

Abstract
The Unfolded Protein Response (UPR) indirectly regulates extracellular proteostasis through transcriptional remodeling of endoplasmic reticulum (ER) proteostasis pathways. This remodeling attenuates secretion of misfolded, aggregation-prone proteins during ER stress. Through these activities, the UPR has a critical role in preventing the extracellular protein aggregation associated with numerous human diseases. Here, we demonstrate that UPR activation also directly influences extracellular proteostasis through the upregulation and secretion of the ER HSP40 ERdj3/DNAJB11. Secreted ERdj3 binds misfolded proteins in the extracellular space, substoichiometrically inhibits protein aggregation, and attenuates proteotoxicity of disease-associated toxic prion protein. Moreover, ERdj3 can co-secrete with destabilized, aggregation-prone proteins in a stable complex under conditions where ER chaperoning capacity is overwhelmed, preemptively providing extracellular chaperoning of proteotoxic misfolded proteins that evade ER quality control. This regulated co-secretion of ERdj3 with misfolded clients directly links ER and extracellular proteostasis during conditions of ER stress. ERdj3 is, to our knowledge, the first metazoan chaperone whose secretion into the extracellular space is regulated by the UPR, revealing a new mechanism by which UPR activation regulates extracellular proteostasis.
Keywords: ER stress, ERdj3, extracellular proteostasis, molecular chaperones, unfolded protein response
See also: TM Buck & JL Brodsky (January 2015)
Introduction
Imbalances in extracellular protein homeostasis (or proteostasis) and consequent protein aggregation are inextricably linked to degenerative phenotypes in over 30 human protein misfolding diseases, including Alzheimer’s disease, Creutzfeldt–Jakob disease and the transthyretin (TTR) amyloidoses (Kelly, 1996; Rochet & Lansbury, 2000; Stefani & Dobson, 2003; Buxbaum, 2004; Haass & Selkoe, 2007). Compelling genetic and pharmacologic evidence supports a causal relationship between protein aggregation (including amyloidogenesis) and the degeneration of post-mitotic tissue in these disorders (Tanzi & Bertram, 2005; Aguzzi et al, 2007; Gotz et al, 2011; Coelho et al, 2012). Primary determinants of extracellular proteostasis capacity include the spectrum and concentration of secreted proteostasis factors (e.g., chaperones) (Wyatt et al, 2013) and the efficiency of protein folding and quality control in the endoplasmic reticulum (ER) (Wiseman et al, 2007).
Extracellular proteostasis capacity is regulated by secreted proteins that prevent the formation of protein aggregates associated with disease. The best characterized secreted chaperones such as clusterin directly bind misfolded proteins in the extracellular environment and prevent their aggregation through an ATP-independent “holdase” mechanism (Wyatt et al, 2012). Deletion of clusterin predisposes mice to aging-dependent progressive glomerulopathy (Rosenberg et al, 2002) and increases Aβ(1–42) aggregation and deposition in mouse models of Alzheimer’s disease (DeMattos et al, 2004). Furthermore, genome-wide association studies implicate clusterin in the development of Alzheimer’s disease (Harold et al, 2009; Lambert et al, 2009; Wijsman et al, 2011). Small populations of some ER chaperones, such as the HSP70 BiP and the lectin calreticulin, can be trafficked to the plasma membrane, particularly under stress or during apoptosis (Martins et al, 2010; Zhang et al, 2010). Protein disulfide isomerases (PDIs) can also be secreted to promote extracellular disulfide exchange (Jordan & Gibbins, 2006; Hahm et al, 2013). A role for these chaperones in extracellular proteostasis maintenance has not been demonstrated to date, rather their surface expression has been implicated in immunological signaling (Peters & Raghavan, 2011; Lee, 2014).
Extracellular proteostasis is also impacted by proteostasis in the ER, which is responsible for the folding and trafficking of the 1/3 of the human proteome that is targeted to the cellular secretory pathway (Fewell et al, 2001; Braakman & Bulleid, 2011). In the ER, nascent polypeptides interact with components of ER protein folding pathways to facilitate their folding into native three-dimensional conformations (Buck et al, 2007). Folded proteins are then packaged into vesicles at the ER membrane and trafficked to downstream compartments of the secretory pathway or the extracellular space. Proteins unable to attain native three-dimensional conformations in the ER are instead targeted to ER degradation pathways such as ER-associated degradation (ERAD) (Benyair et al, 2011). The partitioning of polypeptides between ER protein folding/trafficking and degradation pathways, also referred to as ER quality control, prevents the secretion of misfolded, aggregation-prone proteins (Powers et al, 2009; Araki & Nagata, 2011).
Despite the typical efficiency of ER quality control, exposure to genetic, environmental or aging-related stresses leads to increased protein misfolding within the ER lumen and imbalances in ER proteostasis. Such stresses can increase secretion of misfolding-prone proteins into the extracellular space, directly challenging extracellular proteostasis capacity and facilitating concentration-dependent protein aggregation into proteotoxic oligomeric conformations. As such, ER stress is pathologically associated with numerous extracellular protein aggregation diseases including the systemic amyloidoses and Alzheimer’s disease (Teixeira et al, 2006; Kim et al, 2008).
To restore ER proteostasis following stress, cells activate the Unfolded Protein Response (UPR). The UPR consists of three integrated stress-responsive signaling pathways activated downstream of the ER stress-sensing proteins IRE1, ATF6, and PERK (Schroder & Kaufman, 2005; Walter & Ron, 2011). These stress sensors are activated by the accumulation of misfolded proteins within the ER lumen (a consequence of ER stress) (Bertolotti et al, 2000; Okamura et al, 2000; Shen et al, 2002). The activation of UPR signaling pathways results in the attenuation of new protein synthesis (Harding et al, 1999) and transcriptional remodeling of ER protein folding, trafficking, and degradation pathways (Lee et al, 2003; Yamamoto et al, 2007; Shoulders et al, 2013), thus enhancing ER proteostasis capacity and quality control. Through these mechanisms, UPR activation reduces accumulation of misfolded proteins in the ER and attenuates the aberrant secretion of aggregation-prone proteins into the extracellular space (Adachi et al, 2008; Shoulders et al, 2013).
In contrast, the functional impact of UPR signaling on extracellular proteostasis capacity remains poorly defined. Not only are the established secreted chaperones not transcriptional targets of the UPR, but clusterin secretion is attenuated during conditions of ER stress, indicating that clusterin secretion is not a protective mechanism to regulate extracellular proteostasis in response to pathologic ER insults (Nizard et al, 2007). Similarly, ER stress reduces secretion of ER proteostasis factors such as protein disulfide isomerase (PDI), suggesting reduced extracellular regulation of disulfide integrity during ER stress (Terada et al, 1995). Thus, we sought to study the functional role for UPR signaling in adapting extracellular proteostasis capacity during conditions of ER stress.
Here, we report that UPR activation regulates extracellular proteostasis directly through the secretion of the ER-targeted HSP40 co-chaperone ERdj3. While ERdj3 is established to function as an HSP40 co-chaperone for BiP within the ER HSP70 chaperoning pathway (Shen & Hendershot, 2005), we show that ERdj3 is also a UPR-induced secreted chaperone, whose extracellular levels increase both in response to ER stress and stress-independent activation of the UPR-associated transcription factor ATF6. Secreted ERdj3 binds to misfolded proteins in the extracellular space, prevents the aggregation of amyloidogenic Aβ1–40 at substoichiometric concentrations, and ameliorates the toxic effects of misfolded prion protein on neuronal cells. Furthermore, we demonstrate that ERdj3 can co-secrete with destabilized, misfolding-prone clients under conditions where the ER HSP70 chaperoning pathway is overwhelmed, preemptively chaperoning these misfolding-prone secreted proteins that evade ER quality control in the extracellular environment. Thus, the capacity for ERdj3 to function in both ER and extracellular proteostasis provides an unanticipated direct link between these two environments that is regulated by the UPR during conditions of ER stress.
Results
ER stress increases ERdj3 secretion
We sought to identify UPR-induced secreted chaperones that could increase extracellular proteostasis capacity in response to ER stress. We surveyed previously reported transcriptional and proteomic profiles (Lee et al, 2003; Wu et al, 2007; Yamamoto et al, 2007; Adachi et al, 2008; Shoulders et al, 2013) of UPR stress-responsive signaling to identify a UPR-induced gene(s) whose protein product: (1) is targeted to the ER by an N-terminal signaling sequence, (2) lacks an ER retention motif (e.g., KDEL) or a transmembrane domain (Dancourt & Barlowe, 2010), and (3) selectively binds destabilized, misfolded proteins (i.e., has chaperone activity). Through this analysis, we identified the HSP40 co-chaperone ERdj3 as a potential UPR-induced secreted chaperone that meets all of the above criteria.
ERdj3 is a well-established component of the intracellular ER proteostasis network and is transcriptionally upregulated by the UPR during ER stress (Shen & Hendershot, 2005). In the ER, ERdj3 binds to proteins in non-native conformations through an ATP-independent mechanism and directs these clients to the ER-localized, ATP-dependent HSP70 BiP chaperoning pathway, facilitating the proper folding of client proteins into folded three-dimensional conformations in the ER lumen (Shen & Hendershot, 2005; Jin et al, 2008, 2009; Marcinowski et al, 2011; Guo & Snapp, 2013). ERdj3 contains neither a C-terminal KDEL nor a transmembrane domain, implying that ERdj3 can efficiently traverse the secretory pathway. Consistent with this prediction, extracellular ERdj3 is implicated in neuronal development (Wong et al, 2010).
We evaluated whether ERdj3 is secreted from HEK293T cells overexpressing wild-type ERdj3 (ERdj3WT). Overexpression of ERdj3WT significantly increases extracellular ERdj3 protein levels in conditioned media, while only slightly increasing intracellular ERdj3 levels (Fig 1A). In contrast, overexpression of ERdj3 containing a C-terminal KDEL ER retention motif (ERdj3KDEL) only slightly increases extracellular ERdj3 protein levels, while significantly increasing intracellular ERdj3 (Fig 1A). Furthermore, secretion of overexpressed ERdj3WT is inhibited by the addition of the secretory trafficking inhibitor brefeldin A (Supplementary Fig S1A and B). These results strongly indicate that overexpressed ERdj3WT is secreted through the canonical secretory pathway. Endogenous ERdj3 was also detected in conditioned media collected from a variety of human cell lines, including HepG2, Huh7, SH-SY5Y, and HeLa (Supplementary Fig S1C).
Figure 1. ER stress increases ERdj3 secretion.
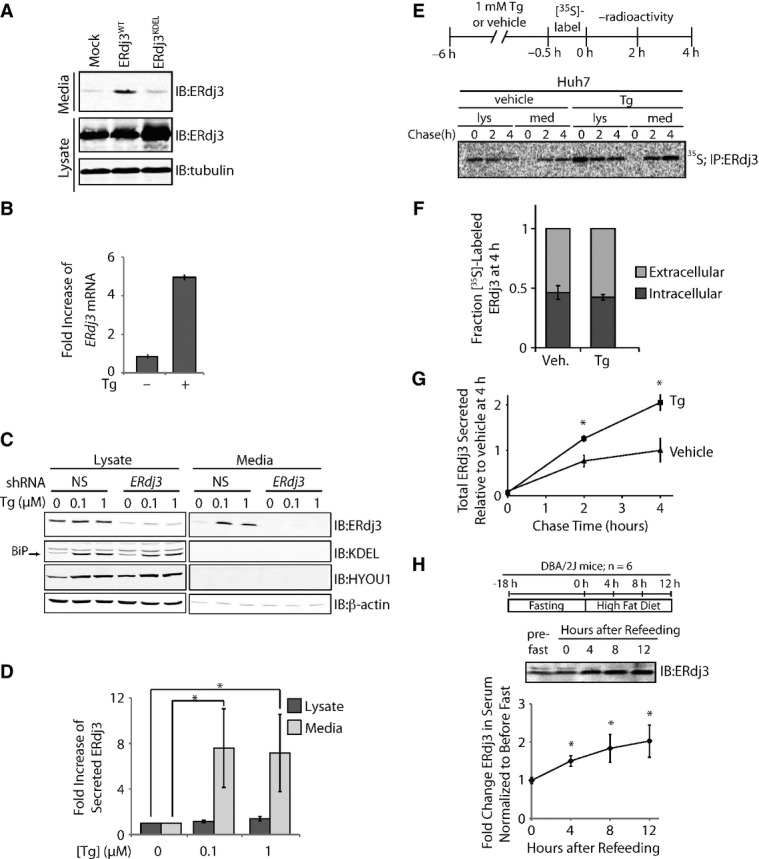
- A Immunoblot of media and lysate collected from HEK293T cells overexpressing ERdj3WT or ERdj3KDEL. Fresh media was conditioned on cells for 24 h prior to harvest.
- B qPCR analysis of ERdj3 mRNA in HEK293T cells treated with or without thapsigargin (Tg; 6 h, 100 nM). Error bars represent the mean ± 95% confidence interval as calculated in DataAssist 2.0 (n = 3).
- C Representative immunoblot of lysates and media collected from HEK293T-Rex cells stably expressing non-silencing (NS) or ERdj3 shRNA. Cells were treated with the indicated concentration of Tg in fresh media for 16 h.
- D Quantification of the fold increase of ERdj3 in lysates and media collected from HEK293T-Rex cells treated with the indicated concentration of Tg in fresh media for 16 h as in (C). *P < 0.05; n = 3, mean ± SEM.
- E Representative autoradiogram of [35S]-labeled ERdj3 in lysates and media collected from Huh7 cells incubated in the presence or the absence of Tg (1 μM). The experimental paradigm for labeling is shown above.
- F Quantification of fraction intracellular (dark gray) and extracellular (light gray) [35S]-labeled ERdj3 following a 4 h chase relative to total [35S]-labeled ERdj3 at 4 h (as shown in E). Error bars represent the standard error from biological replicates (n = 3).
- G Quantification of total [35S]-labeled ERdj3 in media collected from cells pretreated with vehicle or Tg, as shown in (E). The media [35S]-labeled ERdj3 was measured by densitometry and is normalized to the amount of media [35S]-labeled ERdj3 at 4 h from vehicle-treated cells. *P-value < 0.05; n = 3, mean ± SEM.
- H Representative immunoblot and quantification of sera collected at the indicated times from DBA/2J mice fed a high-fat (60% fat) diet after a 18 h fast. A schematic of the fasting/refeeding experimental setup is shown above. The quantification was normalized for each mouse to the amount of ERdj3 in the serum draw immediately prior the fast. *P-value < 0.05; n = 6, mean ± SEM.
Source data are available online for this figure.
We confirmed the ER stress-dependent increase in ERdj3 mRNA in HEK293T cells treated with the small molecule SERCA inhibitor thapsigargin (Tg)—a potent inducer of ER stress (Fig 1B). Upon Tg-induced ER stress, ERdj3 protein levels increased primarily in conditioned media and not intracellularly for HEK293T-Rex (Fig 1C and D). RNAi depletion of ERdj3 reduced intracellular ERdj3 levels > 90% and completely ablated extracellular ERdj3 upon Tg treatment. Tg treatment also selectively increased extracellular, as opposed to intracellular, ERdj3 from Huh7 cells (Supplementary Fig S1D). In stark contrast to ERdj3, BiP and HYOU1, two abundantly expressed, UPR-induced, ER chaperones, were not detected in the conditioned media (Fig 1C), reflecting the presence of ER retention motifs on these proteins. These results are consistent with previous results showing that negligible BiP is secreted to the extracellular space (Munro & Pelham, 1987; Yamamoto et al, 2003; Kern et al, 2009). Rather, BiP that evades the KDEL receptor is typically still retained at the cellular membrane (Wang et al, 2009; Zhang et al, 2010), as is common for other canonical ER-localized chaperones, particularly under apoptotic conditions (Jordan & Gibbins, 2006; Martins et al, 2010; Lee, 2014). HYOU1, the ER resident Hsp110 that both serves as a nucleotide exchange factor for BiP and displays its own chaperone function (Andreasson et al, 2010; Behnke & Hendershot, 2014), has not been implicated in either secretion or presentation at the cellular membrane. Alternatively, intracellular levels of BiP and HYOU1 were significantly increased upon Tg treatment. These data indicate that increased extracellular ERdj3 levels result from constitutive secretion and not from leakage of ER proteins into the extracellular space, as has been proposed for other ER chaperones (Booth & Koch, 1989).
To quantify ERdj3 secretion in the presence or the absence of ER stress, Huh7 cells were pretreated with Tg or vehicle, metabolically pulse labeled with [35S]-Cys/Met, and then incubated in non-radioactive chase media. At the indicated time, [35S]-labeled ERdj3 was immunopurified from both denatured lysates and media and analyzed by autoradiography (Fig 1E). We found that ˜50% of newly synthesized ERdj3 was secreted from Huh7 cells within 4 h, in the presence or the absence of Tg (Fig 1E and F), consistent with ERdj3 being highly secreted from hepatic cells (Supplementary Fig S1C). Tg significantly increased the absolute amount of ERdj3 secreted relative to vehicle-treated controls (Fig 1G), similar to what was observed by immunoblotting (Supplementary Fig S1D). These results further demonstrate that the ERdj3 secretion is not a consequence of cell death or “leaky” release of ER proteins into the cellular media. These results also distinguish ERdj3 from other ER chaperones such as BiP or calreticulin, for which only a minor fraction of newly synthesized protein is released into the extracellular space, even during stress (Dorner et al, 1987; Lodish & Kong, 1990; Peters & Raghavan, 2011). Rather, the efficiency and selectivity of ERdj3 secretion indicates that its secretion is regulated by the canonical secretory pathway.
We next evaluated whether ER stress increases ERdj3 serum levels in mice. In the absence of stress, ERdj3 has a serum concentration of 23 ± 5 nM (Supplementary Fig S1E). Mice subjected to an 18 h fast followed by refeeding on a high-fat diet, which rapidly induces hepatic ER stress (Oyadomari et al, 2008), show a significant twofold increase in serum ERdj3 levels (Fig 1H). ERdj3 serum levels also significantly increased following 7 days on a high-fructose diet (Supplementary Fig S1F and G), which also induces ER stress in hepatic cells (Wang et al, 2012), as indicated by increased BiP, Grp94, and phosphorylated eIF2α in hepatic lysates (Supplementary Fig S1H). These results demonstrate that increased ERdj3 serum levels in mice correlate with hepatic ER stress, strongly indicating that hepatic ER stress increases ERdj3 secretion in vivo.
Stress-independent activation of the UPR-associated transcription factor ATF6 increases ERdj3 secretion
The ER stress-dependent increase in secreted ERdj3 indicates that ERdj3 is induced by the UPR. Consistent with this prediction, stress-independent activation of either of the primary UPR-associated transcription factors, ATF6 or XBP1s, increases ERdj3 transcription (Sriburi et al, 2004; Shoulders et al, 2013). To identify the specific UPR transcription factor(s) responsible for the stress-responsive increase in ERdj3 secretion, we employed HEK293T-Rex cells expressing tetracycline (tet)-inducible XBP1s and a constitutively expressed fusion protein consisting of destabilized bacterial DHFR fused N-terminally to an active ATF6 transcription factor sequence (referred to as HEK293DAX) (Shoulders et al, 2013). In these cells, the addition of the DHFR pharmacologic chaperone trimethoprim (TMP, which stabilizes the DHFR.ATF6 fusion protein) activates the ATF6 transcriptional program, while the addition of doxycycline (dox) activates the XBP1s transcriptional program. These HEK293DAX cells allow for the activation of ATF6 and/or XBP1s in the same cell in the absence of ER stress (Shoulders et al, 2013). In HEK293DAX cells, ERdj3 mRNA is increased by stress-independent activation of ATF6 and/or XBP1s to similar levels observed with Tg (Supplementary Fig S2A), as previously reported (Shoulders et al, 2013). Furthermore, [35S] metabolic labeling demonstrates that ATF6 and/or XBP1s activation increases ERdj3 protein synthesis to similar extents (Supplementary Fig S2B). Hence, we expected that stress-independent activation of either ATF6 or XBP1s would enhance secretion of ERdj3.
Surprisingly, we found that the activation of ATF6, but not XBP1s, selectively increased ERdj3 levels in the media relative to lysates (Fig 2A). Co-activation of ATF6 and XBP1s resulted in a similar amount of secreted ERdj3 to that observed with ATF6 activation alone. We did not identify immunodetectable levels of other abundant ER chaperones induced by ATF6, such as BiP, in conditioned media, further indicating that ATF6 selectively increases ERdj3 secretion (Fig 2A). Longer conditioning under ATF6 activation (48 h) also failed to lead to immunodetectable BiP in conditioned media (Supplementary Fig S2C). Importantly, TMP addition to control cells expressing destabilized DHFR fused N-terminally to YFP (Shoulders et al, 2013) did not increase extracellular ERdj3 (Supplementary Fig S2D), demonstrating that stress-independent activation of the ATF6 transcriptional program is required for the increase in secreted ERdj3 observed in TMP-treated HEK293DAX cells. Overexpression of ATF6, but not XBP1s, similarly increased secreted ERdj3 in HepG2 cells (Supplementary Fig S2E).
Figure 2. ERdj3 is efficiently secreted from cells following stress-independent activation of the UPR-associated transcription factor ATF6.
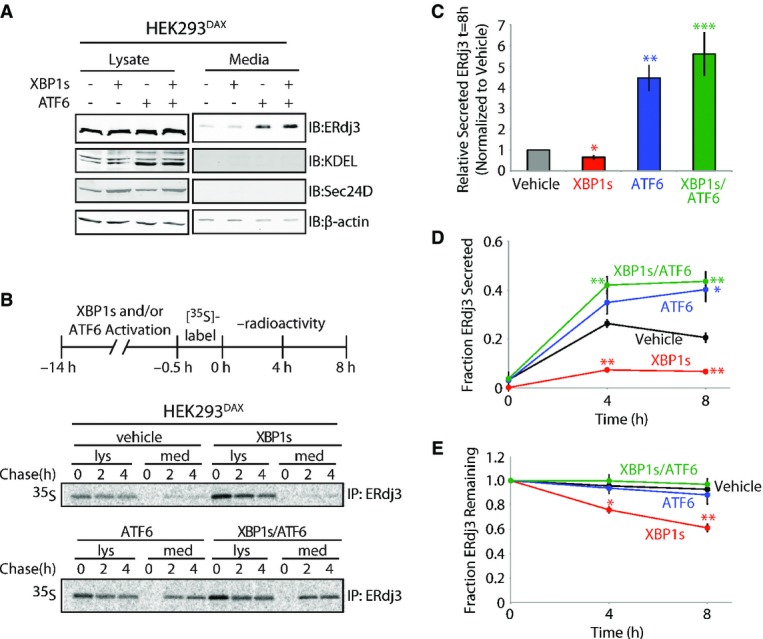
- A Immunoblot of lysates and media collected from HEK293DAX cells following 16 h of activation of XBP1s (by 1 μg/ml dox) and/or ATF6 (by 10 μM TMP), as indicated (Shoulders et al, 2013). Immunoblots of the ATF6 target protein BiP (reactive with the KDEL antibody) and the XBP1s target Sec24D confirm the small-molecule activation of these transcription factors.
- B Representative autoradiogram of [35S]-labeled ERdj3 immunopurified from lysates and media collected from HEK293DAX cells following preactivation of XBP1s (by 1 μg/ml dox) and/or ATF6 (by 10 μM TMP) (Shoulders et al, 2013). The experimental protocol is shown above.
- C Quantification of relative amounts of [35S]-labeled ERdj3 in media at 8 h collected from cells treated as shown in (B) (n = 3). The media [35S]-labeled ERdj3 was measured by densitometry and is normalized to the amount of media [35S]-labeled ERdj3 at 8 h from vehicle-treated cells.
- D, E Quantification of the fraction ERdj3 secreted (D) and the fraction ERdj3 remaining (E) from autoradiograms as shown in (B). The fractions of ERdj3 secreted/ERdj3 remaining were calculated by normalizing the recovered [35S]-labeled ERdj3 signal in the media and in the lysates at t = 4 or 8 h to the total amount of [35S]-labeled ERdj3 signal in the media and lysates at t = 0 h.
Data information: Error bars represent the standard error from biological replicates (n = 3). *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001 as compared to the vehicle-treated condition.
Source data are available online for this figure.
We further characterized the ATF6-dependent increase in ERdj3 secretion using a [35S] metabolic pulse-chase approach. ATF6 preactivation increased the extracellular concentration of newly synthesized ERdj3 fourfold relative to vehicle-treated cells, with nearly 40% of [35S] labeled ERdj3 being secreted following a 4 h incubation in non-radioactive chase media (Fig 2B–D). Despite an increase in ERdj3 synthesis (Supplementary Fig S2B), XBP1s preactivation reduced the fraction of newly synthesized ERdj3 in media, relative to the vehicle treatment (Fig 2D). This decrease in ERdj3 secretion is attributed to an increase in ERdj3 degradation, with ˜40% of newly synthesized ERdj3 being degraded after 4 h in cells following XBP1s preactivation (Fig 2E). ATF6 and XBP1s co-activation demonstrated a similar increase in ERdj3 secretion to that observed with ATF6 preactivation alone, demonstrating that ATF6 activation prevents the increased ERdj3 degradation observed upon XBP1s activation (Fig 2B–E). Collectively, these results show that ERdj3 secretion is increased by stress-independent preactivation of the UPR-associated transcription factor ATF6, directly implicating protective UPR signaling in the regulation of the extracellular ERdj3 concentration.
Secreted ERdj3 increases extracellular proteostasis capacity
To evaluate whether ERdj3 secretion is critical for the maintenance of intracellular ER proteostasis, we overexpressed either ERdj3WT or the non-secreted ERdj3KDEL and then measured expression of UPR target genes and cellular viability, both in the absence and the presence of Tg-induced ER stress (Supplementary Fig S3A and B). Despite the known role of ERdj3 in folding and degradation of specific clients (Shen & Hendershot, 2005; Hoshino et al, 2007; Jin et al, 2008; Buck et al, 2010; Tan et al, 2014), ERdj3 retention does not significantly impair global ER proteostasis maintenance in the absence or the presence of ER stress.
Alternatively, ERdj3 secretion during ER stress could enhance extracellular proteostasis capacity, by binding to misfolded proteins. To test the capacity for secreted ERdj3 to bind misfolded proteins, media collected from cells overexpressing ERdj3WT or ERdj3H53Q—an ERdj3 mutant deficient in delivering protein clients to BiP (Shen & Hendershot, 2005)—was incubated with sepharose resin conjugated to natively folded or denatured RNase A (Petrova et al, 2008). After washing the beads, proteins bound to resin were eluted and analyzed by immunoblotting (Fig 3A). ERdj3WT selectively associates with denatured RNase A, as compared to natively folded RNase A, indicating that secreted ERdj3 selectively binds misfolded protein clients in the extracellular environment. The capacity for ERdj3H53Q to similarly associate with denatured RNase A resin indicates that secreted ERdj3 interacts with denatured substrates through a mechanism largely independent of its intracellular role as an HSP40 co-chaperone that delivers misfolded substrates to BiP for ATP-dependent chaperoning.
Figure 3. Secreted ERdj3 inhibits extracellular protein aggregation.
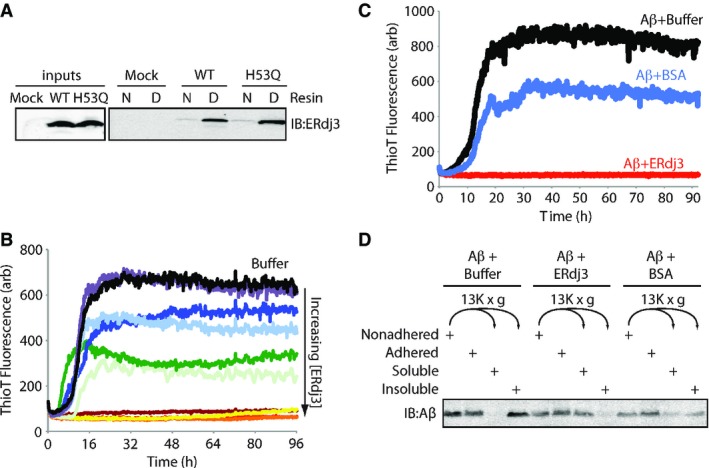
- A Immunoblot of conditioned media collected from HEK293T cells transiently transfected with ERdj3WT or ERdj3H53Q and incubated with an affinity resin consisting of native (N) or guanidinium hydrochloride (GdnHCl)-denatured (D) RNase A covalently coupled to sepharose resin. Resin was incubated with media for 2 h at ambient temperature, followed by washing and then elution in Laemmli reducing buffer.
- B Plot showing the time-dependent increase in ThioT fluorescence (ex: 440 nm, em: 485 nm) of Aβ1–40 incubated (10 μM, 37°C, pH 7.2) with regular agitation in the presence or the absence (black) of recombinant ERdj3 (RERdj3). The RERdj3 concentrations and molar ratios of RERdj3 to Aβ1–40 are as follows: violet: 1.5 nM, 1:6,561; light blue: 4.6 nM, 1:2,187; dark blue: 14 nM, 1:729; dark green: 41 nM, 1:243; light green: 120 nM, 1:81; yellow: 370 nM, 1:27; orange: 1.1 μM, 1:9; red: 3.3 μM, 1:3, or maroon: 10 μM, 1:1. All traces represent the average of three replicates.
- C Plot showing the time-dependent increase in ThioT fluorescence of Aβ1–40 incubated (10 μM, 37°C, pH 7.2) with regular agitation in the presence or the absence of BSA (14 μg/ml) or RERdj3 (14 μg/ml; 370 nM, RERdj3: Aβ = 1:27). All traces represent the average of three replicates.
- D Immunoblot of Aβ1–40 incubated (10 μM, 37°C, pH 7.2) with regular agitation for 90 h in the presence or the absence of RERdj3 (14 μg/ml; 370 nM, RERdj3: Aβ1–40 = 1:27) or BSA (14 μg/ml). The reaction solution in the well was collected (Nonadhered) and separated into soluble and insoluble fractions by centrifugation. These samples, as well as the washed wells of the plate (Adhered), were denatured in 8 M GdnHCl with sonication and analyzed by SDS-PAGE/immunoblotting.
Source data are available online for this figure.
We next prepared recombinant ERdj3 (RERdj3) in E. coli to assess the capacity of ERdj3 to inhibit protein aggregation. We confirmed that, despite the lack of the sole N-glycan present in mammalian ERdj3, RERdj3 is a folded protein as assessed by far-UV circular dichroism (Supplementary Fig S3C), is functional in that it stimulates recombinant BiP ATPase activity (Supplementary Fig S3D), and maintains the capacity to selectively bind denatured RNAse A resin (Supplementary Fig S3E). We monitored the aggregation of the amyloidogenic Aβ1–40 peptide (10 μM) associated with Alzheimer’s disease using thioflavin T (ThioT) fluorescence in the presence of increasing RERdj3 concentrations. ThioT fluorescence sharply increases upon binding to amyloid fibers (Du et al, 2011). RERdj3 completely inhibited the aggregation of Aβ1–40 at concentrations above 370 nM (an ERdj3: Aβ1–40 ratio of 1:27) (Fig 3B). The control serum protein bovine serum albumin (BSA), a protein known to modestly inhibit Aβ aggregation (Bohrmann et al, 1999; Milojevic et al, 2007, 2009; Reyes Barcelo et al, 2009), did not dramatically influence Aβ1–40 aggregation at an equivalent mass concentration (Fig 3C). The ERdj3-dependent decrease in Aβ1–40 aggregation was similarly observed by differential centrifugation, with substoichiometric levels of RERdj3 significantly reducing the population of Aβ1–40 in the pelleted aggregates following a 96 h incubation (Fig 3D). Denaturation of RERdj3 by boiling in the presence of dithiothreitol attenuated the ERdj3-dependent reduction in Aβ1–40 aggregation, demonstrating that the reduction in protein aggregation afforded by RERdj3 requires a native ERdj3 structure (Supplementary Fig S3F).
We next tested whether secreted ERdj3 could impact the cellular phenotype associated with another misfolded protein, the toxic prion protein (TPrP) (Zhou et al, 2012). TPrP is a monomeric, misfolded prion conformer that upon addition to neuronal cells recapitulates the cytoplasmic vacuole formation characteristic of prion diseases in patients (Will et al, 1996). TPrP induces this phenotype at 600 ng/ml, similar to the extracellular concentrations of prion protein associated with prion diseases in patients (Meyne et al, 2009), providing a highly sensitive phenotypic assay for quantifying the protective effects of secreted ERdj3. We treated PK1 mouse neuroblastoma cells with TPrP for 3 days in the presence of media conditioned on HEK293T-Rex cells overexpressing ERdj3WT (Supplementary Fig S4A) and evaluated the extent of vacuole formation (Fig 4A). Strikingly, media conditioned on cells overexpressing ERdj3WT ([ERdj3WT]media = 100–400 nM) significantly attenuated TPrP-induced vacuole formation in PK1 cells (Fig 4B and C), as compared to treatment with control conditioned media ([endogenous ERdj3]media = 2–4 nM), demonstrating that secreted ERdj3WT protects neuroblastoma cells against TPrP toxicity. Alternatively, TPrP incubated with conditioned media prepared on HEK293T-Rex cells RNAi-depleted of ERdj3 significantly aggravated vacuole formation in TPrP-treated PK1 cells, as compared to conditioned media prepared from HEK293T-Rex cells expressing non-silencing shRNA ([endogenous ERdj3]media = 2–4 nM) (Supplementary Fig S4A–C). This indicates that, even the low level of endogenous ERdj3 secreted from HEK293T-Rex in the absence of stress, which is about 10–20% of the measured physiological concentration of ERdj3 in mouse serum (23 nM; Supplementary Fig S1E), increases extracellular proteostasis capacity and protects cells from TPrP proteotoxicity.
Figure 4. Secreted ERdj3 attenuates vacuole formation induced by toxic prion protein in mammalian cells.
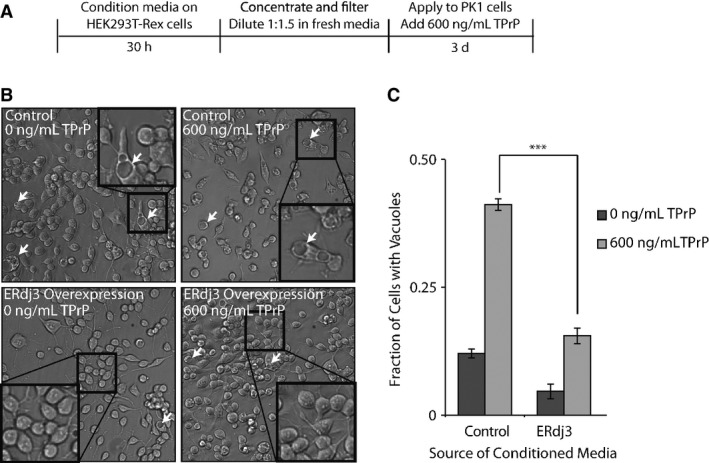
- A Schematic illustrating the treatment of PK1 cells with ERdj3-conditioned media. HEK293T-Rex (Control) and HEK293T-Rex cells stably overexpressing tet-inducible ERdj3WT on poly-D-lysine-coated plates were treated with dox (1 μg/ml) overnight to induce ERdj3WT expression. The media was replaced with fresh OptiMEM I + 5% FBS, which was conditioned for 30 h, sterile-filtered, concentrated 5× against a 3 kD MWCO membrane, and diluted 1:1.5 with fresh OptiMEM I + 5% FBS. PK1 cells were plated in OptiMEM I + 5% FBS and after 24 h were treated with conditioned media and TPrP (600 ng/ml) for 3 days.
- B Representative images under 136× magnification of PK1 cells incubated with control conditioned media collected from HEK293T-Rex or conditioned media prepared from HEK293T-Rex cells overexpressing ERdj3WT in the presence or the absence of TPrP (600 ng/ml), as described in (A). Arrows indicate representative cells containing vacuoles. Insets show 544× magnification of a typical region of the image.
- C Quantification of vacuole formation from images as shown in (B). Equal area images comprising ∼1,200 cells per well were counted for each sample. Counting of cells and of cells with identifiable vacuoles was done using the Fiji image processing package (Schindelin et al, 2012). ***P-value < 0.001 (n = 3); mean ± SEM.
ERdj3 is co-secreted in complex with secreted destabilized mutant proteins
Since ERdj3 binds misfolded, non-native protein conformations in the ER lumen (Shen & Hendershot, 2005), we wondered whether ERdj3 could be co-secreted into the extracellular environment in complex with non-natively folded proteins. To test this prediction, we employed the destabilized, misfolding-prone A25T transthyretin mutant (TTRA25T), a highly amyloidogenic, poorly secreted, disease-associated TTR variant (Sekijima et al, 2003, 2005), previously shown to interact with ERdj3 intracellularly (Shoulders et al, 2013). We immunopurified TTR from conditioned media collected from either a mixed population of HEK293T cells individually overexpressing FLAG-tagged TTRA25T (FTTTRA25T) or ERdj3WT (Fig 5A, “co-incubation”; a condition where FTTTRA25T and ERdj3WT can only interact in the media) or cells co-overexpressing FTTTRA25T and ERdj3WT (Fig 5A; “co-expression”; a condition where FTTTRA25T and ERdj3WT can interact both in the secretory pathway and in the media). Following stringent washing of the immunoisolated secreted TTR in high-detergent RIPA buffer, we eluted and quantified the amount of ERdj3 associated with FTTTRA25T by immunoblotting. Despite similar media levels of both ERdj3WT and FTTTRA25T in the distinct experimental paradigms, ERdj3 efficiently co-immunopurifies with FTTTRA25T only in media collected from cells co-overexpressing both ERdj3WT and FTTTRA25T (Fig 5B). ERdj3 does not co-immunopurify with TTR in media collected from cells co-overexpressing stable, FLAG-tagged wild-type TTR (FTTTRWT) and ERdj3WT, indicating that ERdj3–TTR complex formation is specific to destabilized TTR (Fig 5B). We observe a modest amount of ERdj3 associated with FTTTRA25T, but not FTTTRWT, in the “co-incubation” condition when the immunoisolate is washed less stringently with mild detergent, indicating that free ERdj3 and FTTTRA25T do interact extracellularly, but more weakly than in the co-secreted complex (Supplementary Fig S5A). We obtained similar results in HepG2 and SH-SY5Y cells, where we observed that endogenously secreted ERdj3 selectively co-immunopurifies with overexpressed FTTTRA25T, but not FTTTRWT (Supplementary Fig S5B–D). Interestingly, we do not recover all of the total secreted ERdj3 in these experiments (compare FTTTRA25T immunopurifications and inputs in Supplementary Fig S5B and D), suggesting that even under conditions where a destabilized misfolding-prone protein is overexpressed, ERdj3 can still be secreted as a free protein available for the binding of misfolded proteins in the extracellular environment. Critically, ERdj3WT overexpression does not promote the secretion of the highly destabilized, non-secreted FTTTRD18G variant (Hammarstrom et al, 2003; Sato et al, 2007) (Fig 5C), despite a strong intracellular interaction between ERdj3 and FTTTRD18G (Supplementary Fig S5E). This result indicates that secretion in complex with ERdj3 is not a mechanism for a misfolding-prone protein to evade ER quality control.
Figure 5. ERdj3 is co-secreted with destabilized proteins through the secretory pathway.
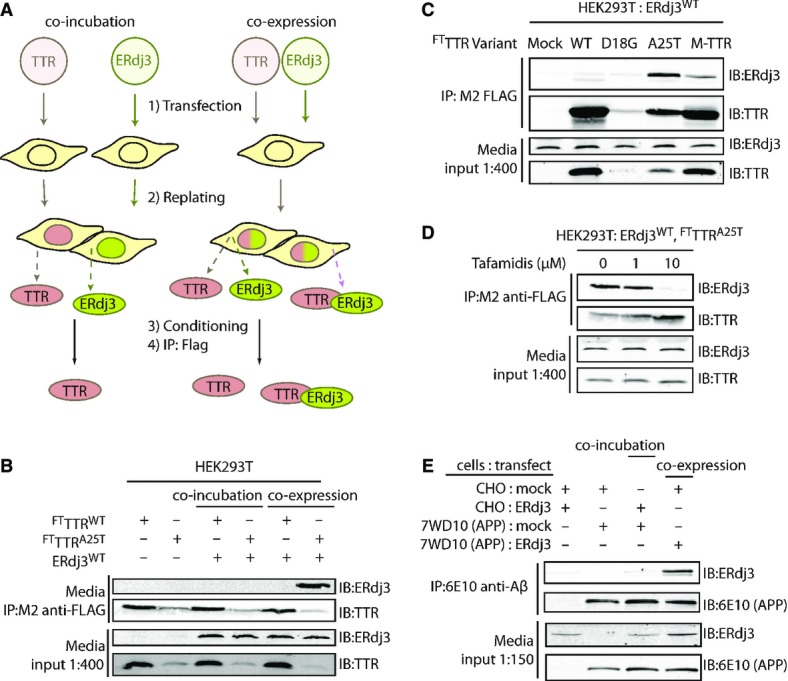
- A Schematic describing the co-incubation and co-expression experiments used to demonstrate ERdj3 co-secretion with destabilized TTR. For co-incubation, HEK293T cells overexpressing FLAG-tag TTR (FTTTR) variants were seeded with an equal number of cells overexpressing ERdj3WT. For co-expression experiments, HEK293T cells are transfected with both FTTTR and ERdj3WT. FTTTR was immunoisolated with M2 anti-FLAG beads from media conditioned overnight on the cells, eluted, and separated by SDS-PAGE for immunoblotting.
- B Immunoblot of M2 anti-FLAG immunopurifications from the conditioned media collected from HEK293T cells overexpressing FTTTRWT, FTTTRA25T, and/or ERdj3WT either as a co-incubation or co-expression experiment as shown in (A). Beads were washed in RIPA buffer prior to elution. Media inputs (1:400) are shown as a control.
- C Immunoblot of M2 anti-FLAG immunopurifications from media collected from HEK293T cells overexpressing ERdj3WT and FTTTR variants as indicated. Media inputs (1:400) are shown as a control.
- D Immunoblot of M2 anti-FLAG immunopurifications from conditioned media of HEK293T cells overexpressing FTTTRA25T and ERdj3WT in the presence of the indicated concentrations of Tafamidis, a kinetic stabilizer of TTR tetramers. Media inputs (1:400) are shown as a control.
- E Immunoblot of 6E10 anti-APP immunopurifications from the conditioned media of CHO cells or CHO-derived APP751-expressing (7WD10) cells overexpressing ERdj3WT as indicated. Cells were replated together as indicated 24 h after transfection at equal stoichiometry, and media conditioned for 36 h. In this case, ERdj3WT-transfected CHO cells replated with mock-transfected 7WD10 cells serves as the co-incubation condition, while mock-transfected CHO cells replated with ERdj3WT-transfected 7WD10 cells comprise the co-expression condition. Media inputs (1:150) are shown as a control.
Source data are available online for this figure.
Interestingly, ERdj3WT co-secretion with FTM-TTR, a stable, secreted monomeric TTR double mutant unable to form native TTR homotetramers (Jiang et al, 2001; Sekijima et al, 2005) is lower than with FTTTRA25T (Fig 5C), indicating that secretion of ERdj3–TTR complexes requires a destabilized TTR. We further explored the dependence of ERdj3–TTR co-secretion on TTR stability using the small molecule TTR kinetic stabilizer Tafamidis (Bulawa et al, 2012). Tafamidis binds to and stabilizes the native TTR tetramer in the ER lumen, acting as a pharmacologic chaperone (Shoulders et al, 2013). The addition of Tafamidis to cells co-overexpressing FTTTRA25T and ERdj3WT increased FTTTRA25T secretion while eliminating the co-immunopurification of ERdj3WT with FTTTRA25T in conditioned media (Fig 5D). These results demonstrate that ERdj3 co-secretion requires client destabilization.
We next evaluated whether ERdj3 could be co-secreted in complex with other destabilized, secreted proteins. Motivated by the substoichiometric inhibition of Aβ1–40 aggregation by ERdj3, we measured ERdj3 co-secretion in a stable CHO-derived cell line (7WD10) that secretes a spectrum of APP751-associated cleavage products, including Aβ1–40 (Xia et al, 1997). We collected conditioned media from plates with either a mixed population of CHO cells overexpressing GFP and 7WD10 cells overexpressing ERdj3WT (mimicking “co-expression”) or a mixed population of CHO cells overexpressing ERdj3WT and of 7WD10 cells, which produce APP, also overexpressing GFP (mimicking “co-incubation”). Aβ-containing species were then immunoprecipitated from the conditioned media with the 6E10 antibody, which recognizes an epitope within the Aβ fragment, and eluates were analyzed by immunoblotting. ERdj3 only co-immunopurifies with APP fragments from media conditioned on 7WD10 cells overexpressing ERdj3WT (i.e., “co-expression” of ERdj3WT and APP) (Fig 5E), demonstrating that the secretion of ERdj3–client complexes is a mechanism not limited to destabilized TTRs.
Other secreted chaperones, most prominently clusterin, also have the capacity to bind misfolded proteins in the extracellular space and prevent extracellular protein aggregation into proteotoxic conformations (Wyatt et al, 2013). Interestingly, while the steady-state plasma concentration of clusterin is reported to be between 500 nM and 2 μM (Schrijvers et al, 2011; Silajdzic et al, 2012; van Dijk et al, 2013), about 20- to 80-fold higher than we report for ERdj3, it is not induced by the UPR; rather, UPR induction reduces clusterin secretion (Nizard et al, 2007). This suggests that ERdj3 has a unique role in regulating extracellular proteostasis in response to ER stress. Since clusterin is not known to contribute to ER proteostasis, we explored whether clusterin can co-secrete from the ER with destabilized proteins using a C-terminally HA-tagged clusterin (CLU3xHA) and FTTTRA25T. While we detected abundant clusterin from FTTTRA25T immunoisolations in the co-expression condition, as compared to the co-incubation condition, we also observed a sharp increase in secreted FTTTRA25T upon clusterin co-overexpression (Supplementary Fig S5F). Furthermore, when we co-express clusterin with the non-secreted FTTTRD18G mutant, we detected a significant increase of FTTTRD18G secretion (Supplementary Fig S5G), an effect not seen with ERdj3 or explainable by variation in FTTTRD18G expression (Supplementary Fig S5H). These results suggest that formation of clusterin–client complexes can allow clients to evade ER quality control, clearly distinguishing the clusterin co-secretion mechanism from that observed with the UPR-regulated secreted chaperone ERdj3.
ERdj3–client co-secretion is regulated by ER proteostasis capacity
The differential capacity of ERdj3 and clusterin to be co-secreted with ER clients could represent the differential involvement of these secreted chaperones in ER proteostasis maintenance. ERdj3 functions intracellularly in a network with the ER HSP70 chaperone BiP, delivering misfolded proteins to BiP for ATP-dependent chaperoning. Alternatively, clusterin is not known to have functional interactions with any ER proteostasis pathway component. Thus, we predicted that the co-secretion of ERdj3 with substrate could be influenced by the activity and capacity of ER proteostasis factors such as BiP.
We tested whether ER stress influences the amount of endogenous ERdj3 co-secreting with FTTTRA25T. In the absence of stress, modest amounts of endogenous ERdj3 co-immunopurified with FTTTRA25T in conditioned media collected from HEK293T-Rex cells overexpressing FTTTRA25T (Supplementary Fig S6A). Despite reducing FTTTRA25T secretion through ER stress-dependent UPR activation (Shoulders et al, 2013), pretreatment with Tg increased the relative recovery of secreted ERdj3 in FTTTRA25T immunopurifications (Supplementary Fig S6A and B). This ER stress-induced change in the association between ERdj3 and its clients suggests that ER proteostasis network components are involved in regulating ERdj3 co-secretion.
Because intracellular ERdj3 delivers destabilized proteins to BiP for ATP-dependent chaperoning, co-secretion of ERdj3 with destabilized mutant proteins could reflect a relative deficit in the ER chaperone capacity in the ER lumen (e.g., BiP levels). To evaluate the role of the ER chaperone environment on ERdj3–client co-secretion, we initially employed our HEK293DAX cell line that allows ligand-regulated activation of the UPR transcription factor ATF6 (Shoulders et al, 2013). ATF6 increases the expression of BiP and numerous BiP co-chaperones including ERdj3 and HYOU1 (Shoulders et al, 2013). Activation of ATF6 significantly reduced the relative recovery of secreted ERdj3 (normalized to recovered TTR) in FLAG immunopurifications of media collected from HEK293DAX cells overexpressing FTTTRA25T and ERdj3WT (Fig 6A and B). Interestingly, ATF6 activation did not influence the ERdj3 recovery in media when FTTTRA25T was co-overexpressed with ERdj3H53Q—an ERdj3 mutant deficient in delivering protein clients to BiP (Shen & Hendershot, 2005)—suggesting that BiP activity mediates the suppression of ERdj3WT–FTTTRA25T complex secretion afforded by ATF6 activation. We directly tested the impact of BiP activity on ERdj3–FTTTRA25T co-secretion in HEK293T cells overexpressing FTTTRA25T, ERdj3 (ERdj3WT or ERdj3H53Q), and BiP. BiP overexpression significantly reduces the relative recovery of secreted ERdj3WT in FTTTRA25T immunopurifications in conditioned media, but does not reduce the relative recovery of secreted ERdj3H53Q (Fig 6C and D). This indicates that a functional ERdj3–BiP interaction is necessary for the reduction in ERdj3WT-FTTTRA25T complex secretion induced by BiP overexpression. Despite a robust interaction between BiP and intracellular FTTTRA25T, BiP does not appear in extracellular FTTTR immunopurifications, consistent with our inability to observe extracellular BiP and supporting our model whereby extracellular ERdj3 functions independent of BiP (Supplementary Fig S6C). Co-overexpression of BiP similarly reduced co-secretion of ERdj3WT with APP cleavage products, indicating that this effect is not limited to destabilized TTRs (Fig 6E). By contrast, and consistent with its lack of interaction with ER proteostasis pathways, the relative recovery of clusterin in FTTTRA25T immunopurifications is insensitive to BiP overexpression, indicating that clusterin co-secretion with FTTTRA25T is not influenced by BiP activity (Supplementary Fig S6D and E). These results are consistent with a model wherein if ERdj3 is unable to deliver a misfolded client protein to BiP, ERdj3 and the client protein will remain in complex and be secreted to the extracellular space.
Figure 6. ERdj3–client protein complexes are co-secreted when BiP activity is limiting.
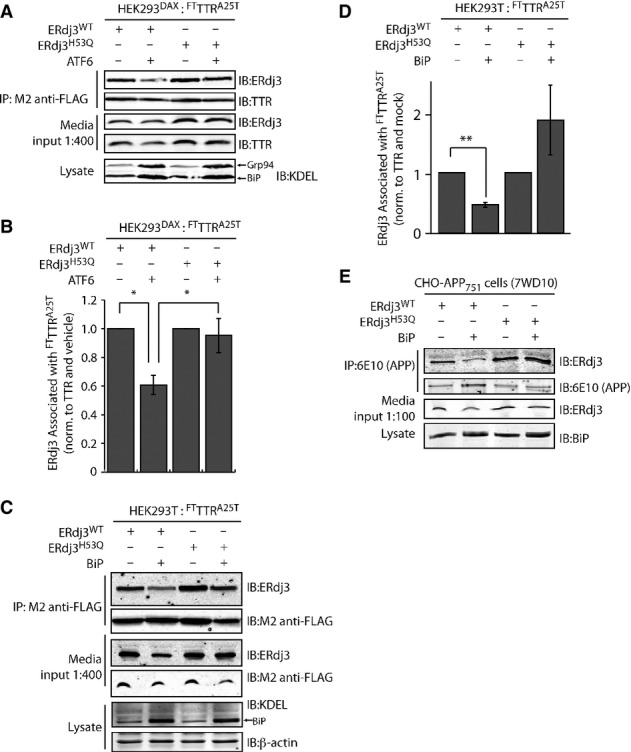
- A Representative immunoblot of M2 anti-FLAG immunopurification of media conditioned for 24 h on HEK293DAX cells co-overexpressing FTTTRA25T and ERdj3WT or ERdj3H53Q. Media contained vehicle or trimethoprim (TMP, 10 μM, 16 h; activates ATF6), as indicated. Media inputs (1:400) are shown as a control. Lysates from these cells are also shown to confirm the increase of the ATF6 target proteins BiP and GRP94 in cells following TMP-dependent activation of ATF6.
- B Bar graph depicting quantification of the change in the amount of ERdj3 co-immunoprecipitating with TTRA25T following activation of ATF6 as in (A). The ERdj3/TTR ratio is normalized to the ratio in the absence of ATF6 activation. *P-value < 0.05; n = 3, mean ± SEM.
- C Representative immunoblot of M2 anti-FLAG immunopurification of media conditioned for 24 h on HEK293T cells co-overexpressing FTTTRA25T, ERdj3WT, ERdj3H53Q, and/or BiP as indicated. Media inputs (1:400) are shown as a control. Lysates from these cells are also shown to confirm the increase of BiP afforded by overexpression.
- D Quantification of immunoblots of M2 anti-FLAG immunopurification of media collected from cells co-overexpressing either ERdj3WT or ERdj3H53Q, FTTTRA25T, and BiP as in (C). The ERdj3/TTR ratio is normalized to the ratio in the absence of BiP overexpression. **P-value < 0.01; n = 5, mean ± SEM.
- E Immunoblot of 6E10 anti-APP immunopurifications from the conditioned media of CHO-derived APP751-expressing (7WD10) cells overexpressing ERdj3WT, ERdj3H53Q, and BiP as indicated. Media inputs (1:100) are shown as a control. Lysates from these cells are also shown to confirm the increase of BiP afforded by overexpression.
Source data are available online for this figure.
Discussion
Protein synthesis is necessarily an intracellular process. Hence, extracellular metazoan environments such as the blood rely on cellular protein secretion to define extracellular protein concentrations and regulate the integrity of the secreted proteome. The presence of misfolding-prone proteins in the ER directly threatens this environment, as imbalances in ER proteostasis can impair the capacity for ER quality control pathways to prevent secretion of misfolded, aggregation-prone client proteins that in turn challenge the integrity of the extracellular proteome (Hetz & Mollereau, 2014). The UPR indirectly regulates extracellular proteostasis by attenuating the secretion of misfolded protein conformations that can both accumulate during and induce ER stress. Here, we demonstrate a direct role for the UPR in regulating extracellular proteostasis through the increased transcription and secretion of ERdj3. The capacity for UPR activation to influence extracellular proteostasis through ERdj3 secretion links protein misfolding in the secretory pathway to extracellular proteostasis, revealing a new mechanism by which cells coordinate intra- and extracellular environments in response to pathologic insults that induce ER stress (Fig 7).
Figure 7. ERdj3 secretion links ER and extracellular proteostasis environments during conditions of ER stress.
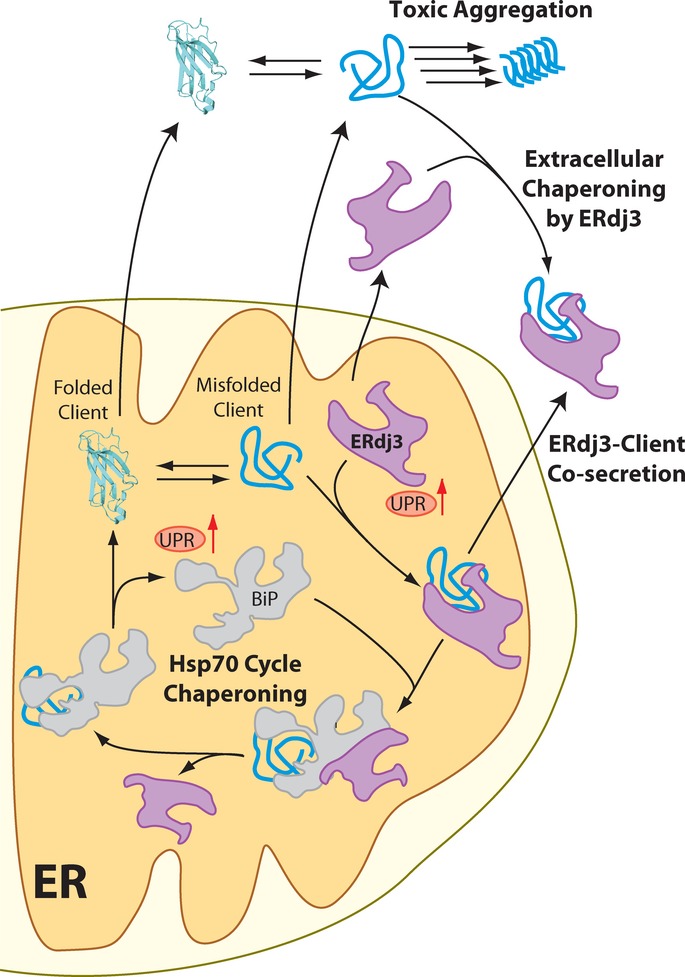
In response to ER stress, newly synthesized ERdj3 binds misfolded ER client proteins and delivers them to BiP for chaperoning in the Hsp70 cycle. When free BiP becomes limiting, or if repeated BiP cycling cannot productively deplete the levels of the misfolded client, the stable ERdj3–client complex is co-secreted to the extracellular environment, preemptively binding the misfolded protein and preventing the aggregation of the misfolded client protein in the extracellular space. Furthermore, stress-induced ERdj3 can be secreted on its own into the extracellular space where it can bind to misfolded, aggregation-prone client proteins and attenuate pathologic protein aggregation in the extracellular environment.
In its role as a traditional HSP40 co-chaperone to BiP, ERdj3 can influence the trafficking of ER client proteins and is exploited for pathogenic toxin internalization (Yu & Haslam, 2005; Massey et al, 2011) and viral infection (Wen & Damania, 2010; Goodwin et al, 2011) (Fig 7). However, we find that at least half of newly synthesized ERdj3 is secreted. Secreted ERdj3 has the capacity to bind misfolded proteins and prevent their extracellular aggregation and proteotoxicity, suggesting that UPR-dependent increases in ERdj3 secretion offer a new potential mechanism to protect the local extracellular environment from toxic protein conformations that can be secreted during ER stress (Fig 7). Interestingly, ERdj3 also can also influence extracellular proteostasis through its co-secretion with destabilized, misfolding-prone client proteins. This capacity of ERdj3 to be co-secreted with misfolding-prone proteins avoids the problem of diffusion-limited encounter in the extracellular space.
ERdj3–client co-secretion serves as a natural bridge between the dual roles of ERdj3 as an ER HSP40 and in regulating extracellular proteostasis (Fig 7). In the ER, ERdj3 delivers misfolded clients (e.g., FTTTRA25T) into the BiP cycle for chaperoning. If inadequate free BiP is available, ERdj3 remains bound to its client protein throughout the secretory pathway and the client-ERdj3 complex can be secreted into the extracellular space, where lack of BiP prevents ERdj3 release from the substrate. When the ER must deal with high, persistent levels of misfolding-prone proteins, the UPR is activated, increasing the levels of ERdj3, BiP and other chaperones to offer misfolding-prone proteins more ER chaperoning capacity. Simultaneously, increased intracellular ERdj3 is available to bind and co-secrete with misfolding-prone protein clients that evade ER quality control and traverse the secretory pathway, providing preemptive chaperone capacity to the extracellular space. Importantly, clusterin co-secretion, unlike ERdj3 co-secretion, assists destabilized clients in evading ER quality control, providing a potential reason for the reduced secretion of clusterin during conditions of ER stress. Thus, ERdj3 offers a unique link between ER and extracellular proteostasis, employing a mechanism that cannot be achieved with other known secreted chaperones.
The capacity of UPR-dependent activation of ATF6 to influence extracellular proteostasis through ERdj3 secretion indicates a potential role for this pathway in extracellular protein aggregation pathologies. UPR signaling is activated in many extracellular protein aggregation diseases, including Alzheimer’s disease and the systemic amyloidoses (Teixeira et al, 2006; Saxena et al, 2009; Hoozemans et al, 2012; Hetz & Mollereau, 2014). Our results showing that secreted ERdj3 can increase the extracellular chaperoning and prevent extracellular aggregation and/or proteotoxicity of disease-associated proteins such as TPrP, TTRA25T, and Aβ suggest that ATF6-dependent regulation of ERdj3 secretion is a potential mechanism to protect the extracellular space from proteotoxicity. Consistent with this prediction, presenilin mutations causatively associated with Alzheimer’s disease significantly impair ATF6 activation during stress (Katayama et al, 1999, 2001). Thus, a decreased capacity to regulate extracellular proteostasis through ATF6 activation and consequent ERdj3 secretion could be a contributing factor in the disease pathology of patients harboring these mutations.
We have established that UPR activation directly and adaptively regulates the composition of the extracellular proteostasis network through ERdj3 secretion. In particular, UPR-induced secretion of ERdj3 offers a mechanism for organisms to adapt to the presence of destabilized proteins in the secretory pathway and increase extracellular chaperoning capacity through two mechanisms: (1) the secretion of free ERdj3 available to bind misfolding-prone proteins in the extracellular environment and (2) the co-secretion of ERdj3–client complexes, which preemptively protects the extracellular environment from proteotoxic protein conformations. This potentially provides an endogenous mechanism to prevent ER stress-induced increases in extracellular protein aggregation and proteotoxicity that can lead to degenerative phenotypes. Our identification of ERdj3 as a UPR-induced secreted chaperone demonstrates that targeting adaptive stress responses, particularly the ATF6 arm of the UPR, can enhance the maintenance of extracellular proteostasis, offering a promising approach to better understand and intervene in diseases characterized by extracellular protein aggregation.
Materials and Methods
Ethics statement
The protocol for the murine fasting/refeeding experiments was approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute (TSRI). Non-nutritional bedding was provided so that these mice did not need to be deprived of bedding during the fast. The protocol for high-fructose diet experiments was reviewed and approved by the Committee on Use and Care of Animals at the Sanford Burnham Medical Research Institute.
Plasmid DNA, shRNA, and antibodies
ERdj3WT, ERdj3H53Q, and ERdj3KDEL overexpression constructs were prepared in the pcDNA3.1(+) vector and Clusterin-HA in the pCI-neo.X3HA vector. Plasmid construction is detailed in Supplementary Methods. FTTTRA25T and FTTTRWT are in the pcDNAI plasmids, as previously reported (Shoulders et al, 2013). The FTBiP overexpression plasmid is in the pCMV1 vector. The ERdj3 shRNA GIPZ shRNAmir and non-silencing constructs were purchased from Open BioSystems. Polyclonal antibodies were used to detect ERdj3 (rabbit, Proteintech) and HYOU1 (rabbit, Genetex), while mouse monoclonal anti-tubulin, anti-β-actin, 6E10 anti-Aβ, and M2 anti-FLAG were from Sigma. M1 anti-FLAG agarose beads and M2 anti-FLAG Dynabeads were from Sigma. Polyclonal rabbit antibody to TTR has been reported previously (Sekijima et al, 2005). Mouse monoclonal antibodies against KDEL (Assay Designs), HA (Covance), and BiP (E-4, Santa Cruz) were used. Secondary antibodies were purchased from Li–Cor.
Mammalian cell culture
HEK293T, Huh7, HeLa, HepG2, and CHO cells and derived lines were cultured in DMEM (Invitrogen) with 10% FBS (Gibco) supplemented with Pen/Strep and glutamine. HEK293T were transfected by calcium phosphate deposition, with a medium change the following day, while CHO and SH-SY5Y cells were transfected using X-tremeGENE 9 (Roche). SH-SY5Y cells were cultured similarly with 50:50 DMEM/F12 (Invitrogen). Activating ligands trimethoprim (TMP) (RPI, Illinois, USA) and doxycycline were used at 10 μM and 1 μg/ml, respectively. Thapsigargin was added to cell media immediately prior to incubation at the indicated concentrations. Stable lines for APP751 or ERdj3 overexpression, or for shRNA knockdown, were established by selection and maintained in 500 μg/ml G418 or 1 μg/ml puromycin, respectively. ATF6, XBP1s, and GFP adenoviruses were propagated and applied as reported previously (Shoulders et al, 2013).
Metabolic labeling and pulse chase
Following the indicated drug treatments, HEK293DAX and Huh7 cells on poly-D-lysine-coated plates were washed well with PBS and metabolically labeled in pulse medium (DMEM without Cys and Met (Mediatech) with [35S]-Cys/Met (MP Biomedicals, ˜0.1 mCi/ml final concentration), 10% 10K MWCO-dialyzed FBS, and supplemented with glutamine and penicillin/streptomycin) for 30 min. Cells were then washed well with pre-warmed media and incubated in pre-warmed complete media for the indicated chase time. After the chase, the media were collected, cells were washed well with PBS and lysed on the plate in RIPA on ice for 20 min, and the lysates centrifuged to remove cellular debris. To ensure complete denaturation, lysates were brought to 1% SDS and 1 mM DTT and boiled at 100°C for 5 min, after which they were diluted to 0.1% SDS and 100 μM DTT, pre-cleared over sepharose 4B (Sigma) and immunoprecipitated using anti-ERdj3 antibody (Proteintech) on Protein A-conjugated sepharose beads (Invitrogen). Elution was performed by boiling 10 min in 6× Laemmli reducing buffer. Eluates were separated by SDS-PAGE, and the gels dried and exposed to an autoradiography cassette overnight. The cassette was scanned on a Typhoon imager, and band intensities were quantified by densitometry in ImageQuant. Error is presented as SEM of at least three replicates, with P-values determined by the Student’s two-tailed t-test.
Quantitative RT–PCR
The quantitative PCR protocol and primers have been previously described (Shoulders et al, 2013). RNA was extracted from cells (RNeasy Mini Kit, Qiagen) and cDNA synthesized from 500 ng of total RNA (QuantiTect Reverse Transcription Kit, Qiagen). qPCR (QuantiTect SYBR Green PCR Kit, Qiagen) on this cDNA was performed (45 cycles of 15 s at 94°C, 30 s at 57°C, 30 s at 72°C) in technical triplicate on an ABI PRISM 7900 System.
Murine experiments
For fasting/refeeding experiments, one male and five female 6-week-old DBA/2J mice, maintained on normal chow (5% fat), were subjected to an 18 h fast with non-nutritional bedding, followed by 12 h on a high-fat (60%) diet (Research Diets) as reported previously (Oyadomari et al, 2008). Immediately prior to the fast and at indicated time points, mice were anesthetized with isoflurane and aliquots of serum extracted retro-orbitally. The same population of mice was used for each time point. For the high-fructose diet experiments, either normal chow or a high-fructose diet (60% fructose, TD. 89247, Harlan Teklad; Madison, WI) was given to C57/BI6 for 2 days, 7 days, and 6 weeks, with three to four mice in each experimental group. At each experimental termination point, mice were sacrificed after a 4 h fast and serum and livers were collected for immunoblotting analysis.
TPrP treatment and image analysis
TPrP toxicity assay was performed as described (Zhou et al, 2012). OptiMEM I (Life Technologies) with 5% FBS was conditioned on HEK293T-Rex-derived cells for 30 h. Collected media were sterile-filtered and concentrated about 5 fold using an Amicon ultra centrifugal unit with 3 kDa MW cutoff (EMD Millipore). About 1,000 PK1 cells (a subclone of N2a neuroblastoma cells) (Klohn et al, 2003) were plated per well in 96-well plates in OptiMEM I (5% FBS), and the media was changed to a mixture of concentrated conditioned media (80 μl) and fresh OptiMEM I with 5% BSA (120 μl, to replenish the nutrients) on the next day. TPrP was prepared as reported (Zhou et al, 2012), and a final concentration of 0.6 μg/ml was added to the treatment group in triplicate. The cells were incubated for 3 days and imaged by phase-contrast microscopy. Three images of each well were taken, and image identities were blinded prior to analysis and counting. Counting of cells and of cells with identifiable vacuoles was performed separately by two individuals, who obtained similar results, using the Fiji image processing package (Schindelin et al, 2012).
Additional experimental details describing previously reported approaches including immunoblotting (Shoulders et al, 2013), preparation of recombinant ERdj3 and BiP, RNase A affinity precipitation (Petrova et al, 2008), Aβ1–40 aggregation assays (Du et al, 2011), and resazurin assays (Shoulders et al, 2013) are available in Supplementary Methods. Error bars from quantitative immunoblotting represent SEM, and associated P-values were determined using the two-tailed Student’s t-test.
Acknowledgments
The authors are grateful to Prof. Evan Powers, Prof. Joel Buxbaum, Prof. Fernando Palhano, and Dr. Xinyi Li for helpful discussions, and to Mike Saure, Gina Dendle, and Eley Wong for technical assistance. We thank the NIH for supportive grants R21NS079882, R01DK102635 (RLW), R37DK046335 (JWK), R21NS081519 (CIL), R37DK042394, R01DK088227, and R01HL052173 (RJK), Arlene and Arnold Goldstein (RLW), the Ellison Medical Foundation (RLW), the Skaggs Institute for Chemical Biology (JWK), the Lita Annenberg Hazen Foundation (JWK), and the Scripps Research Institute for financial support. JCG was supported by the NHLBI (F32-HL099245) and an AHA postdoctoral fellowship. MDS was supported by an American Cancer Society postdoctoral fellowship. SW was supported by an AHA postdoctoral fellowship.
Author contributions
JCG and SQ contributed equally to this work. JCG, SQ, MDS, JWK, and RLW designed experiments. JCG, SQ, and RLW performed cell biology and biochemistry experiments. MZ and CIL designed and performed prion toxicity experiments. SW and RJK prepared sera from mice fed on a high-fructose diet. LMR prepared recombinant BiP and performed experiments. JCG, SQ, and RLW interpreted experimental results and prepared the manuscript, with comments from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Source Data for Supplementary Figure S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Review Process File
Source Data for Figure 1A, C, E, H
Source Data for Figure 2A, B
Source Data for Figure 3A, D
Source Data for Figure 5B, C, D, E
Source Data for Figure 6A, C, E
References
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Andreasson C, Rampelt H, Fiaux J, Druffel-Augustin S, Bukau B. The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J Biol Chem. 2010;285:12445–12453. doi: 10.1074/jbc.M109.096735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2011;3:a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Hendershot LM. The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem. 2014;289:2899–2907. doi: 10.1074/jbc.M113.507491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyair R, Ron E, Lederkremer GZ. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int Rev Cell Mol Biol. 2011;292:197–280. doi: 10.1016/B978-0-12-386033-0.00005-0. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bohrmann B, Tjernberg L, Kuner P, Poli S, Levet-Trafit B, Naslund J, Richards G, Huber W, Dobeli H, Nordstedt C. Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J Biol Chem. 1999;274:15990–15995. doi: 10.1074/jbc.274.23.15990. [DOI] [PubMed] [Google Scholar]
- Booth C, Koch GL. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Buck TM, Wright CM, Brodsky JL. The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:751–761. doi: 10.1016/j.semcdb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Kolb AR, Boyd CR, Kleyman TR, Brodsky JL. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell. 2010;21:1047–1058. doi: 10.1091/mbc.E09-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004;16:67–75. doi: 10.1097/00002281-200401000-00013. [DOI] [PubMed] [Google Scholar]
- Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceicao IM, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem. 2010;79:777–802. doi: 10.1146/annurev-biochem-061608-091319. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- van Dijk KD, Jongbloed W, Heijst JA, Teunissen CE, Groenewegen HJ, Berendse HW, van de Berg WD, Veerhuis R. Cerebrospinal fluid and plasma clusterin levels in Parkinson’s disease. Parkinsonism Relat Disord. 2013;19:1079–1083. doi: 10.1016/j.parkreldis.2013.07.016. [DOI] [PubMed] [Google Scholar]
- Dorner AJ, Bole DG, Kaufman RJ. The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J Cell Biol. 1987;105:2665–2674. doi: 10.1083/jcb.105.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, Kelly JW. A kinetic aggregation assay allowing selective and sensitive amyloid-beta quantification in cells and tissues. Biochemistry. 2011;50:1607–1617. doi: 10.1021/bi1013744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annu Rev Genet. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards AP, Van Goor KE, Paton AW, Paton JC, Atwood WJ, Tsai B, DiMaio D. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio. 2011;2:e00101–e00111. doi: 10.1128/mBio.00101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Eckert A, Matamales M, Ittner LM, Liu X. Modes of Abeta toxicity in Alzheimer’s disease. Cell Mol Life Sci. 2011;68:3359–3375. doi: 10.1007/s00018-011-0750-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Snapp EL. ERdj3 regulates BiP occupancy in living cells. J Cell Sci. 2013;126:1429–1439. doi: 10.1242/jcs.118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hahm E, Li J, Kim K, Huh S, Rogelj S, Cho J. Extracellular protein disulfide isomerase regulates ligand-binding activity of alphaMbeta2 integrin and neutrophil recruitment during vascular inflammation. Blood. 2013;121:3789–3800. doi: 10.1182/blood-2012-11-467985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2012;10:212–215. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Araki W, Suzuki K, Suzuki T, Mizushima T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem J. 2007;402:581–589. doi: 10.1042/BJ20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- Jin Y, Awad W, Petrova K, Hendershot LM. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J. 2008;27:2873–2882. doi: 10.1038/emboj.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhuang M, Hendershot LM. ERdj3, a Luminal ER DnaJ Homologue, Binds Directly to Unfolded Proteins in the Mammalian ER: identification of Critical Residues. Biochemistry. 2009;48:41–49. doi: 10.1021/bi8015923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PA, Gibbins JM. Extracellular disulfide exchange and the regulation of cellular function. Antioxid Redox Signal. 2006;8:312–324. doi: 10.1089/ars.2006.8.312. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–485. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, Tohyama M. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–43454. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, Steurer M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. 2009;114:3960–3967. doi: 10.1182/blood-2009-03-209668. [DOI] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Klohn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci USA. 2003;100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF, Kong N. Perturbation of cellular calcium blocks exit of secretory proteins from the rough endoplasmic reticulum. J Biol Chem. 1990;265:10893–10899. [PubMed] [Google Scholar]
- Marcinowski M, Holler M, Feige MJ, Baerend D, Lamb DC, Buchner J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat Struct Mol Biol. 2011;18:150–158. doi: 10.1038/nsmb.1970. [DOI] [PubMed] [Google Scholar]
- Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, Menger L, Michaud M, Zitvogel L, Kroemer G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann N Y Acad Sci. 2010;1209:77–82. doi: 10.1111/j.1749-6632.2010.05740.x. [DOI] [PubMed] [Google Scholar]
- Massey S, Burress H, Taylor M, Nemec KN, Ray S, Haslam DB, Teter K. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect Immun. 2011;79:4739–4747. doi: 10.1128/IAI.05503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyne F, Gloeckner SF, Ciesielczyk B, Heinemann U, Krasnianski A, Meissner B, Zerr I. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J Alzheimers Dis. 2009;17:863–873. doi: 10.3233/JAD-2009-1110. [DOI] [PubMed] [Google Scholar]
- Milojevic J, Esposito V, Das R, Melacini G. Understanding the molecular basis for the inhibition of the Alzheimer’s Abeta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J Am Chem Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- Milojevic J, Raditsis A, Melacini G. Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism. Biophys J. 2009;97:2585–2594. doi: 10.1016/j.bpj.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Nizard P, Tetley S, Le Drean Y, Watrin T, Le Goff P, Wilson MR, Michel D. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic. 2007;8:554–565. doi: 10.1111/j.1600-0854.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279:445–450. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters LR, Raghavan M. Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J Immunol. 2011;187:919–931. doi: 10.4049/jimmunol.1100690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova K, Oyadomari S, Hendershot LM, Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008;27:2862–2872. doi: 10.1038/emboj.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Reyes Barcelo AA, Gonzalez-Velasquez FJ, Moss MA. Soluble aggregates of the amyloid-beta peptide are trapped by serum albumin to enhance amyloid-beta activation of endothelial cells. J Biol Eng. 2009;3:5. doi: 10.1186/1754-1611-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- Rosenberg ME, Girton R, Finkel D, Chmielewski D, Barrie A, 3rd, Witte DP, Zhu G, Bissler JJ, Harmony JA, Aronow BJ. Apolipoprotein J/clusterin prevents a progressive glomerulopathy of aging. Mol Cell Biol. 2002;22:1893–1902. doi: 10.1128/MCB.22.6.1893-1902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Susuki S, Suico MA, Miyata M, Ando Y, Mizuguchi M, Takeuchi M, Dobashi M, Shuto T, Kai H. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007;26:2501–2512. doi: 10.1038/sj.emboj.7601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA. 2011;305:1322–1326. doi: 10.1001/jama.2011.381. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Hammarstrom P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83:409–417. doi: 10.1097/01.lab.0000059937.11023.1f. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol Biol Cell. 2005;16:40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silajdzic E, Minthon L, Bjorkqvist M, Hansson O. No diagnostic value of plasma clusterin in Alzheimer’s disease. PLoS One. 2012;7:e50237. doi: 10.1371/journal.pone.0050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR, 3rd, Kelly JW. ERdj3 Is an Endoplasmic Reticulum Degradation Factor for Mutant Glucocerebrosidase Variants Linked to Gaucher’s Disease. Chem Biol. 2014;21:967–976. doi: 10.1016/j.chembiol.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Teixeira PF, Cerca F, Santos SD, Saraiva MJ. Endoplasmic reticulum stress associated with extracellular aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. J Biol Chem. 2006;281:21998–22003. doi: 10.1074/jbc.M602302200. [DOI] [PubMed] [Google Scholar]
- Terada K, Manchikalapudi P, Noiva R, Jauregui HO, Stockert RJ, Schilsky ML. Secretion, surface localization, turnover, and steady state expression of protein disulfide isomerase in rat hepatocytes. J Biol Chem. 1995;270:20410–20416. doi: 10.1074/jbc.270.35.20410. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1alpha-XBP1s Induces PDI Expression to Increase MTP Activity for Hepatic VLDL Assembly and Lipid Homeostasis. Cell Metab. 2012;16:473–486. doi: 10.1016/j.cmet.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen KW, Damania B. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene. 2010;29:3532–3544. doi: 10.1038/onc.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, Cheng R, Lee JH, Bird TD, Bennett DA, Diaz-Arrastia R, Goate AM, Farlow M, Ghetti B, Sweet RA, Foroud TM, Mayeux R. Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011;7:e1001308. doi: 10.1371/journal.pgen.1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Koulov A, Powers E, Kelly JW, Balch WE. Protein energetics in maturation of the early secretory pathway. Curr Opin Cell Biol. 2007;19:359–367. doi: 10.1016/j.ceb.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YH, Lu AC, Wang YC, Cheng HC, Chang C, Chen PH, Yu JY, Fann MJ. Protogenin defines a transition stage during embryonic neurogenesis and prevents precocious neuronal differentiation. J Neurosci. 2010;30:4428–4439. doi: 10.1523/JNEUROSCI.0473-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Wyatt AR, Yerbury JJ, Dabbs RA, Wilson MR. Roles of Extracellular Chaperones in Amyloidosis. J Mol Biol. 2012;421:499–516. doi: 10.1016/j.jmb.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Wyatt AR, Yerbury JJ, Ecroyd H, Wilson MR. Extracellular chaperones and proteostasis. Annu Rev Biochem. 2013;82:295–322. doi: 10.1146/annurev-biochem-072711-163904. [DOI] [PubMed] [Google Scholar]
- Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hamada H, Shinkai H, Kohno Y, Koseki H, Aoe T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J Biol Chem. 2003;278:34525–34532. doi: 10.1074/jbc.M304188200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–15075. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Ottenberg G, Sferrazza GF, Lasmezas CI. Highly neurotoxic monomeric alpha-helical prion protein. Proc Natl Acad Sci USA. 2012;109:3113–3118. doi: 10.1073/pnas.1118090109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Source Data for Supplementary Figure S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Review Process File
Source Data for Figure 1A, C, E, H
Source Data for Figure 2A, B
Source Data for Figure 3A, D
Source Data for Figure 5B, C, D, E
Source Data for Figure 6A, C, E