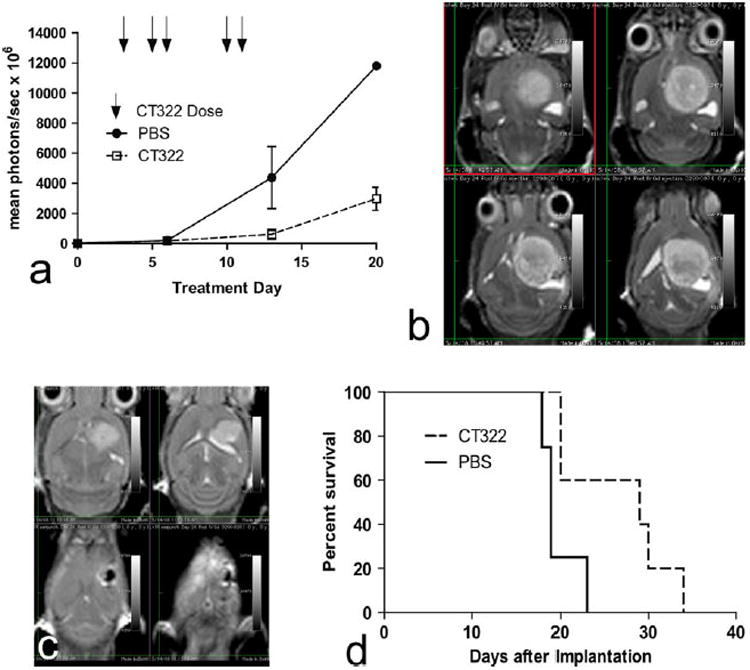
a–d CT322 demonstrates treatment benefit in glioblastoma xenografts. Data represents 4 control mice (PBS, n = 4) and 5 mice in the CT322 treatment group (CT322, n = 5). a CT322 reduces peak bioluminescence (mean photons/s × 106) over the course of treatment, reflecting slowed tumor growth and reduced tumor volume. Days of CT322 doses are represented with arrows. p = 0.08 at day 13 for PBS versus CT322 (unpaired t test). b Multiple image slices from a single MRI of a control mouse on day 24 after xenograft demonstrating bright area of tumor representing a 121 mm3 tumor. The bioluminescence measurement at day 20 for this individual was 11,802 mean photons/s × 106. c Multiple image slices from a single MRI of a CT322-treated mouse on day 24 after xenograft demonstrating bright area of tumor representing a 33 mm3 tumor. The bioluminescence measurement at day 20 for this individual was 3,973 mean photons/s × 106. d Kaplan–Meyer curve demonstrating improved survival of CT322-treated group (p = 0.0336, Gehan-Breslow-Wilcoxon Test). The median survival within the control group of mice was 19 days (sd = 2 days) and 29 days (sd = 6 days) in the CT322 treatment group. Abbreviations: PBS = phosphate buffered saline
Temozolomide increased the therapeutic effect of CT322. Since clinical translation of CT322 would likely involve combination with temozolomide [2], we tested the effect of such combination in our model. When treated with the combination of CT322 and temozolomide, tumor size was reduced and survival improved beyond the results of either drug administered individually (Fig. 2a–e). By day 27, the mean peak bioluminescence (Fig. 2a) was 17,000 photons/s × 106 for the CT322-treated group (n = 5), 4,800 photons/s × 106 for the temozolomide-treated group (n = 6), and 900 photons/s × 106 for the combination CT322 plus temozolomide treated group (n = 6) (p = 0.04 for temozolomide vs PBS at day 13; p = 0.0008 for temozolomide vs CT322 at day 27; p = 0.04 for temozolomide vs combination temozolomide plus CT322 at day 27; p = 0.0001 for CT322 vs combination temozolomide plus CT322 (unpaired t test)). MRI images from a representative subset of mice further confirmed the reduced tumor size with combined CT322 plus temozolomide treatment (Fig. 2b–d). Additionally, mice treated with the combination CT322 plus temozolomide exhibited longer survival relative to both CT322-monotherapy (p = 0.029, Gehan-Breslow-Wilcoxon Test) and temozolomide-monotherapy (p = 0.044, Gehan-Breslow-Wilcoxon Test) (Fig. 2e). There was no statistically significant difference in survival between temozolomidemonotherapy and CT322- monotherapy groups. The median survival was 29 days (sd = 6 days) in the CT322-monotherapy group, 32 days (sd = 2 days) in the tem-ozolomide-monotherapy group, and 47 days (sd = 11 days) in the combination CT322 plus temozolomide treated group.
Fig. 2.
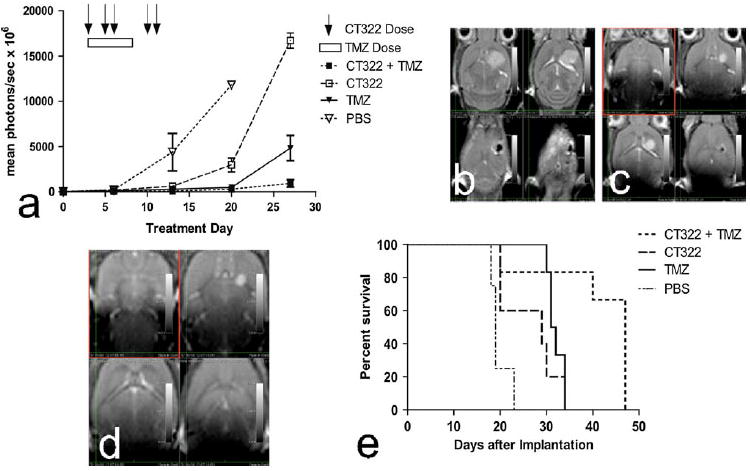
a–e Combination of CT322 with temozolomide demonstrates improved outcomes over monotherapy with each agent. Data represents 4 mice in the PBS control group (n = 4), 5 mice in the CT322 treatment group (CT322, n = 5), 6 mice in the temozolomide group (TMZ, n = 6), and 6 mice in the temozolomide plus CT322 group (CT322 + TMZ, n = 6). a The combination of CT322 with temozolomide resulted in lower peak bioluminescence values than either CT322 or temozolomide as monotherapy agents. p = 0.04 for temozolomide versus PBS at day 13; p = 0.0008 for temozolomide versus CT322 at day 27; p = 0.04 for temozolomide versus combination temozolomide plus CT322 at day 27; p = 0.0001 for CT322 versus combination temozolomide plus CT322 (unpaired t test). b Multiple image slices from a single MRI demonstrating 33 mm3 tumor 24 days after transplant with CT322 treatment. The bioluminescence measurement at day 20 for this individual was 3,973 mean photons/s × 106. c Multiple image slices from a single MRI demonstrating 3.9 mm3 tumor 26 days after transplant with temozolomide treatment. The bioluminescence measurement at day 27 for this individual was 1,273 mean photons/s × 106. d Multiple image slices from a single MRI demonstrating 1.2 mm3 tumor 26 days after transplant with temozolomide and CT322 combination treatment. The bioluminescence measurement at day 27 for this individual was 1,549 mean photons/s × 106. e Kaplan–Meyer curve demonstrating mice treated with the combination CT322 plus temozolomide exhibited longer survival relative to both CT322-monotherapy (p = 0.029, Gehan-Breslow-Wilcoxon Test) and temozolomide-monotherapy (p = 0.044, Gehan-Breslow-Wilcoxon Test). There was no statistically significant difference in survival between temozolomide-monotherapy and CT322 monotherapy groups. The median survival was 29 days (sd = 6 days) in the CT322-monotherapy group, 32 days (sd = 2 days) in the temozolomide-monotherapy group, and 47 days (sd = 11 days) in the combination CT322 plus temozolomide treated group. Abbreviations: TMZ = temozolomide
Addition of CT322 improves efficacy of the current standard glioblastoma treatment regimen. Ultimately, if CT322 were to be tested as a therapeutic for newly diagnosed glioblastoma, it will likely be combined with temozolomide and radiation. Thus, we tested the addition of CT322 to combined temozolomide and radiation therapy. In this experiment, the temozolomide with radiation therapy combination was the control group, and CT322 added to temozolomide with radiation therapy was the experimental group. PBS treatment was also concurrently tested as an additional point of reference, but radiation therapy and temozolomide were not used as variables in the design of this experiment. When added to a simulated regimen of concurrent temozolomide with radiation therapy, CT322 caused further reduction in tumor burden as assessed by bioluminescence (Fig. 3a) and MRI (Fig. 3b, c). By day 35 after transplant, the temozolomide-plus-radiation therapy group's (n = 7) average peak bioluminescence was 16,000 photons/ s × 106, whereas the addition of CT322 to the regimen (n = 7) significantly dropped the bioluminescence to 760 photons/s × 106 (p = 0.05 for temozolomide-plus-radiation vs PBS at day 21; p = 0.01 for temozolomide-plus-radiation-plus-CT322 vs PBS at day 21; p = 0.02 for temozolomide-plus-radiation vs temozolomide-plus-radiation-plus-CT322 at day 35 (unpaired t test)). Accordingly, prolonged survival was observed with the CT322-temozolomide-radiation treated mice relative to the temozolomide-radiation treated mice (p = 0.0014, Gehan-Breslow-Wilcoxon Test) (Fig. 3d). The median survival of the temozolomide-radiation treated mice was 33 days (sd = 4 days), whereas the median survival of the CT322-temozolomide-radiation treated mice was 47 days (sd = 5 days).
Fig. 3.
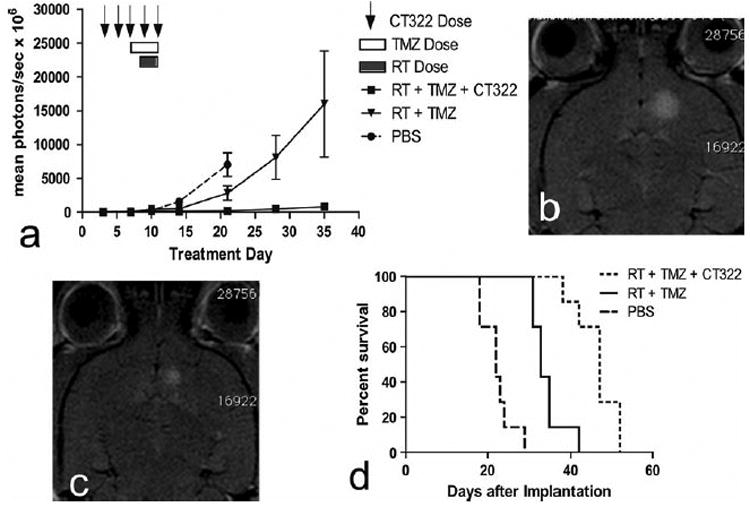
a–d Addition of CT322 to current standard glioblastoma treatment of temozolomide plus radiation therapy improves efficacy of treatment. Data represents 7 mice in each of the treatment groups (PBS, n = 7; RT + TMZ, n = 7, RT + TMZ + CT322, n = 7). a Addition of CT322 to a regimen of temozolomide plus radiation results in decreased peak bioluminescence. p = 0.05 for temozolomide-plus-radiation versus PBS at day 21; p = 0.01 for temozolomide-plus-radiation-plus-CT322 versus PBS at day 21; p = 0.02 for temozolomide-plus-radiation versus temozolomide-plus-radiation-plus-CT322 at day 35 (unpaired t test). b MRI demonstrating tumor after treatment with temozolomide and radiation therapy alone. c MRI demonstrating tumor after treatment with temozolomide and radiation therapy plus CT322. d Kaplan–Meyer curve demonstrating improved survival with the addition of CT322 to temozolomide and radiation therapy (p = 0.0014, Gehan-Breslow-Wilcoxon Test). The median survival of the temozolomide-radiation treated mice was 33 days (sd = 4 days), whereas the median survival of the CT322-temozolomide-radiation treated mice was 47 days (sd = 5 days). Abbreviations: TMZ = temozolomide. RT = radiation therapy
CT322 treatment inhibited angiogenesis and enhanced apoptosis. Immunohistochemical (IHC) analysis of the xenograft obtained after CT322 treatment revealed decreased staining of CD31 compared to control treatment, suggesting decreased micro-vascular density (Fig. 4). This decrease was associated with increased caspase3 staining and decreased Ki67 staining with CT322 compared to control treatment, suggesting increased apoptosis and decreased cellular proliferation. In contrast, temozolomide treatment was associated with increased caspase3 staining and decreased Ki67 staining, without a significant change in the level of CD31 staining. These results suggest that the tumoricidal activity of temozolomide was not derived from inhibition of angiogenesis. Consistent with this observation, the CD31 staining of CT322-monotherapy treated and combination (CT322 plus temozolomide) treated tumor were comparable. Interestingly, though the addition of CT322 to temozolomide significantly delayed tumor growth (Fig. 2a) and prolonged survival (Fig. 2e), this effect was not reflected in Ki-67 or caspase3 staining (Fig. 4). With the addition of CT322 to temozolomide-plus-radiation, IHC analysis of CD31-, Ki67-, and caspase3-staining suggested a trend toward reduced endothelial cell counts, reduced cellular proliferation, and increased apoptosis with CT322 (Fig. 4). However, this trend did not reach statistical significance between these two treatment groups.
Fig. 4.
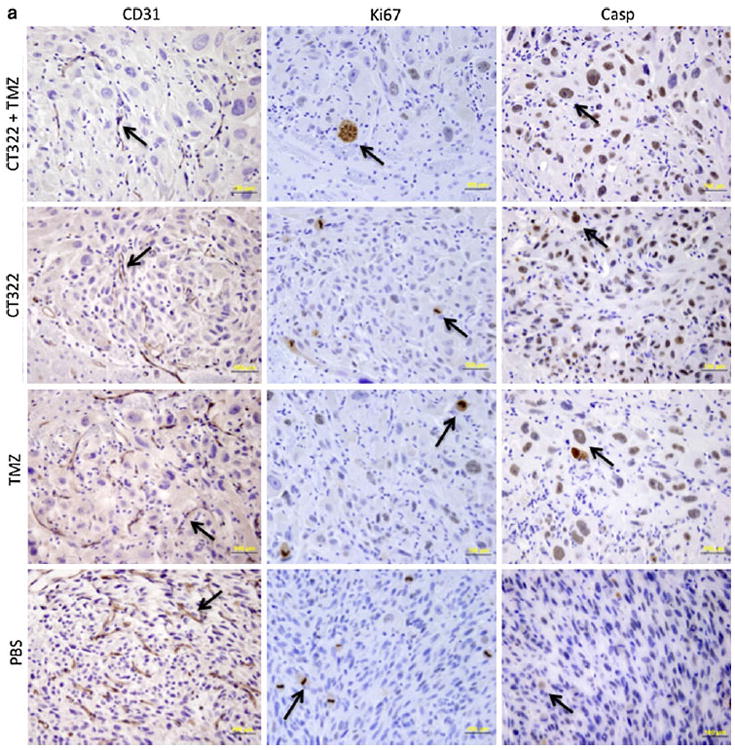
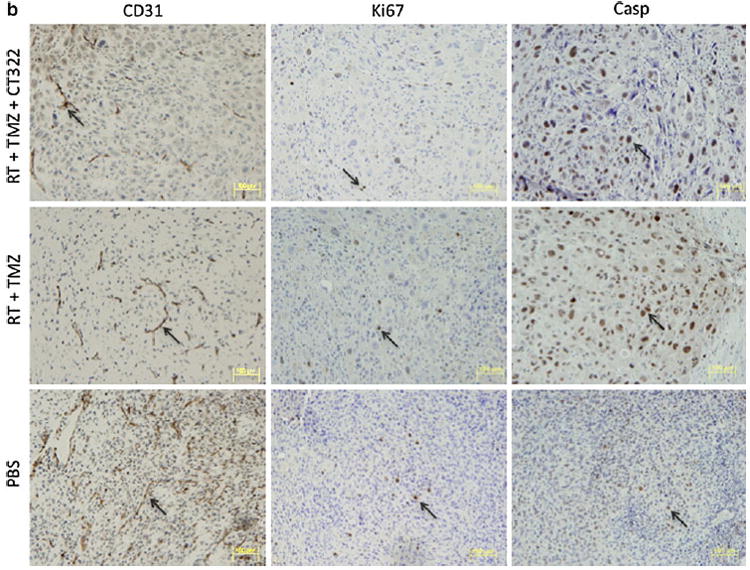
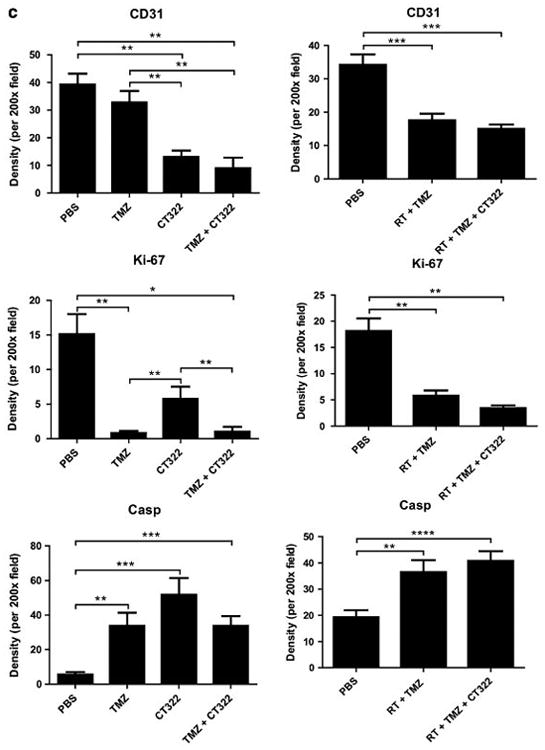
a–c Immunohistochemical (IHC) analysis. a Representative histology images of PBS, CT322 monotherapy, temozolomide monotherapy, and CT322 plus temozolomide therapy groups at day 14 with arrows indicating examples of positive cells at ×400 magnification (bar represents 100 μm). b Representative histology images of PBS, temozolomide plus radiation, and CT322 plus temozolomide and radiation therapy groups at day 14 with arrows indicating examples of positive cells at ×200 magnification (bar represents 100 μm). c Quantification of average CD31, Ki67, and cleaved-Caspase3 positive cell counts per ×200 high powered field upon immunohistochemical staining of tumor samples for control (PBS), temozolomide monotherapy, CT322 monotherapy, and CT322-plus-temozolomide combination therapy, temozolomide with radiation therapy, and CT322 plus temozolomide with radiation therapy. Abbreviations: Casp = caspase3. PBS = phosphate buffered saline. TMZ = temozolomide. RT = radiation therapy
Discussion
This study examined the efficacy of CT322, an Adnectin-based inhibitor of VEGFR-2, in the treatment of glioblastoma using a murine intracranial U87-EGFRvIII luciferase xenograft model. We assessed the effect of tumor growth using independent determinants, including bioluminescence, MRI, and overall survival. Our results indicate that CT322 delayed the growth of the U87-EGFRvIII xenograft and prolonged survival. As a component of combination therapies with temozolomide or with concurrent temozolomide-radiation therapy, CT322 further reduced tumor growth and improved survival. IHC analyses suggest that the mechanism underlying this therapeutic effect is predominantly through inhibition of angiogenesis.
While CT322 has been previously studied in a subcutaneous xenograft model of glioblastoma [19], this is the first report of the efficacy of CT322 in an intracerebral xenograft model of glioblastoma. By using intracerebral grafts, these results more closely model glioblastoma in its native central nervous system [23]. By engineering the U87 cells to express EGFRvIII, which is known to enhance VEGF-secretion [7], we demonstrate effectiveness of CT322 in this setting. Further, our results demonstrate that the CT322-temozolomide-radiation therapy combination is well tolerated and efficacious against EGFRvIII-expressing glioblastomas.
A major limitation of this study involves the use of a single xenograft model, namely that of the U87MG cell over-expressing the EGFRvIII oncogene. We recognize that every experimental model of glioblastoma (xenograft, transgenic, or otherwise) necessarily captures only a subset of the biology inherent within the clinical entity of glioblastoma. In this context, we believe that translational efforts will require testing of any therapeutic strategy using complementary experimental models. We hope that our results provide a sound foundation for such future testing in terms of CT322.
This study demonstrates that CT322 may be combined with radiation and temozolomide for improved survival and reduction in tumor size. Several mechanisms have been proposed to explain the success of anti-angiogenic and anti-VEGF signaling combination therapies in brain tumors [24]. One hypothesis suggests that inhibition of tumor vascularization may decrease nutrient delivery to cells between chemotherapy cycles, thus slowing tumor cell repopulation [25]. This study did not support this hypothesis, as CD31 counts were not significantly reduced with the addition of CT322 to either temozolomide or temozolomide plus radiation therapy. Another hypothesis suggests that anti-angiogenic therapy normalizes tumor vasculature, thereby facilitating delivery of chemotherapeutic agents [26]. Normalization of tumor vasculature may also lead to a reduction in edema, which may account for improved outcomes [27]. Ultimately, the mechanism of therapeutic effect of CT322 is yet to be fully elucidated, and may be a summation of these various effects.
Our results echo previous studies that found improved survival with anti-VEGF therapy, independent of and despite persistent tumor growth [27, 28]. The cause is frequently attributed to normalization of vasculature and reduction of cerebral edema, as reported by Kamoun et al [27]. who demonstrated in an orthotopic mouse model that cediranib alleviated edema as measured by MRI and weight measurements of water content. This improved survival despite tumor growth was seen in our experiments. For example, animals treated with CT322 have tumors that have 3.5-fold more bioluminescence than animals treated with only temozolomide (Fig. 2a). However, the two groups have similar survival (Fig. 2e). We expect this is due to this previously described phenomenon of vascular normalization and reduced edema with anti-VEGF therapy [27, 28].
Importantly, this vascular normalization with anti-VEGF treatment also leads to decreased gadolinium contrast enhancement on MRI [29]. Given this, it may be inappropriate to compare bioluminescence imaging (which is a function of the number of cells metabolizing luciferin) to contrast enhancement on MRI (which is a function of vascular permeability). This is particularly true when testing a drug that is expected to alter vascular permeability. As such, bioluminescence was felt to be a more appropriate measure of tumor growth for our experiments. Thus, MRI was obtained in only a few animals per group for illustrative purposes [29].
Several anti-angiogenic agents have been explored as single agents in the treatment of recurrent glioblastoma, including bevacizumab, cediranib, and aflibercept. Bevacizumab is an antibody directed against human VEGF. In 2009, the United States Food and Drug Administration approved bevacizumab as a single agent for the treatment of glioblastoma after previous treatment failure [30]. This approval is based on two landmark studies of bevacizumab. The first study enrolled 78 patients and showed a partial response rate of 25.9 % with median response duration of 4.2 months. The second study enrolled 56 patients and demonstrated a partial response rate of 19.6 % with a median response duration of 3.9 months [30]. Of note, no overall survival benefit was demonstrated by either study. Cediranib targets all VEGF-receptors [31]. Results of a phase II trial of cediranib as a monotherapy for recurrent glioblastoma were encouraging, demonstrating an “alive and progression free at 6-months” (APF6) rate of 25.8 % [31]. Aflibercept, aka VEGF-trap, is a soluble decoy receptor for VEGF. A phase II trial of aflibercept as a monotherapy for recurrent GBM, which included 42 patients, demonstrated moderate toxicity and a disappointing APF6 rate of 7.7 % [32].
Combination regimens that include antiangiogenic agents have also been studied for newly diagnosed glioblastoma, including bevacizumab and cilengitide. A phase II trial of bevacizumab as part of a regimen of temozolomide and radiation therapy following initial resection of glioblastoma has demonstrated improved progression free survival (13.6 months vs 6.9–7.6 months in control cohorts) with no significant improvement in overall survival (19.6 months vs 14.6–21.1 months in control cohorts) [33]. Cilengitide targets integrins active on endothelial cells during angiogenesis. In a phase I/IIa trial for newly diagnosed glioblastoma, concomitant cilengitide therapy in combination with standard chemoradiotherapy resulted in a median overall survival of 16.1 months. For patients with O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, median survival was longer, at 23.2 months [34]. There are several other anti-angiogenic agents under investigation, and further research is needed to identify optimal agents and define the role of antiangiogenic therapy in the treatment of newly diagnosed or recurrent glioblastoma [35].
In sum, our pre-clinical results support further evaluation of CT322 into the standard temozolomide-radiation regimen in the treatment of glioblastomas over-expressing EGFRvIII. A first step toward this translation was provided by a phase I clinical trial of CT322 confirming safety [21]. Phase II trials of CT322 are underway [36]. Additional preclinical studies focused on time of intervention and treatment schedule will enhance understanding on the value of combining VEGF-axis inhibition with temozolomide chemotherapy and radiation treatment on a sensitive population of GBM patients.
Acknowledgments
Grant Support: Adnexus, a Bristol-Myers Squibb R&D Company.
Footnotes
Conflict of interest None
Contributor Information
J. Dawn Waters, Email: j.dawn.waters@gmail.com, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA.
Carlos Sanchez, Department of Neurosurgery, MGH-HMS Center for Nervous System Repair, MGH, EDR 410 50 Blossom St, Boston, MA 02114, USA.
Ayguen Sahin, Department of Neurosurgery, MGH-HMS Center for Nervous System Repair, MGH, EDR 410 50 Blossom St, Boston, MA 02114, USA.
Diahnn Futalan, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA.
David D. Gonda, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA
Justin K. Scheer, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA
Johnny Akers, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA.
Kamalakannan Palanichamy, Department of Radiation Oncology, Massachusetts General Hospital, 55 Fruit Street, Boston, MA 02114, USA.
Peter Waterman, Center for Molecular Imaging, Massachusetts General Hospital, Building 149, 6th Floor, Charlestown, MA 02129-2060, USA.
Arnab Chakravarti, Department of Radiation Oncology, Massachusetts General Hospital, 55 Fruit Street, Boston, MA 02114, USA.
Ralph Weissleder, Center for Molecular Imaging, Massachusetts General Hospital, Building 149, 6th Floor, Charlestown, MA 02129-2060, USA.
Brent Morse, Adnexus, 100 Beaver Street, Waltham, MA 02453, USA.
Nick Marsh, Adnexus, 100 Beaver Street, Waltham, MA 02453, USA.
Eric Furfine, Adnexus, 100 Beaver Street, Waltham, MA 02453, USA.
Clark C. Chen, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA
Irvith Carvajal, Adnexus, 100 Beaver Street, Waltham, MA 02453, USA.
Bob S. Carter, Email: bobcarter@ucsd.edu, Division of Neurosurgery, University of California-San Diego, 3855 Health Sciences Drive, #0987, La Jolla, CA 92093, USA.
References
- 1.Walker MD, Alexander E, Jr, Hunt WE, MacCarty CS, Mahaley MS, Jr, Mealey J, Jr, Norrell HA, Owens G, Ransohoff J, Wilson CB, Gehan EA, Strike TA. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333–343. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, Bent MJvd, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann Jr, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Quick A, Patel D, Hadziahmetovic M, Chakravarti A, Mehta M. Current therapeutic paradigms in Glioblastoma. Rev Recent Clin Trials. 2010;5:14–27. doi: 10.2174/157488710790820544. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt NO, Westphal M, Hagel C, Ergun S, Stavrou D, Rosen EM, Lamszus K. Levels of vascular endothelial growth factor hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84:10–18. doi: 10.1002/(SICI)1097-0215(19990219)84:1<10:AID-IJC3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 5.Plate K, Breier G, Weich H, Mennel H, Risau W. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 6.Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 7.Feldkamp MM, Lau N, Rak J, Kerbel RS, Guha A. Normoxic and hypoxic regulation of vascular endothelial growth factor (VEGF) by astrocytoma cells is mediated by Ras. Int J Cancer. 1999;81:118–124. doi: 10.1002/(SICI)1097-0215(19990331)81:1<118:AID-IJC20>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhou YH, Tan F, Hess KR, Yung WKA. The expression of PAX6 PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: relationship to tumor grade and survival. Clin Cancer Res. 2003;9:3369–3375. [PubMed] [Google Scholar]
- 9.Leon SP, Folkerth RD, Black PM. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer. 1996;77:362–372. doi: 10.1002/(SICI)1097-0142(19960115)77:2<362:AID-CNCR20>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 10.Wick W, Weller M, Weiler M, Batchelor T, Yung AWK, Platten M. Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro-Oncol. 2011;13:566–579. doi: 10.1093/neuonc/nor039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norden AD, Drappatz J, Muzikansky A, David K, Gerard M, McNamara MB, Phan P, Ross A, Kesari S, Wen PY. An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J Neurooncol. 2008;92:149–155. doi: 10.1007/s11060-008-9745-8. [DOI] [PubMed] [Google Scholar]
- 12.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol. 1999;9:211–220. doi: 10.1006/scbi.1998.0091. [DOI] [PubMed] [Google Scholar]
- 14.Baldwin ME, Catimel B, Nice EC, Roufail S, Hall NE, Stenvers KL, Karkkainen MJ, Alitalo K, Stacker SA, Achen MG. The specificity of receptor binding by vascular endothelial growth factor-D is different in mouse and man. J Biol Chem. 2001;276:19166–19171. doi: 10.1074/jbc.M100097200. [DOI] [PubMed] [Google Scholar]
- 15.Millauer B, Wizigmann-Voos S, Schnürch H, Martinez R, Møller NPH, Risau W, Ullrich A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 16.Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. The EMBO J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, Kunisada T, Mori H, Iwama T. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun. 2007;360:553–559. doi: 10.1016/j.bbrc.2007.06.094. [DOI] [PubMed] [Google Scholar]
- 18.Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Mamluk R, Carvajal IM, Morse BA, Wong H, Abramowitz J, Aslanian S, Lim AC, Gokemeijer J, Storek MJ, Lee J, Gosselin M, Wright MC, Camphausen RT, Wang J, Chen Y, Miller K, Sanders K, Short S, Sperinde J, Prasad G, Williams S, Kerbel R, Ebos J, Mutsaers A, Mendlein JD, Harris AS, Furfine ES. Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2. Monoclon Antib. 2010;2:199–208. doi: 10.4161/mabs.2.2.11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Prot Eng Des Sel. 2010;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC, Sankhala K, Furfine E, Gokemeijer J, Iacono L, Eaton C, Silver BA, Mita M. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin Cancer Res. 2011;17:363–371. doi: 10.1158/1078-0432.ccr-10-1411. [DOI] [PubMed] [Google Scholar]
- 22.Murphy GJ, Mostoslavsky G, Kotton DN, Mulligan RC. Exogenous control of mammalian gene expression via modulation of translational termination. Nat Med. 2006;12:1093–1099. doi: 10.1038/nm1376. [DOI] [PubMed] [Google Scholar]
- 23.Shapiro WR. The chemotherapy of intracerebral vs subcutaneous murine gliomas: a comparative study of the effect of VM 26. Arch Neurol. 1974;30:222–226. doi: 10.1001/archneur.1974.00490330030005. [DOI] [PubMed] [Google Scholar]
- 24.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312:1171–1175. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 25.Hudis CA. Clinical implications of antiangiogenic therapies. Oncology. 2005;19:26–31. [PubMed] [Google Scholar]
- 26.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 27.Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, Batchelor TT, di Tomaso E, Duda DG, Munn LL, Fukumura D, Sorensen AG, Jain RK. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27:2542–2552. doi: 10.1200/jco.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerstner ER, Duda DG, Tomaso Ed, Ryg PA, Loeffler JS, Sorensen AG, Ivy P, Jain RK, Batchelor TT. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229–236. doi: 10.1038/nrclinonc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerstner ER, Frosch MP, Batchelor TT. Diffusion magnetic resonance imaging detects pathologically confirmed, non-enhancing tumor progression in a patient with recurrent glioblastoma receiving bevacizumab. J Clin Oncol. 2010;28:e91–e93. doi: 10.1200/JCO.2009.25.0233. [DOI] [PubMed] [Google Scholar]
- 30.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin(R)) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 31.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T, Louis DN, Cohen KS, Chea H, Exarhopoulos A, Loeffler JS, Moses MA, Ivy P, Sorensen AG, Wen PY, Jain RK. Phase II Study of Cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in Patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/jco.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, Yao J, Jackson EF, Lieberman F, Robins HI, Mehta MP, Lassman AB, DeAngelis LM, Yung WKA, Chen A, Prados MD, Wen PY. Phase II Study of aflibercept in recurrent malignant glioma: a North American brain tumor consortium study. J Clin Oncol. 2011;29:2689–2695. doi: 10.1200/jco.2010.34.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai A, Tran A, Nghiemphu PL, Pope WB, Solis OE, Selch M, Filka E, Yong WH, Mischel PS, Liau LM, Phuphanich S, Black K, Peak S, Green RM, Spier CE, Kolevska T, Polikoff J, Fehrenbacher L, Elashoff R, Cloughesy T. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2010;29:142–148. doi: 10.1200/jco.2010.30.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement Paul MJ, Grabenbauer GG, Ochsenbein AF, Simon M, Dietrich PY, Pietsch T, Hicking C, Tonn JC, Diserens AC, Pica A, Hermisson M, Krueger S, Picard M, Weller M. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28 doi: 10.1200/JCO.2009.26.6650. [DOI] [PubMed] [Google Scholar]
- 35.Beal K, Abrey LE, Gutin PH. Antiangiogenic agents in the treatment of recurrent or newly diagnosed glioblastoma: analysis of single-agent and combined modality approaches. Radiat Oncol. 2011;6:2. doi: 10.1186/1748-717X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiff DRD, Kesari S, Mikkelsen T, De Groot JF, Fichtel L, Coyle TE, Wong EEC, Silver B. Phase II study of CT-322, a targeted biologic inhibitor of VEGFR-2 based on a domain of human fibronectin, in recurrent glioblastoma (rGBM) [abstract] J Clin Oncol. 2010;28:182s. doi: 10.1007/s10637-014-0186-2. [DOI] [PubMed] [Google Scholar]