

Abstract
The final event of the eukaryotic cell cycle is cytokinesis, when two new daughter cells are born. How the timing and execution of cytokinesis is controlled is poorly understood. Here, we show that downregulation of cyclin-dependent kinase (Cdk) activity, together with upregulation of its counteracting phosphatase Cdc14, controls each of the sequential steps of cytokinesis, including furrow ingression, membrane resolution and cell separation in budding yeast. We use phosphoproteome analysis of mitotic exit to identify Cdk targets that are dephosphorylated at the time of cytokinesis. We then apply a new and widely applicable tool to generate conditionally phosphorylated proteins to identify those whose dephosphorylation is required for cytokinesis. This approach identifies Aip1, Ede1 and Inn1 as cytokinetic regulators. Our results suggest that cytokinesis is coordinately controlled by the master cell cycle regulator Cdk together with its counteracting phosphatase and that it is executed by concerted dephosphorylation of Cdk targets involved in several cell biological processes.
Keywords: Cdc14 phosphatase, cytokinesis, functional genomics, mitosis, phospho-proteomics
Introduction
Progression through the cell cycle is governed by oscillations in activity of the cyclin-dependent kinase (Cdk) complexes. Cdk activity is dependent upon periodic expression of cyclin subunits. Cyclin levels are low in G1, gradually increasing to reach a summit in mitosis. The associated high Cdk activity phosphorylates numerous substrates that bring about the mitotic state (Holt et al, 2009). Once chromosomes are bi-oriented and cells enter anaphase, cyclin levels decrease via ubiquitin-mediated proteolysis catalyzed by the anaphase-promoting complex (APC; Glotzer et al, 1991). However, degradation of mitotic cyclins by itself is not sufficient for cell cycle progression out of mitosis. Cdk inhibition and activation of Cdk-counteracting phosphatases are required to reverse the many mitotic phosphorylation events and allow mitotic exit (Visintin et al, 1998; Niiya et al, 2005; Dischinger et al, 2008; Bouchoux & Uhlmann, 2011; Sanchez-Diaz et al, 2012). The major Cdk-counteracting phosphatase in budding yeast is Cdc14. It is kept inactive for much of the cell cycle by sequestration in the nucleolus. In anaphase, activation of a signaling cascade, the mitotic exit network (MEN), results in Cdc14 activation and its release into the nucleus and the cytoplasm (Jaspersen et al, 1998; Shou et al, 1999; Visintin et al, 1999; Lee et al, 2001). Once released, Cdc14 dephosphorylates numerous Cdk targets and contributes to Cdk downregulation by transcriptional activation and stabilization of the Cdk inhibitor Sic1 (Visintin et al, 1998). The role of phosphatases during mitotic exit in higher eukaryotes is a topic of intense current study. Members of the Cdc14, PP1 and PP2A families have been implicated in this process (Mochida et al, 2009; Wu et al, 2009; Wurzenberger & Gerlich, 2011), likely with overlapping roles.
Accurate timing of cell cycle events is critical for faithful completion of the cell division cycle. Thus, the events occurring during mitotic exit need to take place in a strictly defined order. First, chromosome arms reach their state of maximal condensation as the anaphase spindle elongates to pull chromosomes apart. Following this, chromosomes decondense and the spindle disassembles before two new daughter cells are pinched off by cytokinesis as the final event of the cell cycle. We have put forward a model that explains the ordering of mitotic exit events in budding yeast. The gradual change of the Cdk kinase to Cdc14 phosphatase activity ratio over the course of mitotic exit is detected by individual Cdk substrates that respond by dephosphorylation at distinct thresholds (Bouchoux & Uhlmann, 2011). In this model, cytokinesis is under the control of Cdk substrates whose dephosphorylation requires complete Cdk downregulation and the highest level of Cdc14 activity. Whether the regulation of cytokinesis can indeed be included in this paradigm and how the cell cycle timing of cytokinesis is controlled remain poorly understood.
Cytokinesis in budding yeast requires the concerted actions of two interdependent processes, actomyosin ring contraction and deposition of the primary septum. The latter is limited by the localized activity of Chs2, the catalytic subunit of the chitin synthase II complex. The C2 domain protein Inn1 and the SH3 domain proteins Hof1 and Cyk3 are recruited to the bud neck region just before onset of actomyosin ring constriction and have been suggested to coordinate this process with primary septum deposition (Meitinger et al, 2012). Although many players involved in cytokinesis have been uncovered, it is less clear how the cell cycle regulates them.
A role for Cdc14 in cytokinesis has been apparent since the early characterization of the cdc14 temperature-sensitive mutant phenotype, causing cells to arrest after nuclear division but before cytokinesis (Culotti & Hartwell, 1971). It has remained less clear how direct the role of Cdc14 is. A similar late mitotic arrest phenotype is observed after inactivation of MEN components. Cdc14 activates the MEN, which in turn sustains Cdc14 activity (Culotti & Hartwell, 1971; Jaspersen & Morgan, 2000; Lee et al, 2001; Stegmeier et al, 2002; König et al, 2010). Arguments for a contribution of both MEN components and of Cdc14 to cytokinesis have been made in budding and fission yeast as well as in higher eukaryotes (Luca et al, 2001; Gruneberg et al, 2002; Kaiser et al, 2002; Mailand et al, 2002; Hwa Lim et al, 2003; Corbett et al, 2006; Clifford et al, 2008; Meitinger et al, 2010; Sanchez-Diaz et al, 2012). Budding yeast Cdc14 is present at the bud neck at the time of cytokinesis (Bembenek et al, 2005; Palani et al, 2012), suggesting the possibility of a direct contribution. Although a number of cytokinetic proteins have been shown to be Cdc14 targets (Meitinger et al, 2012; Kao et al, 2014), which of these control essential cytokinetic events remains unknown (Breitkreutz et al, 2010; Bloom et al, 2011; Kao et al, 2014).
Here, we further investigate how Cdc14 contributes to cytokinesis. We created cells in which Cdc14 localization remains restricted to the nucleus following its activation in anaphase. These cells show unaltered cell cycle dynamics but fail to complete cytokinesis, with defects at all observable cytokinetic stages, strongly suggestive of direct roles for Cdc14. To identify Cdc14 substrates that regulate cytokinesis, we performed time-resolved phosphoproteome analysis of Cdc14-driven budding yeast mitotic exit. We employed a strategy to conditionally phosphorylate candidate proteins and demonstrate that this is a fertile approach to identify novel cytokinetic regulators.
Results
Cytoplasmic Cdc14 is required for cytokinesis but not cell cycle progression
In order to study the role of the phosphatase Cdc14 in cytokinesis, we aimed to create cells that are defective for Cdc14 activity at the site of cytokinesis. Since the cytokinetic machinery is accessible from the cytoplasm, we postulated that Cdc14 release into this cellular compartment is required for cytokinesis. We therefore reconstituted a cdc14-3 temperature-sensitive budding yeast strain either with wild-type Cdc14 fused to GFP (denoted +CDC14) or with Cdc14 fused to GFP and the strong nuclear localization signal from SV40 large T antigen (+CDC14-NLS: Kalderon et al, 1984). Cytological examination revealed that ectopic wild-type Cdc14 behaved as expected, being constrained to the nucleolus for much of the cell cycle but released throughout the nucleus and cytoplasm in cells containing long anaphase spindles. Cdc14-NLS was similarly sequestered in the nucleolus during interphase, but in contrast to wild-type, Cdc14 was undetectable in the cytoplasm during anaphase, when it maintained a marked nuclear accumulation (Fig 1A and Supplementary Fig S1).
Figure 1. Cytoplasmic Cdc14 is dispensable for mitotic exit.

- cdc14-3 temperature-sensitive cells reconstituted with CDC14 or CDC14-NLS were released from a G1 arrest at the restrictive temperature and stained for Cdc14 and the spindle (Tub1) once cells had reached late anaphase.
- Wild-type, cdc14-3, +CDC14 and +CDC14-NLS cells were released from an α-factor-induced G1 arrest and re-arrested in the next G1. Samples were harvested at the indicated time points and stained with DAPI and for the nucleolar protein Nop1 (n > 100). Photographs of examples of cells without (top) and with (bottom) rDNA segregation defects at 80 min after release are shown together with quantification over time.
- Cells from the experiment in (B) were stained for the spindle (n > 100). Cells without (top) and with (bottom) anaphase spindles at 100 min after release are shown together with their quantification over time.
- FACS analysis of DNA content and Western blot analysis of cell cycle markers of the samples from (B). Tub1 served as a loading control.
Data information: Scale bars, 5 μm.
We first tested whether Cdc14-NLS is able to fulfill its nuclear function in promoting rDNA segregation (Sullivan et al, 2004). We used cells synchronized in G1 using the mating pheromone α-factor and subsequently released these to pass through the cell cycle at the restrictive temperature for the cdc14-3 allele. As described, cdc14-3 cells accumulated in a late anaphase state with bi-lobed nuclei (Culotti & Hartwell, 1971). The rDNA locus, visualized by staining against the nucleolar protein Nop1, was often stretched across the bud neck region or segregated unequally (Fig 1B). rDNA segregation was rescued in both +CDC14 and +CDC14-NLS cells, as indicated by equally segregated Nop1 signals in opposite cell halves during anaphase. This suggests that Cdc14-NLS is capable of fulfilling its nuclear function in rDNA segregation.
We next compared markers of cell cycle progression in +CDC14 and +CDC14-NLS cells. Staining for tubulin confirmed late mitotic arrest of the cdc14-3 parental strain with elongated anaphase spindles. Both +CDC14 and +CDC14-NLS cells elongated and then disassembled their spindles with similar kinetics to a wild-type control that was included in the experiment for comparison, indicative of unhindered cell cycle progression out of mitosis (Fig 1C). Western blotting confirmed that Cdc14-dependent degradation of the major budding yeast mitotic cyclin Clb2 occurred in both +CDC14 and +CDC14-NLS cells with kinetics similar to wild-type (Fig 1D). In addition, the Cdc14-dependent appearance of the Cdk inhibitor Sic1, indicating completion of mitotic exit and return to a G1-like cell cycle state, occurred with wild-type kinetics in both +CDC14 and +CDC14-NLS cells. However, FACS analysis of the DNA content showed that only +CDC14 cells completed cytokinesis and returned to a 1C DNA content. In striking contrast, +CDC14-NLS cells persisted as large-budded cells with 2C DNA content. This suggests that cytoplasmic Cdc14 is not required for most aspects of cell cycle progression out of mitosis, but that it is required for cytokinesis.
To confirm that cytoplasmic Cdc14 is required for cytokinesis but not cell cycle progression, we grew +CDC14 and +CDC14-NLS cells at a restrictive temperature for 5 h. After brief sonication, +CDC14 cells were all individualized, while +CDC14-NLS cells had formed large chains and aggregates of connected cells (Fig 2A and Supplementary Fig S2A). This phenotype is consistent with cytokinetic failure but continued cell cycle progression.
Figure 2. Cytoplasmic Cdc14 is required for cytokinesis.
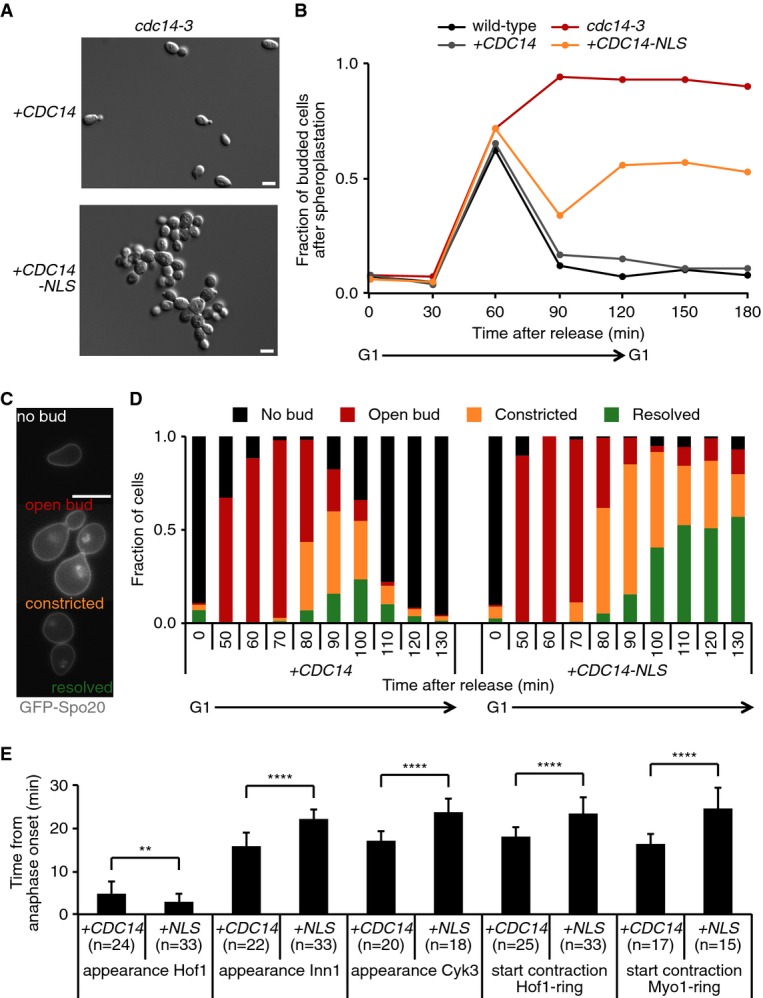
- +CDC14 and +CDC14-NLS cells were grown at 35.5°C for 5 h and sonicated, and differential interference contrast (DIC) images were acquired.
- Cells from a synchronous culture (as in Fig 1B) were treated with zymolyase to remove the cell wall. The fraction of remaining budded cells is shown over time (n = 100).
- Cells expressing the plasma membrane marker GFP-Spo2051–91, exemplifying the indicated bud neck phenotypes and stages of cytokinesis.
- Bud neck phenotypes of synchronized GFP-Spo2051–91-expressing +CDC14 or +CDC14-NLS cells during the progression of a cell cycle (n > 75).
- +CDC14 or +CDC14-NLS cells expressing Spc42-YFP and GFP fusions with the indicated cytokinetic protein were synchronized in G1 using α-factor. Upon release at 35.5°C, cells were imaged every 2 min and the time from anaphase onset to the indicated events was recorded. Error bars represent SD; Student’'s t-test was applied to analyze differences between the indicated conditions; **P ≤ 0.01; ****P ≤ 0.0001. For representative image sequences, see Supplementary Fig S2C.
Data information: Scale bars, 10 μm.
Cytoplasmic Cdc14 promotes sequential stages of cytokinesis
To investigate how Cdc14 contributes to cytokinesis, we repeated a time course and compared cell separation in wild-type, cdc14-3, +CDC14 and +CDC14-NLS backgrounds, but this time after enzymatic removal of the cell wall using zymolyase. Over half of +CDC14-NLS cells failed to separate, while no budded +CDC14 spheroplasts persisted above background levels (Fig 2B). Thus, cytoplasmic Cdc14 is required for cytokinesis at stages preceding plasma membrane separation.
To analyze in more detail at which stage of cytokinesis Cdc14 acts, we visualized the plasma membrane using a GFP-Spo2051–91 fusion protein (Nakanishi et al, 2004). This allowed us to identify four stages of cytokinesis: (i) cells with an open bud neck, (ii) a constricted plasma membrane that still connects the daughter cells, (iii) resolved plasma membranes after abscission while cells remain connected by their cell wall, and (iv) unbudded cells after cell separation (Fig 2C; Norden et al, 2006). Quantification of the four stages during cell cycle progression of synchronized cells expressing +CDC14 or +CDC14-NLS (now without fusion to GFP) is shown in Fig 2D, revealing delayed progression in each of the stages of cytokinesis. The “open bud” configuration persisted in 11% of +CDC14-NLS cells until the end of the time course, which was never observed in the +CDC14 control. An even greater percentage of cells persisted with “constricted” and “resolved” plasma membranes. Virtually none of the +CDC14-NLS cells completed cytokinesis, while almost all +CDC14 control cells completed cell separation by the end of the experiment. The NLS fusion greatly reduces, but might not completely abolish cytoplasmic Cdc14, due to the dynamic process of nuclear shuttling. This could explain the residual slow cytokinetic progression in +CDC14-NLS cells. Alternatively, Cdc14-independent pathways might compensate for Cdc14-dependent cytokinesis, albeit inefficiently. In either event, these observations suggest that cytoplasmic Cdc14 promotes several sequential steps during cytokinesis.
To address the molecular functions affected by cytoplasmic Cdc14, we used live cell imaging of cells expressing GFP fusions with cytokinetic proteins. We measured the timing of several early cytokinetic events, namely the appearance of the SH3 domain-containing proteins Hof1 and Cyk3 and of the C2 domain protein Inn1, at the bud neck, as well as the onset of Hof1 and Myo1 ring contraction. The timing of these events was recorded relative to the start of anaphase spindle elongation, monitored by Spc42-YFP-marked spindle pole bodies (Fig 2E and Supplementary Fig S2B). While the recruitment of Hof1 to the bud neck occurred with slightly earlier timing in +CDC14-NLS than in +CDC14 cells, subsequent recruitment of Inn1 and Cyk3 and the start of cytokinetic ring contraction were each delayed by approximately 8 min in cells lacking cytoplasmic Cdc14. This suggests that one role of Cdc14 lies in a step after Hof1 recruitment and before Inn1 and Cyk3 appearance at the bud neck.
We next took a genetic approach to characterize the contribution of cytoplasmic Cdc14 to cytokinesis. In budding yeast, cytokinetic factors have been categorized into three pathways (Korinek et al, 2000; Meitinger et al, 2010). Defects in either of the pathways are compensated by the others, but deficiencies in any two of the pathways are synthetically lethal. We therefore introduced deletions in genes representing two of these pathways, hof1Δ and cyk3Δ, into a +CDC14-NLS strain. The absence of cytoplasmic Cdc14 had a detrimental effect on the growth of hof1Δ cells, while cyk3Δ cells were not affected (Supplementary Fig S2C). These observations are consistent with a role for cytoplasmic Cdc14 in the Cyk3-dependent cytokinetic pathway. Analysis of the third, myosin-dependent, pathway in this manner was inconclusive, as cells lacking Myo1 grow very poorly and rapidly accumulate genetic alterations to compensate for the absence of myosin (Rancati et al, 2008).
Cdk downregulation and Cdc14 activation drive cytokinesis
Our above results confirm that Cdc14 plays an important role during budding yeast cytokinesis. We next wanted to address whether dephosphorylation of Cdk target proteins by Cdc14 is sufficient to drive cytokinesis. We used cells that are arrested in metaphase by Cdc20 depletion. Reducing Cdk activity by expression of the Cdk inhibitor Sic1, or ectopic Cdc14 expression, promoted cytokinesis relatively inefficiently (Fig 3A). Since dephosphorylation especially of late Cdc14 targets is synergistically driven by Cdk downregulation and Cdc14 activation (Bouchoux & Uhlmann, 2011), we combined both treatments. In line with a previous report (Sanchez-Diaz et al, 2012), co-expression of Sic1 and Cdc14 greatly enhanced the efficiency of cytokinesis (Fig 3A). We observed plasma membrane resolution in virtually all cells within 2 h of induction.
Figure 3. Cdk downregulation and Cdc14 drive MEN-independent cytokinesis.
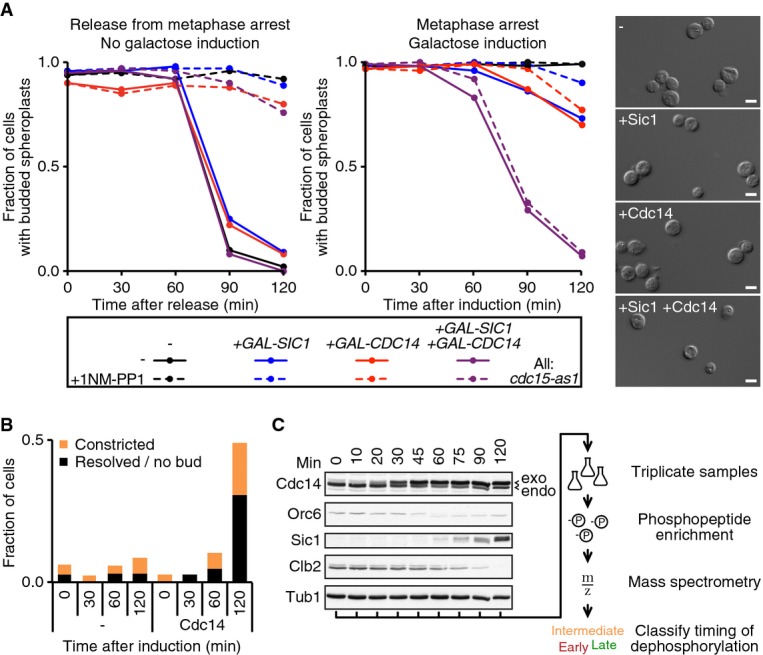
- MET3pr-CDC20 cdc15-as1 cells were arrested in metaphase by Cdc20 depletion and subsequently treated with 1NM-PP1 to inhibit Cdc15-as1. Cells were released from the metaphase arrest by Cdc20 re-induction (left graph), or Cdc14 and/or Sic1 expression were induced from the GAL1 promoter while cells remained arrested (right graph). The fraction of large-budded cells after spheroplastation (n = 100) was quantified. Representative photographs 120 min after galactose addition are shown. Scale bar, 10 μm.
- As (A), the plasma membrane marker GFP-Spo2051–91 was visualized following Cdc14 induction (n = 100).
- Samples from the experiment in (B) were processed for Western blot analysis of the indicated cell cycle proteins. Arrowheads indicate endogenous and exogenous forms of Cdc14. Tub1 served as a loading control. A schematic of downstream processing for the phosphoproteome analysis is shown on the right.
Efficient cytokinesis driven by Cdk inhibition and Cdc14 expression allowed us to address the contribution of the MEN. We inactivated the MEN using an ATP analogue (1NM-PP1)-sensitive allele of the essential MEN kinase Cdc15 (D'Aquino et al, 2005). As expected, this efficiently blocked mitotic exit when releasing cells from the mitotic block. In contrast, MEN inhibition did not reduce the efficiency of cytokinesis following Sic1 and Cdc14 induction (Fig 3A). These observations suggest that Cdk downregulation and Cdc14 phosphatase activation, and therefore most likely the dephosphorylation of Cdk target proteins, drive cytokinesis. We therefore hypothesize that the main contribution of the MEN to cytokinesis is that of promoting Cdk downregulation and Cdc14 activation.
Phosphoproteome analysis of mitotic exit and cytokinesis
To identify Cdk substrates that regulate cytokinesis, we performed mass spectrometric phosphoproteome analysis of budding yeast mitotic exit. To achieve sufficient synchrony within this short period of the budding yeast cell cycle, we utilized mitotically arrested cells and overexpressed Cdc14. Using the GFP-Spo2051–91 membrane marker, we found that cells constricted their plasma membrane within 2 h of induction, with half of the cells completing membrane resolution (Fig 3B). The dephosphorylation of known Cdk substrates occurs with good synchrony during this time course of Cdc14-induced mitotic exit (Bouchoux & Uhlmann, 2011; Fig 3C). We harvested triplicate samples at eight time points after Cdc14 induction. Whole cell extracts were prepared, and phospho-peptides were enriched from tryptic protein digests and subjected to label-free quantitative mass spectrometry. This allowed us to follow the abundance of over 2,100 phospho-peptides, representing more than 800 proteins (Fig 4A; for details, see Supplementary Materials and Methods, Supplementary Dataset S1).
Figure 4. Phosphoproteome analysis of Cdc14-induced mitotic exit.
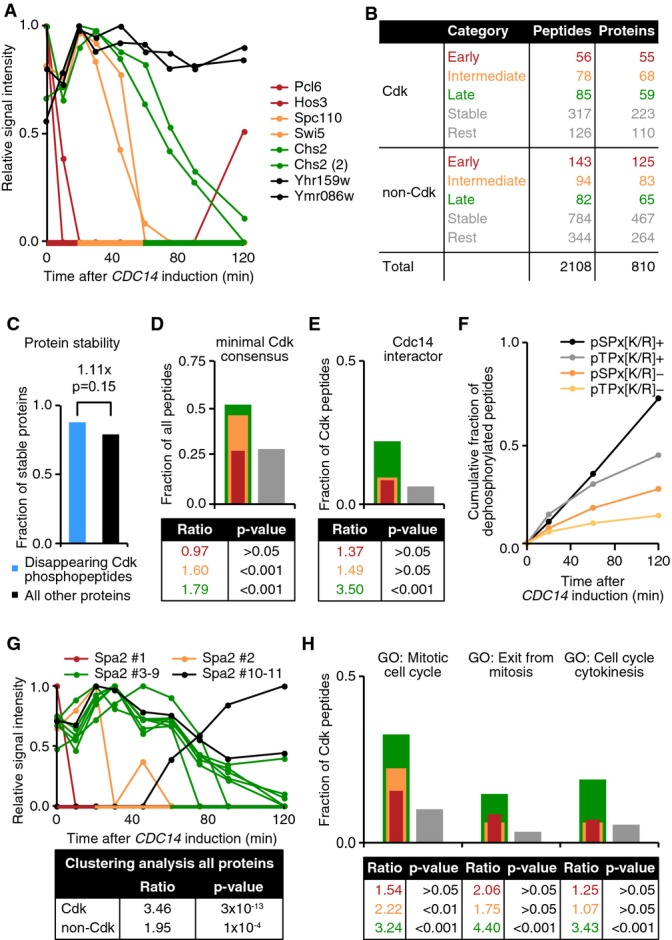
- Phospho-peptide abundance during Cdc14-induced mitotic exit. Examples of phospho-peptides that disappear with early (red), intermediate (orange) and late (green) timing or that remain stable (black) are shown. This color coding is used throughout all panels of this figure.
- Numbers of unique peptides and corresponding proteins that do or do not contain a minimal Cdk consensus motif are shown, categorized according to timing of disappearance as described in (A).
- Protein stability data for proteins corresponding to disappearing phospho-peptides (blue) and for all other proteins (black).
- The fractions of peptides that contain a minimal Cdk consensus motif ([S/T]-P) within the timing categories shown in (B) were calculated. The ratio and P-value of enrichment of each category relative to the combined stable/rest category (gray) are shown.
- Similar to (D), the enrichment for reported physical Cdc14 interactors is assessed within [S/T]-P-containing phospho-peptides.
- Peptides that contain the indicated Cdk consensus sequence motifs and that have disappeared over time are represented as a fraction of all phospho-peptides containing that motif.
- Unique phospho-peptides of Spa2, as an example, are categorized based on their dephosphorylation timing (graph). Enrichment of clustered timing for all proteins covered by phospho-peptides was compared to random for Cdk or non-Cdk consensus site-containing peptides (table). Fisher’'s exact tests were used to determine significance.
- Enrichment of the indicated gene ontologies among Cdk consensus site-containing peptides as in (D).
We first classified the identified phospho-peptides into those that contain the minimal Cdk phosphorylation consensus site, [S/T]-P, and those that do not. Of the peptides with a Cdk consensus site, about a third disappeared over the course of mitotic exit (Fig 4B). Depending on the timing of disappearance, we classified the peptides into five categories: those whose abundance decreased with early (≤ 20 min), intermediate (20–60 min) or late (> 60 min) timing, those that remained stable and a rest category that could not be unambiguously attributed to one of the former categories. The dephosphorylation timing of Cdc14 targets Fin1 (early; Supplementary Fig S3), and Orc6 and Sic1 (late; Fig 3C) have previously been determined (Bouchoux & Uhlmann, 2011) and were used to define the time boundaries for these categories. Disappearance of a phospho-peptide could be due to dephosphorylation, or it could be due to degradation of the target protein during mitotic exit. To obtain an estimate of the fraction of peptides that disappeared due to proteolysis, we performed shotgun mass spectrometry of our time course samples without phospho-peptide enrichment. This provided protein stability data for 1,808 proteins (see Materials and Methods). Among those were 49 proteins for which we found at least one disappearing phosphorylated Cdk peptide, and of those 43 (88%) were stable proteins. This is an equal, if not greater, degree of stability as observed for all other 1,759 proteins (79%, Fig 4C). We conclude that protein degradation makes only a small contribution to the disappearance of Cdk phospho-peptides during mitotic exit and that the majority of the observed phosphorylation changes are most likely due to protein dephosphorylation.
Protein dephosphorylation in response to Cdc14 expression could be due to direct dephosphorylation by Cdc14 or due to dephosphorylation by other phosphatases that might be indirectly activated by Cdc14. A Cdk consensus phosphorylation site is also the preferred sequence for dephosphorylation by Cdc14 (Gray et al, 2003; Bremmer et al, 2012). Disappearing phospho-peptides, especially those found in the intermediate and late categories, were significantly enriched for minimal Cdk consensus sites (Fig 4D). Moreover, late dephosphorylated peptides that contain Cdk sites were enriched for known physical Cdc14 interactors as annotated in the Saccharomyces Genome Database (SGD; Fig 4E). Together, these observations make it likely that many of the dephosphorylation events recorded in our survey are catalyzed by Cdc14.
In vitro, Cdc14 has been observed to favor phospho-serine (pS) over phospho-threonine (pT) residues, as well as sites with a positively charged residue (K/R) at the +3 position (Bremmer et al, 2012). When plotting cumulative peptide dephosphorylation over time, we observed that pS-containing peptides were almost twice as likely than pT-containing peptides to disappear by the end of the experiment. Similarly, K/R-containing peptides were 2.8 times more likely to disappear compared to peptides lacking a positive charge at the +3 position (Fig 4F). These observations are consistent with phospho-serines and a basic residue at the +3 position being determinants for Cdc14 substrate preference in vivo.
Our large phospho-peptide dataset generated in this study offers additional insights into protein dephosphorylation dynamics. Proteins are often phosphorylated at multiple sites. In cases where we could follow more than one phospho-peptide per protein, they were often but not always dephosphorylated with similar timing (Fig 4G). Two Cdk phospho-peptides of the same protein were 3.5 times more likely to be member of the same dephosphorylation timing category than expected by chance. Non-Cdk peptides were also often co-regulated, albeit less stringently. Thus, multiple modified residues, especially Cdk phosphorylation sites, within the same protein tend to be coordinately dephosphorylated. Exceptions to this tendency, that we observed (Fig 4G), could point to distinct modes of regulation affecting individual phosphorylation sites. Our dataset is a valuable resource to delineate protein features that determine dephosphorylation dynamics.
The primary goal of these measurements was the discovery of proteins whose dephosphorylation controls cytokinesis. We therefore tested whether protein dephosphorylation timing in our dataset is associated with biological function, based on gene ontologies (GO). We used unbiased DAVID analysis (Huang et al, 2009a,b) to identify GO terms associated with dephosphorylation timing. This revealed the GO term “mitotic cell cycle” as significantly enriched among both intermediate and late dephosphorylated proteins with Cdk sites. “Exit from mitosis” and “cell cycle cytokinesis” were significantly enriched in the late category (Fig 4H). Thus, dephosphorylation timing in our experiment is associated with relevant physiological processes, which justifies the search of our dephosphorylation dataset for cytokinesis regulators.
Identification of phospho-regulated cytokinesis factors
Having identified likely Cdc14 substrates whose dephosphorylation coincides with cytokinesis, we sought to establish a causal relationship between the observed dephosphorylation events and cytokinesis. We designed a PCR-based gene targeting strategy, depicted in Fig 5A, based on the idea that covalent fusion to a cyclin can maintain constitutive Cdk target protein phosphorylation (Lyons & Morgan, 2011). We used the mitotic cyclin Clb2 for the fusions, from which we removed degradation and localization signals (denoted Clb2m) to avoid interfering with target protein function. Following gene targeting, fusion of Clb2m to a protein of interest was conditional, pending estradiol-activated Cre recombinase-mediated excision of the selection marker. As a control, we constructed a similar targeting vector in which Clb2m contained three additional point mutations that prevent its interaction with the Cdk kinase subunit (Bailly et al, 2003; denoted Clb2mΔCdk). Tests with two established Cdk substrates, Ask1 and Sic1, validated the conditional fusion strategy and the effect of the fusions on the target proteins (Supplementary Fig S4A–C).
Figure 5. Identification of phospho-regulated cytokinetic factors.
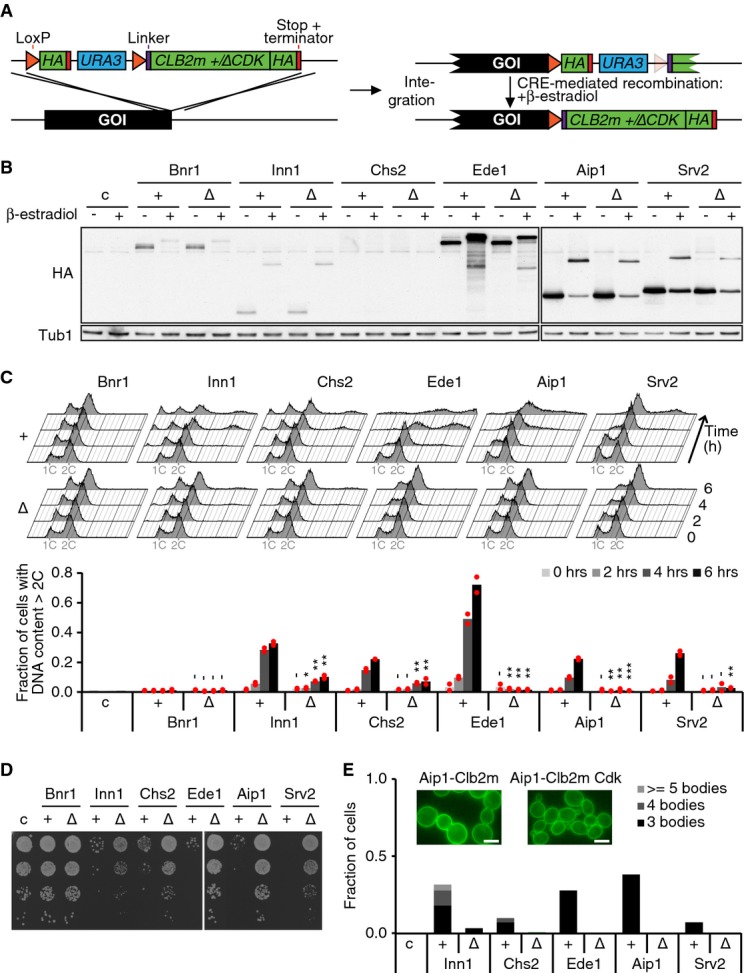
- Targeting strategy used for creating conditionally phosphorylated proteins. Upon integration of the targeting cassette and expression of the gene of interest (GOI), the targeted protein is C-terminally tagged with an HA epitope. After Cre recombinase-mediated excision, Clb2m-HA is fused to the targeted protein.
- Western blot analysis of cells expressing conditional Clb2m (+) or Clb2mΔCdk (Δ) fusions to the indicated proteins, before (−) and 5 h after (+) β-estradiol treatment. Tub1 served as a loading control. c, control cells.
- FACS analysis of DNA content after conditional Clb2m or Clb2mΔCdk fusion to the indicated proteins. Quantification of the fraction of cells with > 2C DNA content is shown in the bar graph for two independent biological replicates, with red dots representing individual and bars representing average values. Bnr1 fusions were included as controls for cytokinetic defects. Student’'s t-test was applied to compare matched Clb2m versus Clb2mΔCdk fusion strains. -P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
- Serial dilutions of cells from (C) were plated 6 h following β-estradiol treatment and grown for 1 day at 36°C.
- The length of cell chains containing a common cytoplasm was scored 6 h after β-estradiol treatment using the GFP-Spo2051–91 plasma membrane marker (n > 150). c, control cells; scale bars, 10 μm.
We then targeted 26 candidate cytokinetic regulators with the conditional Clb2m fusion, and the Clb2mΔCdk control. These included proteins of intermediate or late dephosphorylation timing in our phosphoproteome analysis, as well as additional potential cytokinetic regulators (Supplementary Fig S4D). Following Cre recombinase activation and consequent Clb2m fusion (Fig 5B), we monitored cellular DNA content in exponentially proliferating cell cultures over 6 h by fluorescence-activated cell scanning (FACS; Fig 5C top). Clb2m fusion to five of the 26 candidates, Inn1, Chs2, Ede1, Aip1 and Svr2, elicited an increased cellular DNA content, suggestive of cytokinetic defects. This was less pronounced or absent after fusion to Clb2mΔCdk (Fig 5C bottom). In addition, the five Clb2m-fusion strains displayed growth retardation, compared to their Clb2mΔCdk controls (Fig 5D), consistent with the expected long-term effects of cytokinetic failure.
Our FACS-based screening protocol would be unable to distinguish erroneous DNA over-replication from cytokinetic failure. We therefore microscopically examined cells containing the Inn1, Chs2, Ede1, Aip1 and Svr2-Clb2m fusions. This revealed cell chain formation in all strains containing the Clb2m fusions, but not their controls (Supplementary Fig S4E). We next utilized the GFP-Spo2051–91 plasma membrane marker to assess cytokinesis (Fig 5E). This confirmed that the cell chains remained connected by a common cytoplasm in all cases. Thus, fusion of Inn1, Chs2, Ede1, Aip1 and Svr2 to Clb2m, but not to Clb2mΔCdk, leads to cytokinetic defects. In addition, we observed strikingly widened bud necks among the Clb2m fusion cells (Supplementary Fig S4F). These could be related to impaired septin and/or chitin ring formation (Blanco et al, 2012), or more generally to prolonged persistence of bud necks following cytokinetic failure.
Chs2 dephosphorylation by Cdc14 is known to be required for cytokinesis (Chin et al, 2011). The cytokinetic defect in Chs2-Clb2m cells is therefore expected and we regard Chs2 as a confirmation that our screening method is able to identify Cdk-regulated cytokinetic regulators. Dephosphorylation of Inn1 during mitotic exit has also been observed. However, phospho-mimetic point mutations in Inn1, changing five Cdk phospho-acceptor sites to glutamate (inn1-5E), did not cause a noticeable cytokinetic defect (Palani et al, 2012). This could be because our approach to phosphorylate Inn1 more closely resembles true phosphorylation than phospho-mimetic amino acid replacements. Alternatively, it could be that the Inn1-Clb2m fusion causes a cytokinetic defect other than through maintaining Inn1 phosphorylation.
To differentiate between these two possibilities, we mutated the five known Cdk phosphorylation sites in Inn1 at its endogenous chromosomal locus to alanine (INN1-5A) and repeated the Clb2m fusion experiment. Clb2m fusion to Inn1-5A no longer caused a cytokinetic defect (Fig 6A and B). We noticed that a small cytokinetic defect caused by Inn1-Clb2mΔCdk fusion was also rescued using the INN1-5A allele. This suggests that the cytokinetic defect in the Inn1-Clb2m strain and also the small defect in the Inn1-Clb2mΔCdk strain are due to persistent Inn1 phosphorylation at its five known Cdk phosphorylation sites. This confirms Inn1 as a Cdk target whose dephosphorylation by Cdc14 is crucial for successful cytokinesis.
Figure 6. Ede1 and Aip1 are phospho-regulated cytokinesis factors.
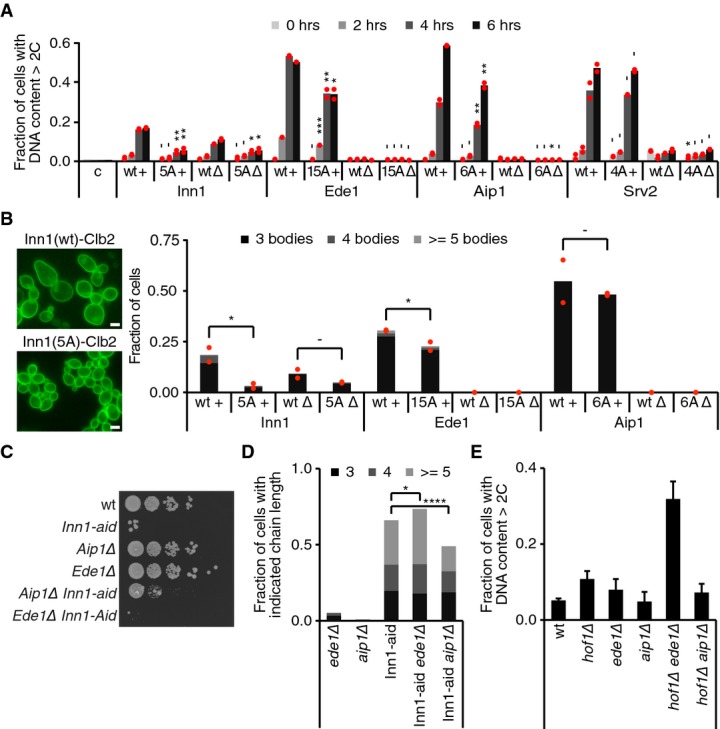
- Genes corresponding to the indicated proteins were replaced by wild-type (wt) or Cdk phospho-site mutant (xA) sequences and then conditionally fused to Clb2m or Clb2mΔCdk. Cytokinetic defects were scored by FACS analysis of DNA content as in Fig 5C. The individual values from two independent clones (red dots) as well as their average (bars) are shown. Student’'s t-test was applied to compare matched wt and xA strains. c, control cells.
- Cell chain formation in the samples from (A). The fraction of cells with > 2 cell bodies for each clone are represented with red dots and their averages by the black bar (n > 150). Significance is determined by Student’'s t-tests for the indicated conditions. Scale bars, 5 μm.
- The indicated strains were spotted in serial dilutions onto auxin-containing plates and grown for 2 days.
- Auxin was added to cultures of the indicated strains, and after 6 h, cells were sonicated and the number of attached cell bodies was counted (n > 200). Significance was calculated using a Wilcoxon rank-sum test.
- The fraction of cells in asynchronously proliferating cultures of the indicated strains with a DNA content of > 2C was determined by FACS analysis (n = 3). Error bars represent SD.
Data information: -P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
To confirm the causative role of Cdk phosphorylation for the cytokinetic defect also in strains expressing a fusion of Ede1, Aip1 or Srv2 with Clb2m, we created Cdk phosphorylation site mutant alleles of these three proteins. Ede1-15A and Aip1-6A both partially rescued cytokinesis in the Clb2m fusion strains, suggesting that persistent Cdk phosphorylation contributed to the observed cytokinetic defects (Fig 6A and B and Supplementary Fig S5). However, mutation of the four Cdk consensus phosphorylation sites in Srv2 did not alter the cytokinetic defect following Clb2m fusion. It could be that fusion of Clb2m to Srv2 leads to phosphorylation of Srv2 residues other than the Cdk consensus sites or that Clb2m fused to Srv2 leads to Cdk phosphorylation of another protein in spatial proximity to Srv2. Taken together, we found that in three out of the four cases analyzed, the cytokinetic defect due to a Clb2m fusion is at least in part due to persistent Cdk phosphorylation of the target protein.
Ede1 and Aip1 have not been previously implicated in cytokinesis. To analyze a possible cytokinetic role for these proteins, we deleted their genes in a strain background in which Inn1 can be depleted using an auxin-inducible degron (inn1-aid; Nishimura et al, 2009). inn1-aid cells show compromised growth on medium containing auxin, an effect that was augmented in the absence of Ede1 (Fig 6C). In contrast, removing Aip1 led to a partial rescue of inn1-aid cell growth. We confirmed these effects by measuring cell chain formation 6 h following auxin addition to exponentially growing cultures of the same strains. Inn1 depletion resulted in extensive chain formation. While deletion of ede1 by itself led to the occurrence of occasional cell chains, its deletion in the inn1-aid background significantly enhanced chain formation and chain length (Fig 6D). Removing Aip1 did not result in noticeable cell chain formation and, on the contrary, significantly reduced cell chains in inn1-aid cells. We observed similar genetic interactions between either Ede1 or Aip1 and Hof1. Deletion of ede1 augmented cytokinetic defects in a hof1Δ strain, while aip1 deletion partially rescued the defects (Fig 6E). We also noticed that the reduced spore viability following sporulation of a hof1Δ strain is efficiently rescued in the absence of Aip1 (data not shown). This suggests that Ede1 is a positive and Aip1 a negative regulator of cytokinesis. A possible scenario is that Ede1 dephosphorylation activates a function of this endocytic protein in cytokinesis, while the phosphorylated form of the actin regulator Aip1 may act as an inhibitor of actomyosin ring contraction.
Phospho-regulation of Inn1 localization and Chs2 activation
We set out to address the mechanism by which Inn1 dephosphorylation promotes cytokinesis. Based on experiments using an INN1-5E, it has been suggested that Inn1 dephosphorylation does not affect Inn1 localization but regulates Cyk3 recruitment to the cytokinetic furrow (Palani et al, 2012). In contrast, the dynamics and extent of Cyk3 accumulation were unaltered between Inn1-Clb2m, Inn1-Clb2mΔCdk and control cells before Clb2m fusion (Fig 7A). In addition, when directly measuring the time from anaphase onset to Cyk3 recruitment to the bud neck, we did not observe any difference between Inn1-Clb2m and Inn1-Clb2mΔCdk cells (Supplementary Fig S6B). Thus, Cyk3 recruitment is unlikely to explain the cytokinetic defect observed upon persistent Inn1 phosphorylation. We revisited the possibility that phosphorylation of Inn1 regulates its localization. Using disassembly of the anaphase spindle as an internal marker for mitotic exit progression, we found that wild-type Inn1 appears at the bud neck at a time when the anaphase spindle is still intact and remains associated with the bud neck during spindle disassembly (Fig 7B and Supplementary Fig S6A). Inn1-Clb2m was never seen at the bud neck, while spindles were intact and only appeared after spindles had been largely disassembled. This suggests that Inn1 dephosphorylation promotes its timely recruitment to the site of cytokinesis. Inn1-Clb2mΔCdk, which causes an intermediate cytokinetic defect, showed intermediate localization timing. To measure Inn1 bud neck recruitment timing directly, we used cells expressing an Inn1-GFP fusion and containing YFP-marked SPBs (Fig 7C). This confirmed that Inn1 recruitment is delayed in Inn1-Clb2m cells, thus corroborating that Inn1 dephosphorylation promotes its timely bud neck localization.
Figure 7. Inn1 phosphorylation regulates bud neck localization and Chs2 activation.

- Cyk3-GFP signal intensity at the bud neck relative to the onset of actomyosin ring contraction (t = 0). The mean and SEM from at least 20 cells are shown.
- Inn1-Clb2m, Inn1-Clb2mΔCdk and control cells before Clb2m fusion were arrested in G1 using α-factor, released and harvested 90–100 min later at the time of cytokinesis. Indirect immunofluorescence was used to determine Inn1 bud neck staining and the spindle status. Representative images of full spindles and partially and fully disassembled spindles are shown. nr, non-recombined cells. Scale bars, 5 μm.
- The time from anaphase onset until the bud neck appearance of Inn1 was determined by filming synchronized Inn1-Clb2m-GFP and Inn1-Clb2mΔCdk-GFP cells containing Spc42-YFP-labeled spindle pole bodies. Error bars represent SD.
- As in (C), now the persistence of a full actomyosin ring was measured in cells expressing Inn1-Clb2m, Inn1-Clb2mΔCdk or their respective non-phosphorylatable mutants together with Myo1-GFP and Spc42-YFP. Error bars represent SD.
- Montage of medial view projections of actomyosin rings in Inn1-Clb2m and Inn1-Clb2mΔCdk cells. Frames are shown in 2-min intervals.
- Actomyosin ring contraction phenotypes from at least 21 cells filmed as in (E) were blindly scored as having a normal (“constriction”) or aberrant (“disintegration”) cytokinetic phenotype.
- Cytokinetic defect and cell viability of Inn1-Clb2m or Inn1-Clb2mΔCdk cells in the absence or presence of Chs2V377I. In the bar graph, red dots represent values of duplicate biological replicates with bars representing the average. Significance is calculated using Student’'s t-test. 10-fold spot dilution assays are represented, together with the Western blotting analysis for Inn1 before and after its fusion to Clb2m. Tub1 served as a loading control.
Data information: -P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
To study the consequences of delayed Inn1 recruitment, we visualized Chs2 and measured the timing of its appearance at the bud neck. Chs2 appeared with indistinguishable kinetics in Inn1-Clb2m and Inn1-Clb2mΔCdk cells (Supplementary Fig S6B). Thus, Chs2 recruitment occurred independently of Inn1 dephosphorylation. However, the full Myo1 ring persisted for longer in Inn1-Clb2m compared to Inn1-Clb2mΔCdk or Inn1(5A)-Clb2m control cells (Fig 7D). Instead of constricting in a homogeneous centripetal fashion as in Inn1-Clb2mΔCdk control cells, the Myo1 signal in Inn1-Clb2m cells collapsed into one or a few spots on the original ring circumference, but the ring never contracted (Fig 7E). This phenotype was very penetrant among many cytokinetic events recorded (Fig 7F and Supplementary Fig S6C). We conclude that Inn1 dephosphorylation promotes actomyosin ring constriction.
When looking for a molecular explanation of how Inn1 dephosphorylation promotes actomyosin ring constriction, we noticed a greater variability in bud neck widths in Inn1-Clb2m cells compared to control cells (Supplementary Fig S6D). Similar increased variability has been observed in other budding yeast cytokinetic mutants, notably ones affecting cell wall synthesis (Korinek et al, 2000; Schmidt et al, 2003; Wright et al, 2008). When we measured the bud neck diameter of Chs2-Clb2m cells, we observed a similarly extended width distribution as in Inn1-Clb2m cells. A phenotype of disintegrating Myo1 rings, similar to what we observed in Inn1-Clb2m cells, has been described in cells lacking Chs2 (VerPlank & Li, 2005; Nishihama et al, 2009). This prompted us to further investigate the relationship between Inn1 and Chs2.
Inn1 has been postulated to activate Chs2 (Nishihama et al, 2009; Devrekanli et al, 2012). We therefore addressed whether the cytokinetic defect caused by constitutive Inn1 phosphorylation is due to insufficient Chs2 activity. Expression of a dominant active Chs2 variant (Chs2V377I; Devrekanli et al, 2012) in Inn1-Clb2m cells almost completely rescued cytokinesis and restored wild-type cell growth (Fig 7G). These results support a model in which Inn1 dephosphorylation promotes its timely recruitment to the bud neck region. There it activates Chs2 to initiate primary septum formation, which in turn supports normal actomyosin ring contraction.
Discussion
It has long been known that cytokinesis is linked to cell cycle progression. Budding yeast cells that are unable to downregulate mitotic Cdk activity, for example, due to expression of a non-degradable cyclin, or that are defective in Cdk-counteracting phosphatase activity, arrest in a late anaphase-like state and do not undergo cytokinesis (Culotti & Hartwell, 1971; Wäsch & Cross, 2002). The molecular basis for the coupling of cell cycle progression to cytokinesis remained less clear. Arguments have been made for both a direct contribution of Cdk substrate dephosphorylation and protein phosphorylation by late mitotic protein kinases of the MEN. Using two approaches, in which we either deplete cytoplasmic Cdc14 or provide ectopic phosphatase activity, we show that Cdc14 is required for the proper execution of multiple cytokinetic steps and that Cdc14 is able to drive cytokinesis independently of the MEN. These results do not exclude a contribution to cytokinesis from the MEN, or other late mitotic kinases, but they suggest that dephosphorylation of Cdk targets by the Cdk-counteracting phosphatase plays a key role in cytokinetic control.
Phosphoproteome analysis of mitotic exit identified Cdk substrates that are dephosphorylated at the time of cytokinesis. In our experiment, mitotic exit and cytokinesis were induced by ectopic Cdc14. This provided the necessary time resolution during an otherwise very short part of the cell cycle. Dephosphorylation of numerous known Cdk substrates under these experimental conditions closely matches that observed during normal mitotic exit (Bouchoux & Uhlmann, 2011). This approach was validated by the identification of several dephosphorylation events of physiological significance. Nevertheless, Cdc14-induced mitotic exit might not recapitulate all aspects of substrate dephosphorylation in an undisturbed cell cycle. For example, phospho-serines were preferred substrates for dephosphorylation during Cdc14-induced mitotic exit. Whether this is indeed a feature of mitotic exit, or whether phospho-threonines are the preferred substrate of another mitotic exit phosphatase, needs to be addressed in the future. Our comprehensive method to survey phospho-peptide abundance should in the future allow time course analysis of undisturbed mitotic exit with the required temporal resolution.
Having identified proteins that are dephosphorylated at the right time, the challenge became to pinpoint those proteins whose dephosphorylation controls cytokinesis. The observation of a dephosphorylation event does not by itself contain information about how important that event is. A traditional approach to address this is the use of phospho-mimetic variants of a protein to study the concomitant phenotype. This was not practical in our case for several reasons. Our phosphoproteome analysis often identified more than one phospho-peptide per protein, but is unlikely to have reached full coverage of each protein’'s phosphorylation status. Without that knowledge, it is hard to design a meaningful phospho-mimetic. In addition, cloning phospho-mimetic mutants is laborious, and in many cases, they present poor reflections of the phosphorylated state. Aspartate or glutamate residues are typically used to mimic phospho-serine or phospho-threonine, yet there remain considerable discrepancies in the size and charge of these side chains. We therefore employed a novel strategy to screen for phenotypes of the phosphorylated state, based on the idea that phosphorylation can be maintained by covalent fusion of a protein of interest to a cyclin (Lyons & Morgan, 2011). Although this approach does not confirm the identity of the phosphatase responsible for dephosphorylation, it did allow us to conduct a phenotypic screen on a medium-sized scale.
This screen identified several proteins that cause a cytokinetic defect following fusion to Clb2m, but not Clb2mΔCdk. An important control in this approach remains confirmation that the phenotype caused by Clb2m fusion is indeed due to phosphorylation of the target substrate. In all but one of the cases reported here (and others that we have studied in the laboratory; M. Godfrey, unpublished results), the defect caused by Clb2m fusion was at least in part alleviated by Cdk phospho-site mutations. This validates Clb2m fusion as a valuable approach for identifying phospho-regulated substrates. The screen identified Chs2, the so far only known gene whose dephosphorylation is required for cytokinesis (Chin et al, 2011). We also identified Inn1, whose dephosphorylation has been postulated to contribute to cytokinesis, but for which conventional phospho-mimetic mutations failed to reveal a clear morphological cytokinesis defect (Palani et al, 2012). We identified two additional targets whose Clb2m fusion phenotype depended on the Cdk phosphorylation sites to a varying degree. There are reasons why the effect of Clb2m fusion may only in part depend on the target’'s Cdk phosphorylation sites. Clb2m may phosphorylate unintended substrates in the vicinity of the target, or non-canonical phosphorylation sites on the substrate of interest might be targeted. With regard to the latter, we note that in a large-scale survey of Cdk targets, < 45% of all Cdk-dependent phosphorylation sites conformed to the minimal [S/T]-P Cdk consensus (Holt et al, 2009). Overall, we present a valuable discovery tool for proteins regulated by dephosphorylation that is transferable to other organisms including human cultured cells.
Our analysis of Inn1 phosphorylation, using the Clb2m fusion allele, led to a picture of its regulation that differs from what has been proposed (Palani et al, 2012). Inn1 dephosphorylation is required for its timely recruitment to the site of cytokinesis. There it appears to be required for Chs2 activation, which in turn supports furrow ingression during cytokinesis. Without Inn1 dephosphorylation, or without Chs2 (VerPlank & Li, 2005), the actomyosin ring is unable to constrict and instead disintegrates. Other Cdk targets that must be dephosphorylated may play a role in promoting additional cell physiological events relevant to cytokinesis. Ede1 is an endocytotic protein, suggesting that regulated membrane fusion events partake in cytokinesis. Aip1 in turn impinges on the actin depolymerization factor cofilin, a factor of importance to actomyosin ring constriction (Mendes Pinto et al, 2012). Together, our results exemplify that cytokinesis is controlled by the master cell cycle regulator Cdk kinase and its counteracting phosphatase Cdc14 and that cytokinesis involves the coordination of numerous cell biological processes through dephosphorylation of Aip1, Ede1 and Inn1 to successfully conclude the cell division cycle.
Materials and Methods
Yeast strains, plasmids and techniques
All strains used in this study are isogenic to W303 and are listed in the Supplementary Materials and Methods. Unless indicated otherwise, cells were grown in rich medium (YP) supplemented with 2% glucose as the carbon source. The cdc15-as1 strain was a gift from A. Amon (D'Aquino et al, 2005), and 5 μM 1NM-PP1 was used for kinase inhibition. Epitope tagging of endogenous genes was performed by polymerase chain reaction (PCR)-based gene targeting (Knop et al, 1999). The plasmid used to generate +CDC14 strains was a derivative of pRS416-Cdc14-GFP, a kind gift from H. Yu (Bembenek et al, 2005). To create the +CDC14-NLS strains, the SV40 virus large T antigen NLS (AGAPPKKKRKVAGI; Kalderon et al, 1984) was inserted before the stop codon. The CHS2V377I allele in plasmid pRS313 was a kind gift from K. Labib (Devrekanli et al, 2012). For cell wall removal, formaldehyde-fixed cells were treated with 40 μg/ml zymolyase T20 (MP Biomedicals) for 15 min. For conditional Clb2m fusions, a mutant lacking its destruction and KEN boxes (Wäsch & Cross, 2002) and its NLS (Eluère et al, 2007) was used. In addition to these alterations, the Clb2mΔCdk mutant lacks Cdk-binding capacity owing to three point mutations (Bailly et al, 2003). For Clb2 fusions, three repeats of the unstructured 10-mer GGSGTGGSGT were used as a spacer between the tagged protein and Clb2m. Strains harboring conditional Clb2m fusion cassettes were routinely propagated on medium lacking uracil to prevent spontaneous recombination between loxP sites. To obtain non-phosphorylatable mutants of Inn1, Ede1, Aip1 and Svr2, all [S/T]-P sites in these proteins were mutated to A-P, except for Inn1 where the previously described Inn1-5A mutant was used (Palani et al, 2012). The genes were cloned into integrative targeting plasmids that allow one-step gene replacement at the endogenous locus. As a control, similar targeting constructs were generated harboring the wild-type genes. Recombination resulting in Clb2m fusion was induced by β-estradiol-dependent activation of Cre recombinase fused to an estradiol-binding domain (Cre-EBD78; Lindstrom & Gottschling, 2009) by the addition of 1 μM β-estradiol to the growth medium. To deplete cells of Inn1 using the auxin-inducible degron, 500 μM indole-3-acetic acid (IAA) was added to the growth medium. Cell synchronization in G1 was performed by adding 0.8 μg/ml α-factor to cultures every 45 min for ∼1.5 population doubling times. For re-arresting cells in the following G1, 6 μg/ml α-factor was added to the culture once buds had formed. Arrest of cells in metaphase by depletion of Cdc20 under control of the MET3 promoter was performed as previously described (Uhlmann et al, 2000). In experiments using fluorescence-activated cell scanning (FACS), 10,000 cells were counted. Antibodies used for Western detection were: α-Cdc14 (rabbit serum raised against recombinant Cdc14; López-Avilés et al, 2009); α-Clb2 (Santa Cruz, sc9071); α-HA (clone 12CA5); α-Orc6 (clone SB49); and α-Sic1 (Santa Cruz, sc50441). α-Tub1, clone YOL1/34 (AbD Serotec), was used as a loading control in all experiments, except in Supplementary Fig S5 where α-Nuclear Pore Complex Proteins (Abcam, Mab414) was used. All error bars represent standard deviations, except where indicated. The following symbols are used to indicate significance: -P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Microscopy
Indirect immunofluorescence was performed on formaldehyde-fixed cells using the following antibodies: α-GFP, clone TP401 (Torrey Pines Biolabs); α-HA, clone 16B12 (Babco); α-Nop1, clone 28F2 (EnCor Biotechnology); and α-tubulin, clone YOL1/34 (AbD Serotec). Fluorescent images were acquired using an Axioplan 2 imaging microscope (Zeiss) equipped with a 100× (NA = 1.45) Plan-Neofluar objective and an ORCA-ER camera (Hamamatsu). To analyze the state of the plasma membrane in the bud neck region, we expressed eGFP-Spo2051–91, a gift from A. Neiman (Nakanishi et al, 2004), and captured 20 z-sections at 200-nm intervals. Time-lapse imaging was performed on cells grown in synthetic medium (YNB) and imaged on a DeltaVision Olympus IX70 inverted microscope with a 100× (NA = 1.40) PlanApo objective every 2 min. For analyzing the timing of cytokinetic events, the spindle pole body (SPB) was labeled using Spc42-YFP, and timings were expressed relative to the start of anaphase spindle elongation. The Kymograph plugin (http://www.embl.de/eamnet/html/body_kymograph.html) was used for creating side projections of Myo1-GFP rings. For statistical analysis of the spread of bud neck diameter, an R package calculating bootstrapped Kolmogorov–Smirnov P-values was used (http://sekhon.berkeley.edu/matching/ks.boot.html). For quantification of Cyk3-GFP fluorescence intensity at the bud neck, a threshold was applied on a fixed-size rectangular selection around the bud neck and signal intensities of particles were analyzed using ImageJ software. Mean-normalized values are used for representation in graphs.
Mass spectrometric analysis
Triplicate cultures were grown in synthetic medium supplemented with 2% raffinose and arrested in metaphase by Cdc20 depletion at an OD600 of 0.45. Cdc14 expression was induced by the addition of 2% galactose. 100 ml of the cultures was fixed at the indicated times by the addition of 6.7 ml 100% (w/v) TCA and left overnight on ice. Cells were collected by centrifugation, washed three times with ice-cold acetone and stored at −80°C. Cells were broken with glass beads in urea buffer (8 M urea, 100 mM ammonium bicarbonate and 5 mM EDTA), and an equivalent of 3 mg protein of the supernatant was reduced with 10 mM Tris(2-carboxyethyl)phosphine (TCEP) and alkylated with 10 mM iodoacetamide. The solution was diluted and digested overnight at 37°C with sequencing-grade modified trypsin (Promega) at a protein-to-enzyme ratio of 50:1. Phospho-peptides were isolated using TiO2 beads as previously described (Bodenmiller & Aebersold, 2010). Details describing the MS approach and phosphoproteome analysis are included in the Supplementary Materials and Methods.
Acknowledgments
We would like to thank A. Amon, F. Cross, K. Labib, A. Neiman, F. Van Leeuwen, M. Simon and H. Yu for strains and plasmids; G. Kelly for statistical analysis; M. Petronczki and M. Godfrey for critical reading of the manuscript; and the members of our laboratory for helpful discussions. This work was supported by a Netherlands Organization for Scientific Research (NWO) Rubicon fellowship and a Dutch Cancer Society long-term fellowship to TK.
Author contributions
TK and FU conceived the project. TK, MG and NS designed and performed the experiments. TK, MG, NS and FU analyzed the data. AM and RA performed the phosphoproteome analysis, which was analyzed by TK and AM. TK and FU wrote the paper with input from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Dataset S1
Review Process File
References
- Bailly E, Cabantous S, Sondaz D, Bernadac A, Simon M-N. Differential cellular localization among mitotic cyclins from Saccharomyces cerevisiae: a new role for the axial budding protein Bud3 in targeting Clb2 to the mother-bud neck. J Cell Sci. 2003;116:4119–4130. doi: 10.1242/jcs.00706. [DOI] [PubMed] [Google Scholar]
- Bembenek J, Kang J, Kurischko C, Li B, Raab JR, Belanger KD, Luca FC, Yu H. Crm1-mediated nuclear export of Cdc14 is required for the completion of cytokinesis in budding yeast. Cell Cycle. 2005;4:961–971. doi: 10.4161/cc.4.7.1798. [DOI] [PubMed] [Google Scholar]
- Blanco N, Reidy M, Arroyo J, Cabib E. Crosslinks in the cell wall of budding yeast control morphogenesis at the mother-bud neck. J Cell Sci. 2012;125:5781–5789. doi: 10.1242/jcs.110460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J, Cristea IM, Procko AL, Lubkov V, Chait BT, Snyder M, Cross FR. Global analysis of Cdc14 phosphatase reveals diverse roles in mitotic processes. J Biol Chem. 2011;286:5434–5445. doi: 10.1074/jbc.M110.205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Aebersold R. Quantitative analysis of protein phosphorylation on a system-wide scale by mass spectrometry-based proteomics. Meth Enzymol. 2010;470:317–334. doi: 10.1016/S0076-6879(10)70013-6. [DOI] [PubMed] [Google Scholar]
- Bouchoux C, Uhlmann F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell. 2011;147:803–814. doi: 10.1016/j.cell.2011.09.047. [DOI] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin Z-Y, Breitkreutz B-J, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras A-C, Nesvizhskii AI, Tyers M. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremmer SC, Hall H, Martinez JS, Eissler CL, Hinrichsen TH, Rossie S, Parker LL, Hall MC, Charbonneau H. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J Biol Chem. 2012;287:1662–1669. doi: 10.1074/jbc.M111.281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CF, Bennett AM, Ma WK, Hall MC, Yeong FM. Dependence of Chs2 ER export on dephosphorylation by cytoplasmic Cdc14 ensures that septum formation follows mitosis. Mol Biol Cell. 2011;23:45–58. doi: 10.1091/mbc.E11-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford DM, Wolfe BA, Roberts-Galbraith RH, McDonald WH, Yates JR, Gould KL. The Clp1/Cdc14 phosphatase contributes to the robustness of cytokinesis by association with anillin-related Mid1. J Cell Biol. 2008;181:79–88. doi: 10.1083/jcb.200709060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett M, Xiong Y, Boyne JR, Wright DJ, Munro E, Price C. IQGAP and mitotic exit network (MEN) proteins are required for cytokinesis and re-polarization of the actin cytoskeleton in the budding yeast, Saccharomyces cerevisiae. Eur J Cell Biol. 2006;85:1201–1215. doi: 10.1016/j.ejcb.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Culotti J, Hartwell LH. Genetic control of the cell division cycle in yeast. 3. Seven genes controlling nuclear division. Exp Cell Res. 1971;67:389–401. doi: 10.1016/0014-4827(71)90424-1. [DOI] [PubMed] [Google Scholar]
- D'Aquino KE, Monje-Casas F, Paulson J, Reiser V, Charles GM, Lai L, Shokat KM, Amon A. The protein kinase Kin4 inhibits exit from mitosis in response to spindle position defects. Mol Cell. 2005;19:223–234. doi: 10.1016/j.molcel.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Devrekanli A, Foltman M, Roncero C, Sanchez-Diaz A, Labib K. Inn1 and Cyk3 regulate chitin synthase during cytokinesis in budding yeasts. J Cell Sci. 2012;125:5453–5466. doi: 10.1242/jcs.109157. [DOI] [PubMed] [Google Scholar]
- Dischinger S, Krapp A, Xie L, Paulson JR, Simanis V. Chemical genetic analysis of the regulatory role of Cdc2p in the S. pombe septation initiation network. J Cell Sci. 2008;121:843–853. doi: 10.1242/jcs.021584. [DOI] [PubMed] [Google Scholar]
- Eluère R, Offner N, Varlet I, Motteux O, Signon L, Picard A, Bailly E, Simon M-N. Compartmentalization of the functions and regulation of the mitotic cyclin Clb2 in S. cerevisiae. J Cell Sci. 2007;120:702–711. doi: 10.1242/jcs.03380. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Gray CH, Good VM, Tonks NK, Barford D. The structure of the cell cycle protein Cdc14 reveals a proline-directed protein phosphatase. EMBO J. 2003;22:3524–3535. doi: 10.1093/emboj/cdg348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruneberg U, Glotzer M, Gartner A, Nigg EA. The CeCDC-14 phosphatase is required for cytokinesis in the Caenorhabditis elegans embryo. J Cell Biol. 2002;158:901–914. doi: 10.1083/jcb.200202054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009a;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009b;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa Lim H, Yeong FM, Surana U. Inactivation of mitotic kinase triggers translocation of MEN components to mother-daughter neck in yeast. Mol Biol Cell. 2003;14:4734–4743. doi: 10.1091/mbc.E03-04-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Tinker-Kulberg RL, Morgan DO. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2803–2817. doi: 10.1091/mbc.9.10.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Morgan DO. Cdc14 activates cdc15 to promote mitotic exit in budding yeast. Curr Biol. 2000;10:615–618. doi: 10.1016/s0960-9822(00)00491-7. [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Zimmerman ZA, Charbonneau H, Jackson PK. Disruption of centrosome structure, chromosome segregation, and cytokinesis by misexpression of human Cdc14A phosphatase. Mol Biol Cell. 2002;13:2289–2300. doi: 10.1091/mbc.01-11-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- Kao L, Wang Y-T, Chen Y-C, Tseng S-F, Jhang J-C, Chen Y-J, Teng S-C. Global analysis of Cdc14 dephosphorylation sites reveals essential regulatory role in mitosis and cytokinesis. Mol Cell Proteomics. 2014;13:594–605. doi: 10.1074/mcp.M113.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- König C, Maekawa H, Schiebel E. Mutual regulation of cyclin-dependent kinase and the mitotic exit network. J Cell Biol. 2010;188:351–368. doi: 10.1083/jcb.200911128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek WS, Bi E, Epp JA, Wang L, Ho J, Chant J. Cyk3, a novel SH3-domain protein, affects cytokinesis in yeast. Curr Biol. 2000;10:947–950. doi: 10.1016/s0960-9822(00)00626-6. [DOI] [PubMed] [Google Scholar]
- Lee SE, Frenz LM, Wells NJ, Johnson AL, Johnston LH. Order of function of the budding-yeast mitotic exit-network proteins Tem1, Cdc15, Mob1, Dbf2, and Cdc5. Curr Biol. 2001;11:784–788. doi: 10.1016/s0960-9822(01)00228-7. [DOI] [PubMed] [Google Scholar]
- Lindstrom DL, Gottschling DE. The mother enrichment program: a genetic system for facile replicative life span analysis in Saccharomyces cerevisiae. Genetics. 2009;183:413–422. doi: 10.1534/genetics.109.106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Avilés S, Kapuy O, Novák B, Uhlmann F. Irreversibility of mitotic exit is the consequence of systems-level feedback. Nature. 2009;459:592–595. doi: 10.1038/nature07984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca FC, Mody M, Kurischko C, Roof DM, Giddings TH, Winey M. Saccharomyces cerevisiae Mob1p is required for cytokinesis and mitotic exit. Mol Cell Biol. 2001;21:6972–6983. doi: 10.1128/MCB.21.20.6972-6983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons NA, Morgan DO. Cdk1-dependent destruction of eco1 prevents cohesion establishment after s phase. Mol Cell. 2011;42:378–389. doi: 10.1016/j.molcel.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Lukas C, Kaiser BK, Jackson PK, Bartek J, Lukas J. Deregulated human Cdc14A phosphatase disrupts centrosome separation and chromosome segregation. Nat Cell Biol. 2002;4:317–322. doi: 10.1038/ncb777. [DOI] [PubMed] [Google Scholar]
- Meitinger F, Petrova B, Lombardi IM, Bertazzi DT, Hub B, Zentgraf H, Pereira G. Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J Cell Sci. 2010;123:1851–1861. doi: 10.1242/jcs.063891. [DOI] [PubMed] [Google Scholar]
- Meitinger F, Palani S, Pereira G. The power of MEN in cytokinesis. Cell Cycle. 2012;11:219–228. doi: 10.4161/cc.11.2.18857. [DOI] [PubMed] [Google Scholar]
- Mendes Pinto I, Rubinstein B, Kucharavy A, Unruh JR, Li R. Actin depolymerization drives actomyosin ring contraction during budding yeast cytokinesis. Dev Cell. 2012;22:1247–1260. doi: 10.1016/j.devcel.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Ikeo S, Gannon J, Hunt T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009;28:2777–2785. doi: 10.1038/emboj.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, de los Santos P, Neiman AM. Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol Biol Cell. 2004;15:1802–1815. doi: 10.1091/mbc.E03-11-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiya F, Xie X, Lee KS, Inoue H, Miki T. Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J Biol Chem. 2005;280:36502–36509. doi: 10.1074/jbc.M508007200. [DOI] [PubMed] [Google Scholar]
- Nishihama R, Schreiter JH, Onishi M, Vallen EA, Hanna J, Moravcevic K, Lippincott MF, Han H, Lemmon MA, Pringle JR, Bi E. Role of Inn1 and its interactions with Hof1 and Cyk3 in promoting cleavage furrow and septum formation in S. cerevisiae. J Cell Biol. 2009;185:995–1012. doi: 10.1083/jcb.200903125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Meth. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell. 2006;125:85–98. doi: 10.1016/j.cell.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Palani S, Meitinger F, Boehm ME, Lehmann WD, Pereira G. Cdc14-dependent dephosphorylation of Inn1 contributes to Inn1-Cyk3 complex formation. J Cell Sci. 2012;125:3091–3096. doi: 10.1242/jcs.106021. [DOI] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell. 2008;135:879–893. doi: 10.1016/j.cell.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Diaz A, Nkosi PJ, Murray S, Labib K. The Mitotic Exit Network and Cdc14 phosphatase initiate cytokinesis by counteracting CDK phosphorylations and blocking polarised growth. EMBO J. 2012;31:3620–3634. doi: 10.1038/emboj.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Varma A, Drgon T, Bowers B, Cabib E. Septins, under Cla4p regulation, and the chitin ring are required for neck integrity in budding yeast. Mol Biol Cell. 2003;14:2128–2141. doi: 10.1091/mbc.E02-08-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Shevchenko A, Charbonneau H, Deshaies RJ. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207–220. doi: 10.1016/s0092-8674(02)00618-9. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Higuchi T, Katis VL, Uhlmann F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell. 2004;117:471–482. doi: 10.1016/s0092-8674(04)00415-5. [DOI] [PubMed] [Google Scholar]
- Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- VerPlank L, Li R. Cell cycle-regulated trafficking of Chs2 controls actomyosin ring stability during cytokinesis. Mol Biol Cell. 2005;16:2529–2543. doi: 10.1091/mbc.E04-12-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- Wäsch R, Cross FR. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418:556–562. doi: 10.1038/nature00856. [DOI] [PubMed] [Google Scholar]
- Wright DJ, Munro E, Corbett M, Bentley AJ, Fullwood NJ, Murray S, Price C. The Saccharomyces cerevisiae actin cytoskeletal component Bsp1p has an auxiliary role in actomyosin ring function and in the maintenance of bud-neck structure. Genetics. 2008;178:1903–1914. doi: 10.1534/genetics.107.082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Guo JY, Tang W, Yang C-S, Freel CD, Chen C, Nairn AC, Kornbluth S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol. 2009;11:644–651. doi: 10.1038/ncb1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Dataset S1
Review Process File