

Significance
We have shown previously that dipeptides can support efficient in vitro folding of the MHC class I molecules. Here, we describe the discovery of dipeptides that catalyze the dissociation of low- and high-affinity peptides from class I and their replacement for exogenous peptides of interest, on both recombinant and cell surface HLA-A*02:01, HLA-B*27:05, and H-2Kb molecules. Understanding peptide exchange on class I will help us understand peptide optimization in live cells. We demonstrate that the peptide-exchange technology can be used to produce epitope-specific MHC tetramers much faster and more easily than possible with techniques currently available and to enhance peptide loading onto live cells for cancer immunotherapy. Further applications include allotype specific peptide epitope elution from tumor cells.
Keywords: peptide exchange, MHC tetramers, tapasin, immunotherapy, dipeptides
Abstract
Peptide ligand selection by MHC class I molecules, which occurs by iterative optimization, is the centerpiece of immunodominance in antiviral and antitumor immune responses. For its understanding, the molecular mechanisms of peptide binding and dissociation by class I molecules must be elucidated. To this end, we have investigated dipeptides that bind to the F pocket of class I molecules. We find that they accelerate the dissociation of prebound peptides of both low and high affinity, suggesting a mechanism of action for the peptide-exchange chaperone tapasin. Peptide exchange on class I molecules also has practical uses in epitope discovery and T-cell monitoring.
MHC class I proteins, which are expressed on the surface of nucleated cells of higher vertebrates, present intracellular peptides to cytotoxic T lymphocytes (CTLs). To bind to class I, a peptide must fulfill allotype-specific length and sequence requirements: Hydrogen bond networks that hold the N and C termini usually restrict peptide length to 8–10 amino acids, and side chains at defined positions (anchor residues) must fit into specificity pockets at the bottom of the binding groove (1).
Cellular selection of antigenic peptides that conform to these requirements is an important step in immunodominance, the focusing of the adaptive immune system on a few epitopes in an immune response to viruses and cancer (2). This selection is thought to occur via iterative optimization, usually with the help of the peptide-exchange chaperone tapasin (3). Despite longstanding investigations, the molecular mechanism of peptide binding, dissociation, and tapasin-mediated exchange has remained elusive (4), in part because of the lack of structural information about peptide-free class I proteins.
To explore the molecular mechanism of peptide binding and dissociation, we have searched for small molecules that can bind to class I and modulate the binding of high-affinity peptides. We have demonstrated previously that dipeptides of appropriate sequence bind to class I molecules, presumably to the F pocket, and support their folding (5), and we now show that they also dramatically accelerate the dissociation of prebound peptides and their exchange for exogenous peptides in a function analogous to that of tapasin.
Results
On MHC class II molecules, short dipeptides that bind into the P1 pocket catalyze the exchange of prebound peptides for exogenous ones (6, 7). Because we found previously that certain dipeptides bind to and stabilize the MHC class I proteins H-2Kb (Kb) and HLA-A*02:01 (A2) in the absence of high-affinity full-length peptides (5), we asked whether such dipeptides also promote peptide exchange on MHC class I. We expressed recombinant A2 in Escherichia coli, folded it in vitro with the peptide NLVPMVATA, removed the free peptide by gel filtration, and added NLVPKFITCVATV (in which the lysine side chain is labeled with FITC in a position that does not interfere with binding to A2). The binding of NLVPKFITCVATV to A2, which we followed by real-time fluorescence anisotropy, was very slow, probably because of the slow dissociation of the prebound NLVPMVATA. Strikingly, when 10 mM of the dipeptide glycyl-methionine (GM) was added to the reaction, the binding of NLVPKFITCVATV proceeded about 25-fold faster (Fig. 1, Left and Fig. S1E). A similar acceleration was observed with another peptide, ILKEKFITCVHGV (Fig. S1D). GM also accelerated the dissociation of prebound fluorescent peptide, demonstrating that the incoming peptide is replacing prebound peptide and is not binding to empty unstructured molecules (Fig. 1, Center). To see whether the exchange reaction was complete, we performed thermal denaturation measured by tryptophan fluorescence (TDTF) assays (8) in which the minimum of the curve corresponds to the midpoint of thermal denaturation (Tm) (Fig. 1, Right). After a 3-h incubation with GM and NLVPMVATV, the complex of A2 with prebound NLVPMVATA (Tm, 45 °C) was no longer detected but was replaced by the A2–NLVPMVATV complex (Tm, 56 °C). This result shows that the prebound NLVPMVATA had been exchanged for the externally added NLVPMVATV.
Fig. 1.

The dipeptide GM induces peptide exchange on A2. (Left) A2–NLVPMVATA monomers were incubated with or without 10 mM of the dipeptide GM, and the association rate of added NLVPKFITCVATV was measured using fluorescence anisotropy. Association rate constants (kon ± SEM) are 0.15 ± 0.02 × 103 M−1⋅s−1 without GM and 3.93 ± 0.86 × 103 M−1⋅s−1 with GM. Another exchange reaction is shown in Fig. S1D. (Center) An analogous experiment the measuring dissociation rate of NLVPKTAMRAVATA peptide in the presence or absence of GM, with 10 µM NLVPMVATV peptide. The dissociation rate constant (koff ± SEM) is 2.16 ± 0.03 × 10−6⋅min−1 with GM. (Right) Exchange of A2-bound NLVPMVATA peptide for NLVPMVATV. A2–NLVPMVATA was incubated for 180 min with 10 µM NLVPMVATV peptide in the presence or absence of 10 mM GM. Thermal stability of the resulting A2–peptide complexes was measured by TDTF; the minimum of the each curve indicates the melting temperature (Tm) of the complex. The TDTF curves of A2 folded with the respective peptides are shown in Fig. S1 A and B.
Because we estimated from systematic structural variations in the experiments, and also from in silico docking studies, that the C-terminal side chain of the dipeptide binds into the F pocket of class I (see fig. 4E in ref. 5), we compared several dipeptides that have hydrophobic second residues with respect to their ability to release prebound NLVPMVATA or another peptide, ILKEPVHGV, and replace it with incoming NLVPKFITCVATV (Fig. 2). We found that for both leaving peptides, GM performed best, followed by glycyl-leucine (GL). In contrast, glycyl-alanine (GA), with the shortest side chain, did not support peptide exchange significantly. GM also catalyzed the displacement of the NLVPMVATV peptide, which we consider a high-affinity peptide for A2 because it is listed as a positive-high affinity peptide by most quantitative measurements listed on the Immune Epitope Database (Fig. S2A) (9, 10). We conclude that dipeptides with a hydrophobic second residue can catalyze peptide exchange on A2, even for high-affinity prebound peptides, and that dipeptides with longer hydrophobic side chains have a stronger effect.
Fig. 2.

Dipeptides differ in peptide-exchange dependence on their second hydrophobic residue. A2/β2m/peptide complexes were prepared and incubated with 100 nM exogenous NLVPKFITCVATV plus 10 mM dipeptides as indicated. Left panels show representative experiments. Right panels show kon values (average ± SEM of three or more independent experiments). (A) A2–NLVPMVATA. (B) A2–ILKEPVHGV. Numerical values are shown in Table S1.
When we tested GM for peptide exchange on Kb, the effect was only minor. Therefore we tested other amino acids in the second position of the dipeptide and found that the nonproteinogenic amino acid dipeptides glycyl-cyclohexylalanine (GCha) and glycyl-homoleucine (GHle) were both effective (Fig. 3 A and B). When the binding affinity of the prebound peptide FAPGNYPAL to Kb was lowered by replacing the C-terminal anchor residue with alanine (FAPGNYPAA), a strong exchange function still could be seen for GCha and GHle, and now for GL, GM, GV, and glycyl-phenylalanine (GF) also, but GA was still ineffective (Fig. 3C). Likewise, the exchange of the SIINFEKL peptide [which forms a high-affinity Kb complex (8)] by GL or GM (as measured by binding of exogenous SIINFEKTAMRAL) was very weak, but once the C-terminal leucine was replaced by glycine (SIINFEKG) or alanine (SIINFEKA), dipeptide-mediated exchange proceeded readily (Fig. 4A). In contrast, GCha was able to catalyze the exchange of unmodified prebound SIINFEKL (Fig. S2B). We conclude that peptide-exchange reactions on Kb and A2 require different dipeptides and that the strength of C-terminal binding of the prebound peptide codetermines the exchange efficiency of individual dipeptides. Exchange of prebound high-affinity peptides is possible, but optimal dipeptides may be required.
Fig. 3.

GCha and GHle effectively catalyze peptide exchange on Kb. Kb–FAPGNYPAL and Kb–FAPGNYPAA were prepared by folding Kb/hβ2m with the indicated peptides. To these complexes, 10 mM dipeptides were added as indicated, and exchange was monitored by measuring the association rate (kon) of SIINFEKTAMRAL (100 nM) peptide in a fluorescence anisotropy assay. (A) A representative experiment shows exchange of the Kb-bound high-affinity peptide FAPGNYPAL for SIINFEKTAMRAL in the presence and absence of indicated dipeptides. (B) Structure of dipeptides with nonnatural amino acids GCha and GHle. (C, Left) Representative experiment showing exchange of FAPGNYPAA bound to Kb in the presence of indicated dipeptides and in the absence of any dipeptide. (Right) kon values (average ± SEM of three or more independent experiments). All association rates are shown in Table S2.
Fig. 4.

Dipeptides target the C terminus of the peptide to catalyze peptide exchange. (A) Exchange of Kb-bound high-affinity peptide SIINFEKL and its C-terminally modified variants SIINFEKG and SIINFEKA were measured in the presence of 10 mM GL or GM or without any dipeptide by measuring the association rate (kon) of SIINFEKTAMRAL in a fluorescence anisotropy assay. (B) Analogous experiment comparing the exchange of Kb–SIINFEKL-Cdel, Kb–SIINFEKL-Ndel, and Kb–SIINFEKL for SIINFEKTAMRAL. All numerical values are shown in Table S2. (C) Mechanistic model of dipeptide-facilitated peptide exchange (see text).
To compare the importance of the N and C termini of the prebound peptide in the exchange reaction directly, we used SIINFEKL-derived peptides without the C-terminal carboxyl group (replacing the leucine with isopentylamine, SIINFEKL-Cdel) or without the N-terminal amino group (replacing the serine with 3-hydroxypropionic acid, SIINFEKL-Ndel) as leaving peptides. Both modifications were good leaving peptides, and for both the enhancement of SIINFEKTAMRAL binding by GL and GM was significant (Fig. 4B). Thus, decreasing the binding affinity of a peptide to class I seems to be a universal way to make it a better leaving peptide in the exchange reaction.
Our previously published experiments suggest that the GX dipeptides bind to the F pocket of class I: To be efficient, they must have a free C terminus and the L configuration, and they can stabilize empty folded class I molecules; that stabilization requires engagement of the F pocket (5). We found, in addition, that the C-terminal amino acid of the dipeptide must match the F-pocket specificity of the respective allotype, with B*27:05 preferring GR over GL (Fig. S3).
In the most parsimonious model for dipeptide mediated peptide exchange (Fig. 4C, Right), a peptide-bound class I molecule is in equilibrium (step 1) with a state in which the C terminus, but not the N terminus, of the peptide is temporarily unbound. To this state, a dipeptide can bind and occupy the F pocket such that the long peptide cannot rebind (step 2). Dissociation of the N terminus (step 3) leads to loss of the leaving peptide to the environment, and binding of an exogenous peptide (shown in green in Fig. 4C, Right) becomes possible (step 4). Upon removal of the dipeptide (step 5), tight binding of the new peptide ensues.
We next tested dipeptide-mediated peptide exchange on live cells. To obtain a uniform population of Kb–FAPGNYPAL complexes, we accumulated peptide-receptive Kb molecules on the surface of TAP-deficient RMA-S cells by overnight incubation at 25 °C (11) and then added FAPGNYPAL to the medium to allow it to bind to surface Kb. We washed the cells and incubated them with SIINFEKFITCL and dipeptides, and we measured bound SIINFEKFITCL by flow cytometry (Fig. 5A). The addition of GCha resulted in a fourfold increase in SIINFEKFITCL binding, whereas GA was ineffective. Detection with the monoclonal antibody 25-D1.16 (specific for Kb–SIINFEKL) showed that, in the presence of GCha, SIINFEKL indeed was binding to Kb (Fig. 5B). On wild-type lymphocytes, GCha also facilitated the exchange of natural endogenous peptides for SIINFEKFITCL (RMA cells, Fig. 5C), whereas GM mediated exchange on A2 (T1 cells, Fig. 5D). The newly formed Kb–SIINFEKL complexes elicited a response from the B3Z hybridoma (Fig. 5E) (12), demonstrating that they were functionally indistinguishable from endogenously formed complexes. When RMA cells were incubated with SIINFEKL, GCha addition caused a twofold increase in overall B3Z activation (Fig. 5F).
Fig. 5.

Dipeptides catalyze peptide exchange on the cell-surface class I molecules. (A) Exchange of Kb-bound FAPGNYPAL peptide on the surface of RMA-S cells. RMA-S cells were incubated overnight at 25 °C with 10 µM FAPGNYPAL or SIINFEKFITCL (as a positive control, dashed black line). Then excess peptide was washed off, and 1 µM SIINFEKFITCL was added in the presence of 10 mM GA or GCha or without dipeptide. Binding of SIINFEKFITCL was detected by flow cytometry; the shadow curve is the cell autofluorescence background. (B) An analogous experiment; instead of SIINFEKFITCL, SIINFEKL (10 µM) was used. Binding of SIINFEKL was detected by flow cytometry using the Kb-SIINFEKL–specific monoclonal antibody 25D1.16; the shadow curve is the control (no primary antibody). (C) Exchange of Kb-bound endogenous peptides on the surface of RMA cells. Cells were incubated with 5 µM SIINFEKFITCL in the presence of 10 mM GCha or without any dipeptide. Binding of SIINFEKFITCL was detected by flow cytometry; the shadow curve is the control without SIINFEKFITCL. (D) Exchange of A2-bound endogenous peptide on the surface of T1 cells. Cells were incubated with the A2-specific peptide NLVPKFITCVATV (5 µM) in the absence or presence of 10 mM GA or GM. Binding of NLVPKFITCVATV was detected by flow cytometry; the shadow curve is the control without NLVPKFITCVATV. Data in A–D are representative of at least three independent experiments. (E) Activation of the B3Z hybridoma after cell-surface exchange of Kb–FAPGNYPAL for SIINFEKL on RMA-S cells (with or without the indicated dipeptides). After the exchange reaction, RMA-S cells were incubated with B3Z cells overnight at 37 °C. On the next day, activation of B3Z was detected by a β-galactosidase assay. Data (average of three independent experiments ± SD) were normalized to the positive control (SIINFEKL added to RMA-S cells after overnight incubation at 25 °C). (F) Activation of B3Z hybridoma after peptide exchange on the surface of RMA cells. Data shown are the average of three independent experiments ± SD.
Finally, we tested peptide exchange as a method for generating MHC multimers (13). We used monomeric recombinant biotinylated A2 molecules folded with ILKEPVHGV, and we performed an exchange reaction with GM and YVLDHLIVV peptide (Peptide exchange using fluorescence anisotropy is shown in Fig. S4). MS analysis showed that the resulting complexes contained 94.5% YVLDHLIVV (Fig. 6A). [In other exchange reactions with monomers, we found 91.9–99.1% exchange efficiency (Table 1)]. We then assembled the exchanged monomers into tetramers by incubation with streptavidin-phycoerythrin and used those tetramers to stain T cells from donors. Intriguingly, tetramers made from peptide-exchanged monomers performed indistinguishably from the positive controls, i.e., tetramers made from monomers folded with the same peptide (Fig. 6B). We conclude that dipeptide-mediated peptide exchange is a fast and easy method to generate MHC multimers of a desired specificity.
Fig. 6.
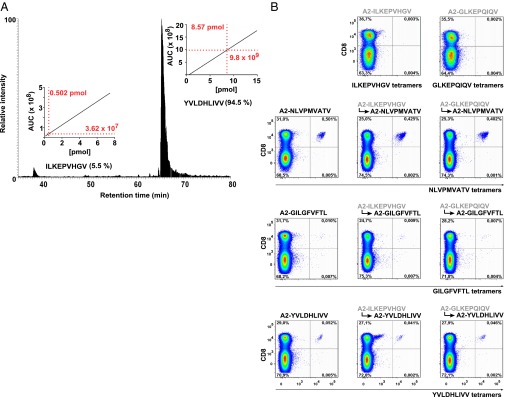
Quantification of peptide exchange and production of viral epitope-specific tetramers. (A) Relative quantification of peptide abundance after exchange reaction of A2–ILKEPVHGV monomer for YVLDHLIVV in the presence of 10 mM GM. Peptides were eluted from monomers by acid treatment and analyzed by LC-MS. (Insets) The peptide concentration was calculated by peak integration (area under the curve, AUC) using predetermined standard curves created for each individual peptide. (B) Dot plots of MHC tetramer staining from a representative donor (staining data from a different donor are shown in Fig. S5). Tetramers were prepared after exchanging the A2-monomers (indicated in Table 1) in the presence of 10 mM GM for viral epitopes.
Table 1.
Exchange efficiency of A2 monomers in the presence of 10 mM GM
| A2 monomers folded with | Peptide exchanged for | Efficiency of exchange, % |
| GLKEPQIQV | YVLDHLIVV | 91.9 |
| GLKEPQIQV | GILGFVFTL | 93.6 |
| GLKEPQIQV | NLVPMVATV | 93.2 |
| ILKEPVHGV | YVLDHLIVV | 94.5 |
| ILKEPVHGV | GILGFVFTL | 99.1 |
| ILKEPVHGV | NLVPMVATV | 96.8 |
Discussion
Procedures for the cleavage of chemically modified peptides on MHC class I molecules and their subsequent replacement by an exogenous peptide already exist, but they have not contributed to the mechanistic understanding of peptide binding and exchange (14, 15). Here, we describe a novel, simple, and nondestructive protocol for peptide exchange that supports a mechanistic suggestion: By binding into the F pocket of a partially dissociated class I–peptide complex, they may prevent the rebinding of the C terminus of the prebound peptide (Fig. 4C). Because we have not formally shown the binding of the dipeptides into the F pocket, the possibility remains that the dipeptides bind elsewhere; only crystal structures will provide this clarification. Still, our findings support the mechanistic hypothesis that the ends of a peptide can dissociate from their bound states individually (16). Even though “breathing” (i.e., short-lived detachment) of the N terminus of the peptide, which is not shown in Fig. 4C, is also possible, we expect it to be of no consequence for dipeptide-mediated exchange, if, as we assume, the dipeptides bind to the F pocket of class I. The low affinity of the dipeptides to class I, in the millimolar range, facilitates their removal after the exchange reaction, along with the trapping of the incoming peptide (Fig. 4C, step 5).
Optimization of the class I peptide load in live cells over time has been observed previously (17), but its molecular mechanism is elusive, despite much experimentation (18). In live cells, the peptide-exchange chaperone tapasin accelerates the dissociation of prebound peptide (19–21). Because tapasin appears to bind to class I close to the F pocket (22), it might interfere with the binding of the C terminus in a manner analogous to the dipeptides, perhaps by inserting an amino acid side chain into the F pocket. We now have demonstrated that such an exchange mechanism is mechanistically feasible. In vitro competition assays with tapasin and dipeptides may resolve this question.
Peptide exchange on MHC class II molecules is triggered by HLA-DM (in humans) and H-2M (in mice) through the displacement of a peptide anchor side chain from the P1 pocket (23). The action of DM can be mimicked by small molecules, such as dipeptides, phenol, or adamantane, that occupy the P1 pocket (6, 7, 24). Because small-molecule peptide-exchange mechanisms on class I and class II are similar (Fig. 4C), we propose that tapasin and DM might use analogous mechanisms for peptide exchange on class I and class II, respectively (25).
The controlled exchange of peptides on MHC class I molecules, as described here, can be used in the production of MHC multimers for immunodiagnostics, as we have demonstrated (Fig. 6) (26). Exchange of peptides at the cell surface (Fig. 5) can be used to enhance loading of exogenous peptides on patient dendritic cells (DCs) for DC vaccination or in vitro restimulation of patient CTLs in cancer immunotherapy (27). Because the high-affinity incoming peptide is supplied in abundance during the reaction (Materials and Methods), and the dipeptide is washed out rapidly because of its very low binding affinity, the chance of a full-length or truncated endogenous peptide binding to class I and causing an aberrant immune reaction during therapy is minimal. The removal of prebound peptides from the cell surface in an allotype-specific fashion, under physiological conditions, also might be used in the future to elute tumor-specific peptide antigens for immunotherapy. This approach is generally feasible, because we have demonstrated that both low-affinity and high-affinity (Figs. 2B, 3A, 5, and 6B) peptides may be exchanged with dipeptides. Still, it is conceivable that some prebound peptides with very tight binding to the F pocket may resist exchange on short time scales.
Materials and Methods
Expression and Purification of MHC Class I Heavy Chains and Human β2 Microglobulin.
The heavy chain of A2 and H-2Kb and human β2 microglobulin (hβ2m) were expressed in E. coli and purified from inclusion bodies (as denatured proteins) as described previously (8, 28). Purified proteins were stored at −80 °C until use.
In Vitro Folding of MHC Class I Heavy Chain and hβ2m.
Heavy chain and β2m were folded in the presence of peptides as described previously (5, 8). Briefly, 100–200 µg of denatured heavy chain and an equal amount of β2m were diluted in 2 mL folding buffer [100 mM Tris·Cl (pH 8), 0.5 M arginine, 2 mM EDTA, 0.5 mM oxidized glutathione, 5 mM reduced glutathione] along with 10 µM peptide and were incubated at 4 °C for 2–7 d. After folding reaction, heavy chain–β2m–peptide complexes were harvested by ultracentrifugation at 100,000 × g for 20 min. The supernatant was collected, and the protein concentration was measured by the Bradford assay. To obtain monomers for peptide-exchange experiments, excess peptide was removed using Vivaspin 2 (Sartorius) filter columns.
Peptide Exchange Measured by Fluorescence Anisotropy Assay.
Fluorescence anisotropy measurements were performed as described previously (5). For peptide exchange on A2, 1 µM A2 monomers (A2–hβ2m–peptide complex, prepared by removing excess peptides after in vitro folding) were incubated with 10 mM dipeptide (or no dipeptide) for 10 min in a 96-well plate (Greiner Bio-One); to this preparation 100 nM of fluorophore-labeled peptide (NLVPKFITCVATV, FITC λex = 494 nm, λem = 517 nm or ILKEKTAMRAVHGV, TAMRA λex = 536 nm, λem = 580 nm) was added, and binding kinetics were measured in real time by fluorescence anisotropy for 180 min using a Tecan Infinite M1000 PRO plate reader (Tecan). Association rates (kon) were calculated using GraphPad Prism (GraphPad). Similarly, for Kb, peptide exchange was monitored by measuring the binding kinetics of 100 nM SIINFEKTAMRAL for 10 min after Kb monomers (1 µM) were incubated for 10 min with 10 mM dipeptides (or no dipeptide). All reactions were performed in 100 mM Hepes buffer (pH 7), at room temperature (22–25 °C).
For peptide-dissociation experiments, A2–NLVPKTAMRAVATA monomers were incubated in a 96-well plate with 10 mM GM or without dipeptide for 10 min. Peptide dissociation was initiated by adding 10 µM NLVPMVATV peptide, and dissociation kinetics was measured for 180 min. Data were fitted using GraphPad Prism.
TDTF Measurements.
The thermal stability of the class I–peptide complex (A2 with NLVPMVATA, NLVPMVATV, and ILKEPVHGV) was measured by TDTF experiments as explained in ref. 8. TDTF measurements are represented as local regression (LOWESS) fits (GraphPad Prism) of first derivatives derived from plots of tryptophan fluorescence vs. temperature. The minimum of these curves corresponds to the melting temperature, Tm, of the complex (29).
For peptide exchange measured by TDTF, 2.5 µM A2–NLVPMVATA monomers were incubated with 20 µM NLVPMVATV peptide in the presence or absence of 10 mM GM for 3 h. After 3 h, free peptides were removed from each reaction by Vivaspin 2 filter columns (10-kDa cutoff), and the thermal stability of resulting complexes was measured by TDTF.
Cell-Surface Peptide Exchange.
For exchange of Kb-bound peptide, RMA-S cells were cultured at 25 °C overnight in CO2-independent medium to accumulate peptide-receptive class I molecules on the cell surface. On the next day, 0.5–0.8 × 105 cells per well were allowed to bind 10 µM FAPGNYPAL or 1 µM SIINFEKFITCL (positive control) in the presence of 10 µg/mL Brefeldin A (BFA) (Invitrogen) to block surface delivery of newly synthesized class I for 60 min at 37 °C. After incubation, excess peptide was removed by washing (2×, PBS at 200 × g 4 °C). The FAPGNYPAL-bound cells then were incubated with 10 mM dipeptide (or without any dipeptide) and 1 µM SIINFEKFITCL for 60 min at 37 °C in 25 mM Hepes-RPMI medium (pH 7.0) with 10 µg/mL BFA. Cells then were washed twice by 0.02% NaN3 in PBS, and the binding of SIINFEKFITCL was measured using a flow cytometer (CyFlow Space). Kb-specific binding of SIINFEKL was confirmed in a similar experiment in which unlabeled SIINFEKL was used instead of SIINFEKFITCL and binding was detected by the Kb–SIINFEKL-specific monoclonal antibody 25D1.16. Data were analyzed using FlowJo (Tree Star).
For cell-surface exchange of endogenous peptide on Kb, RMA cells were cultured at 37 °C and incubated for 60 min in 25 mM Hepes-RPMI medium with 10 µg/mL BFA and with 10 mM GCha or without any dipeptide in the presence of 5 µM SIINFEKFITCL. Cells then were washed two times, and SIINFEKFITCL binding was detected by flow cytometry.
For cell-surface exchange of endogenous peptide on A2 molecules, T1 cells cultured at 37 °C were incubated (in CO2-independent medium with 10 µg/mL BFA) with 10 mM GM or GA or without dipeptide in the presence of 5 µM NLVPKFITCVATV for 180 min. Cells then were washed, and binding of NLVPKFITCVATV was detected by flow cytometry.
T-Cell Activation Assay.
After cell-surface exchange on RMA-S or RMA cells by SIINFEKL in the presence of GCha or GA, these cells (150,000 per well) were incubated in 96-well plates with the B3Z hybridoma (100,000 cells per well) for 24 h at 37 °C (RPMI + 10% FBS, 5% CO2). Because B3Z cells produce β-galactosidase upon activation (30), the cells then were washed twice with PBS, treated with lysis buffer containing 0.15 mM CPRG (chlorophenol red β-d-galactopyranoside; Sigma) and 0.5% Nonidet P-40, and incubated for 20–24 h at room temperature. Activity was measured as absorption at 570 nm using a Tecan Infinite M1000 PRO plate reader. B3Z cells incubated with untreated RMA-S/RMA cells served as background control.
Generation of MHC Multimers and Staining of Peripheral Blood Mononuclear Cells.
MHC multimers were produced as described previously (13). Briefly, MHC light and modified heavy chain [in which the transmembrane domain was replaced by a biotin ligase (BirA) enzymatic biotinylation sequence] were expressed separately in E. coli BL21(DE3)pLysS (Promega). Bacteria were lysed by sonification/lysozyme treatment, and released inclusion bodies were purified. Refolding was performed by limited dilution of MHC heavy and light chain together with the binding peptide. The refolded complexes were purified by gel filtration on an Äkta fast protein liquid chromatography system (Superdex G-75; GE Healthcare), biotinylated by BirA, and the product was repurified by gel filtration (Superdex G-75). MHC multimers were assembled from biotinylated monomers, directly or after exchange reactions, by the addition of streptavidin-phycoerythrin (Life Technologies) to the complexes at a molecular ratio of 4:1. For T-cell staining, MHC multimers were diluted to 2.5 µg/mL in staining buffer containing 50% (vol/vol) FCS and 2 mM EDTA in PBS. Multimer staining was performed with peripheral blood mononuclear cells from healthy A2-positive donors previously screened and selected to show positive ex vivo multimer staining with Influenza M158–66(GILGFVFTL), CMV pp65495–503(NLVPMVATV), and EBV BRLF1109–117(YVLDHLIVV). Cells were stained additionally at 4 °C for the surface marker CD8 (PerCP; BioLegend) and for the viability marker Aqua Live/Dead (Life Technologies). All washing steps were performed with FACS buffer (PBS with 2% FCS, 2 mM EDTA). Finally, samples were acquired on a BD FACS Canto II Analyzer (BD Biosciences). Analysis and assessment of the percentage of multimer-positive single viable lymphocytes was performed with FlowJo (Tree Star).
Relative Quantification of Exchange Efficiency by MS.
Before analysis, exchanged monomers were purified twice by gel filtration using PD SpinTrap G25 columns (GE Healthcare) according to the manufacturer’s instructions to remove unbound excess peptide. MHC-bound peptides then were eluted by the addition of 10 µL of 10% trifluoroacetic acid and sonification in a water bath at 45 °C for 5 min. Eluted peptides were separated from empty monomers by gel filtration (SpinTrap G25 columns; GE Healthcare), and the volume was adjusted to 100 µL by vacuum centrifugation. For the analysis, 5 µL of each sample was injected into an Ultimate 3000 RSLC Nano UHPLC system (Dionex) coupled to the nanoelectron spray ion source of an LTQ Orbitrap XL (Thermo Fisher Scientific) mass spectrometer. Samples first were loaded on a 2-cm PepMAP100 C18 Nano-Trap column (Dionex) within 6 min at a flow rate of 4 µL/min and 5% acetonitrile. Afterward, peptides were separated on a 25-cm PepMap C18 column (2-µm particle size) running in a column oven at 50 °C with a gradient of 5–65% acetonitrile within 90 min and a flow rate of 175 nL/min.
MS identification and relative quantitation were performed in direct data-dependent acquisition mode using a top-five method in which the five most intense ions were selected for fragmentation during each scan cycle. Survey scans were performed with 60-k resolution and a scan range between 400 and 650 m/z. Fragmentation of peptides was achieved by collision-induced dissociation (collision energy 35%, activation time 30 ms, isolation width 1.3 m/z) with fragment ions analyzed in the linear ion trap.
Peptides’ identity and relative quantity were assessed by Xcalibur Qual Browser v. 2.07 (Thermo Fisher). Peptide abundance was determined by peak integration of precursor masses over time with a mass window of 2 ppm. Standard curves were created with respective synthetic peptides to calculate their concentration within the sample. For methionine-containing NLVPMVATV, the native and the oxidized species (NLVPM(ox)VATV) were evaluated separately.
Supplementary Material
Acknowledgments
We thank Ursula Wellbrock for excellent technical assistance and for drawing Fig. 4C; Hubert Kalbacher for kindly donating reagent; and Martin Zacharias, Katja Ostermeir, and Florian Kandzia for discussions. The work was supported by Deutsche Forschungsgemeinschaft Grant SP 583/6-1 (to S. Springer).
Footnotes
Conflict of interest statement: S. Springer and S.K.S. hold the patent for the dipeptide-mediated peptide-exchange technology.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1418690112/-/DCSupplemental.
References
- 1.Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell JW. Confronting complexity: Real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25(4):533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Elliott T, Williams A. The optimization of peptide cargo bound to MHC class I molecules by the peptide-loading complex. Immunol Rev. 2005;207:89–99. doi: 10.1111/j.0105-2896.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- 4.Hulpke S, Tampé R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem Sci. 2013;38(8):412–420. doi: 10.1016/j.tibs.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Saini SK, et al. Dipeptides promote folding and peptide binding of MHC class I molecules. Proc Natl Acad Sci USA. 2013;110(38):15383–15388. doi: 10.1073/pnas.1308672110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S, et al. Anchor side chains of short peptide fragments trigger ligand-exchange of class II MHC molecules. PLoS ONE. 2008;3(3):e1814. doi: 10.1371/journal.pone.0001814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou CL, et al. Short peptide sequences mimic HLA-DM functions. Mol Immunol. 2008;45(7):1935–1943. doi: 10.1016/j.molimm.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 8.Saini SK, et al. Not all empty MHC class I molecules are molten globules: Tryptophan fluorescence reveals a two-step mechanism of thermal denaturation. Mol Immunol. 2013;54(3-4):386–396. doi: 10.1016/j.molimm.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Vita R, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38(Database issue):D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sylvester-Hvid C, et al. Establishment of a quantitative ELISA capable of determining peptide - MHC class I interaction. Tissue Antigens. 2002;59(4):251–258. doi: 10.1034/j.1399-0039.2002.590402.x. [DOI] [PubMed] [Google Scholar]
- 11.Ljunggren HG, et al. Empty MHC class I molecules come out in the cold. Nature. 1990;346(6283):476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 12.Sanderson S, Shastri N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int Immunol. 1994;6(3):369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- 13.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274(5284):94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 14.Rodenko B, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1(3):1120–1132. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- 15.Rodenko B, et al. Class I major histocompatibility complexes loaded by a periodate trigger. J Am Chem Soc. 2009;131(34):12305–12313. doi: 10.1021/ja9037565. [DOI] [PubMed] [Google Scholar]
- 16.Wright CA, Kozik P, Zacharias M, Springer S. Tapasin and other chaperones: Models of the MHC class I loading complex. Biol Chem. 2004;385(9):763–778. doi: 10.1515/BC.2004.100. [DOI] [PubMed] [Google Scholar]
- 17.Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity. 2002;16(4):509–520. doi: 10.1016/s1074-7613(02)00304-7. [DOI] [PubMed] [Google Scholar]
- 18.Van Hateren A, et al. The cell biology of major histocompatibility complex class I assembly: Towards a molecular understanding. Tissue Antigens. 2010;76(4):259–275. doi: 10.1111/j.1399-0039.2010.01550.x. [DOI] [PubMed] [Google Scholar]
- 19.Wearsch PA, Cresswell P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol. 2007;8(8):873–881. doi: 10.1038/ni1485. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. 2007;26(6):1681–1690. doi: 10.1038/sj.emboj.7601624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Praveen PV, Yaneva R, Kalbacher H, Springer S. Tapasin edits peptides on MHC class I molecules by accelerating peptide exchange. Eur J Immunol. 2010;40(1):214–224. doi: 10.1002/eji.200939342. [DOI] [PubMed] [Google Scholar]
- 22.Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity. 2009;30(1):21–32. doi: 10.1016/j.immuni.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pos W, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell. 2012;151(7):1557–1568. doi: 10.1016/j.cell.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson MJ, et al. Small molecules that enhance the catalytic efficiency of HLA-DM. J Immunol. 2006;176(7):4208–4220. doi: 10.4049/jimmunol.176.7.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaneva R, Schneeweiss C, Zacharias M, Springer S. Peptide binding to MHC class I and II proteins: New avenues from new methods. Mol Immunol. 2010;47(4):649–657. doi: 10.1016/j.molimm.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Newell EW, Davis MM. Beyond model antigens: High-dimensional methods for the analysis of antigen-specific T cells. Nat Biotechnol. 2014;32(2):149–157. doi: 10.1038/nbt.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: Refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992;89(8):3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eftink MR. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys J. 1994;66(2 Pt 1):482–501. doi: 10.1016/s0006-3495(94)80799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci USA. 1992;89(13):6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.