

Significance
Pneumonia caused by bacterial coinfection with influenza virus is the leading cause of mortality in influenza pandemics. TGF-β is known to be activated by influenza virus. In this study we demonstrated that cellular adhesins for bacteria, such as fibronectin and α5 integrin, are up-regulated in influenza viral infection. Inhibition of TGF-β impeded the up-regulation of these cellular adhesins and also influenza viral-enhanced bacterial adherence. In addition, we found that influenza viral-promoted bacterial adherence was dependent on bacterial fibronectin-binding protein. The results indicate that up-regulated expression of cellular adhesins by TGF-β, which is activated in influenza viral infection, increases host susceptibility to bacterial coinfection and suggest that TGF-β and host adhesion molecules are potential pharmaceutical targets for prevention of coinfection.
Keywords: influenza A virus, TGF-beta, coinfection, fibronectin binding protein, bacterial adherence
Abstract
Influenza infection predisposes the host to secondary bacterial pneumonia, which is a major cause of mortality during influenza epidemics. The molecular mechanisms underlying the bacterial coinfection remain elusive. Neuraminidase (NA) of influenza A virus (IAV) enhances bacterial adherence and also activates TGF-β. Because TGF-β can up-regulate host adhesion molecules such as fibronectin and integrins for bacterial binding, we hypothesized that activated TGF-β during IAV infection contributes to secondary bacterial infection by up-regulating these host adhesion molecules. Flow cytometric analyses of a human lung epithelial cell line indicated that the expression of fibronectin and α5 integrin was up-regulated after IAV infection or treatment with recombinant NA and was reversed through the inhibition of TGF-β signaling. IAV-promoted adherence of group A Streptococcus (GAS) and other coinfective pathogens that require fibronectin for binding was prevented significantly by the inhibition of TGF-β. However, IAV did not promote the adherence of Lactococcus lactis unless this bacterium expressed the fibronectin-binding protein of GAS. Mouse experiments showed that IAV infection enhanced GAS colonization in the lungs of wild-type animals but not in the lungs of mice deficient in TGF-β signaling. Taken together, these results reveal a previously unrecognized mechanism: IAV NA enhances the expression of cellular adhesins through the activation of TGF-β, leading to increased bacterial loading in the lungs. Our results suggest that TGF-β and cellular adhesins may be potential pharmaceutical targets for the prevention of coinfection.
Secondary bacterial pneumonia or coinfection is the leading cause of viral-associated mortality during influenza A virus (IAV) pandemics (1, 2). The synergistic lethality of IAV and bacterial coinfection has been observed in animal models (3), suggesting a causative relationship between IAV infection and secondary bacterial pneumonia. Increased bacterial adherence post-IAV has been well recognized (4); however, the underlying mechanisms remain elusive. It has been demonstrated that IAV neuraminidase (NA) promotes the adherence of Streptococcus pneumoniae to lung epithelial cells, and viral NA activity has been associated with the levels of bacterial adherence and mortality in coinfected mice (5). In addition, inhibitors of NA, such as oseltamivir, reversed the effects of NA on bacterial adherence (6). These findings suggest that IAV NA contributes substantially to coinfection.
ECM proteins, such as fibronectin (Fn), collagen, and laminin, interact with integrins, which transduce signals to regulate cell growth, differentiation, migration, and other cellular activities. ECM proteins and integrins are receptors that bind to microbial surface components recognizing adhesive matrix molecules (MSCRAMM) for bacterial adherence and invasion (4, 7). The expression of these cellular adhesion molecules can be up-regulated through TGF-β (8). This cytokine is secreted as an inactive or latent protein that subsequently is activated through various mechanisms (9). Schultz-Cherry and Hinshaw (10) reported that latent TGF-β is activated through IAV NA, and recently these authors demonstrated that viral NA triggers TGF-β activation through the removal of sialic acid motifs from latent TGF-β (11). These findings suggest that TGF-β might play a role in IAV-enhanced bacterial adherence.
Adherence to host tissue is a critical initial step to establish infection. The most frequently observed bacteria in coinfections are S. pneumoniae, group A Streptococcus pyogenes (GAS), Staphylococcus aureus, and Haemophilus influenza (1, 12, 13). These bacteria require ECM components or integrins as receptors for adherence (14–17). We previously demonstrated that the invasion of host cells by GAS is promoted through the TGF-β–enhanced expression of integrin and Fn (8). These observations suggest that the activation of TGF-β through IAV NA might promote the expression of cellular receptors, facilitating bacterial adherence and leading to increased host susceptibility to coinfection.
The goal of the present study was to define the mechanisms underlying the increased bacterial adherence post-IAV infection. We showed that expression of α5 integrin/Fn was up-regulated in response to IAV infection or viral NA treatment and reversed through the inhibition of TGF-β signaling, indicating that IAV increased the expression of host receptors through NA-activated TGF-β. In addition, IAV-mediated bacterial adherence required the Fn-binding protein of GAS, and the adherence of coinfective pathogens to IAV-infected cells was impeded by TGF-β inhibitors, suggesting that the bacteria commonly observed in coinfection likely share a similar mechanism for initiating an infection. Interventions targeting these mechanisms might reduce the incidence and severity of postinfluenza bacterial pneumonia.
Results
IAV Increased TGF-β Activity and Enhanced GAS Adherence to Human Lung Epithelial Cells.
TGF-β is secreted from virtually all cells in a biologically inactive form. The infection of mice or Madin–Darby canine kidney (MDCK) cells with IAV increases TGF-β activity (10). To determine whether TGF-β also is activated in human lung cells through IAV, A549 cells were infected with IAV strain H1N1 influenza virus A/Puerto Rico/08/1934 (PR8), and the supernatants were assayed for TGF-β activation using Mv1Lu reporter cells (8). The activity of TGF-β was three times higher in the supernatant from A549 cells treated with PR8 than from those treated with PBS alone (Fig. 1A), suggesting that the activity of TGF-β in human lung epithelial cells is increased following IAV infection. The M1 protein of GAS mediates the binding of bacteria to integrin via Fn. Exogenous active TGF-β significantly enhances GAS invasion through the up-regulation of α5 integrin/Fn (8). These results prompted our using GAS as a model to investigate IAV-mediated bacterial adherence. A549 cells were infected with PR8, and then an adhesion assay was performed with the GAS M1 strain. The bacterial numbers associated with viral-infected cells were two times higher than in PBS-treated control cells (Fig. 1B). The concomitant enhancement of TGF-β activity and GAS adherence by IAV suggests a potential correlation.
Fig. 1.

IAV enhanced TGF-β1 activity and GAS adherence to human lung epithelial cells. (A) Cell-culture supernatant was collected after A549 cells were infected with PR8 (5,000 TCID50) for 2 h. Active TGF-β in the supernatant was measured by Mv1Lu luciferase assay, and the results were extrapolated from a standard curve run in parallel. (B) A549 cells were infected with PR8 as described in Materials and Methods followed by an adhesion assay with GAS strain 90226 M1 at an MOI of 10. The bacterial adherence to cells without PR8 infection (PBS) was considered as 100%. Data are presented as means ± SE of two to four independent determinants. **P ≤ 0.01.
GAS Adherence Was Enhanced by Recombinant Viral NA and Was Prevented by the Inhibition of TGF-β Signaling.
To determine whether GAS adherence is enhanced through IAV NA, an adhesion assay was performed after A549 cells were treated with a recombinant NA (Materials and Methods). Similar to the effect of NA on S. pneumoniae adherence (5, 6), higher GAS cfus were obtained with NA-treated cells than with untreated cells (Fig. 2A), and the cfu counts were increased in an NA dose-dependent manner (Fig. 2B), indicating that viral NA contributes to the PR8-mediated increase in GAS adherence. Next, we determined whether NA-enhanced GAS adherence is mediated through the activation of TGF-β. A549 cells were infected with PR8 or were treated with NA in the presence of SB 431542, a specific inhibitor of the TGF-β receptor. The adherence assay showed that GAS cfus with the inhibitor-treated cells (Fig. 2C) were significantly reduced compared with inhibitor carrier control cells. An examination of the viability of the cells and GAS did not show differences between the groups. These results suggest that the activation of TGF-β through NA increases GAS binding to IAV-infected cells.
Fig. 2.
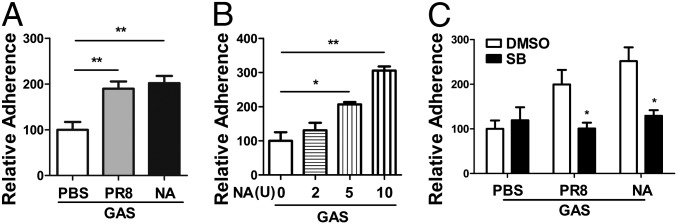
Recombinant viral NA enhanced GAS adherence in a TGF-β signaling-dependent manner. Adherence assays were performed with the GAS M1 strain on A549 cells infected with PR8 as in Fig. 1B or treated with recombinant NA at 5 U (A) or indicated doses (B) for 18 h. For the adherence inhibition assay, cells were infected with PR8 or were treated with recombinant NA at 10 U in the presence of the TGF-β inhibitor SB 431542 (50 μM) followed by an adherence assay (C). Data are presented as means ± SEM of two or three independent experiments (n = 4–6). *P ≤ 0.05, **P ≤ 0.01.
The Expression of Cellular Receptors Was Enhanced Through PR8 or NA and Was Prevented by Inhibitors of TGF-β1 and NA.
TGF-β promotes GAS adherence through the up-regulation of α5 integrin/Fn (8). Thus, we speculated that these molecules are increased after IAV infection. The expression of α5 integrin/Fn on PR8-infected A549 cells was examined by flow cytometric analysis. High levels of α5 integrin were detected on a few of the noninfected A549 cells (Fig. 3A, gray area and column), but after PR8 infection the number of cells with high levels of α5 integrin was increased dramatically, by 18-fold compared with cells without viral infection (Fig. 3A, solid line and open column). Similar results were obtained for Fn: The number of cells with the higher levels of Fn expression was increased by 6.5-fold (Fig. 3B). The increased expression of α5 integrin/Fn was reversed significantly after cells infected with PR8 were treated with SB431542 (Fig. 3 A and B, dashed line and black column). To confirm this result, SIS3, a different inhibitor of TGF-β signaling that specifically inhibits Smad3 was used. Similarly, the expression of α5 integrin/Fn was repressed after SIS3 treatment (Fig. S1 A and B, dashed line and black column). These results suggest that the expression of α5 integrin/Fn is up-regulated after PR8 infection and that this effect is mediated by TGF-β.
Fig. 3.

Expression of cellular adhesion molecules was enhanced by PR8 or NA and was prevented by TGF-β1 and NA inhibitors. (A and B) Cells were infected with PR8 in the presence of DMSO (carrier control) (PR8/DMSO, black lines) or of SB 431542 (PR8/SB, dashed lines). PBS/DMSO is a control without PR8 infection (gray areas). Cells then were stained and analyzed for α5 integrin (A) or Fn (B) by flow cytometry. Ten thousand live cells were acquired; results are expressed as the percentage of cells expressing high levels of α5 integrin or Fn among the total acquired cells. (C) Cells were treated with 10 U of recombinant NA (black line) or same amount of NA buffer (gray area) for indicated times followed by flow cytometric analysis of Fn expression. (D) Cells were treated with PBS/PBS (gray area), NA/PBS (black line), or NA/zanamivir (ZMV) dissolved in PBS (dashed line) and were analyzed as in B. The bar graphs in A, B, and D show summarized data of flow cytometry (n = 4). (E) Cells were treated as in D, and adherence assay was performed as in Fig. 1B. Adherence data are presented as means ± SEM of two independent experiments (n = 6). **P ≤ 0.01, ***P ≤ 0.0001.
ECM synthesis increases for tissue repair following viral infection, and Fn, a major component of the ECM, is produced during this process. To determine whether cellular receptors are up-regulated primarily through NA, Fn expression was examined in cells treated with NA. Cells with higher levels of Fn were observed in a time-dependent manner, showing a significant increase at 20 h after NA treatment (Fig. 3C). The Fn expression induced by NA or PR8 was inhibited using the specific NA inhibitor zanamivir (Fig. 3D and Fig. S2A), which also suppressed NA- or PR8-enhanced GAS adherence (Fig. 3E and Fig. S2B). Although zanamivir was less effective in inhibiting Fn expression, its effect on adherence was more evident. The possible causes are explained in Discussion. These results indicate that IAV NA promotes the expression of cellular adhesion receptors, thereby increasing the susceptibility of host cells to GAS attachment.
IAV-Enhanced Bacterial Adherence Is Dependent on the Bacterial Fn-Binding Protein.
Klebsiella pneumoniae is a human respiratory pathogen that uses mannose for attachment to the host cell (18). The requirement of ECM/integrin for K. pneumoniae adherence has not been reported previously. Coinfection with K. pneumoniae in influenza patients is rare. Thus, we postulated that IAV infection would have less effect on K. pneumoniae adherence. Without PR8 infection, the adherence of K. pneumoniae was very low compared with the integrin-dependent adherence of GAS at the same multiplicity of infection (MOI) (Fig. 4A, PBS) and was similar to that reported in the literature (19). After PR8 infection, GAS adherence increased up to 60% of the inoculum, but the K. pneumoniae adherence stayed unchanged (≤0.04% of the inoculum) (Fig. 4A, PR8). These results suggest that IAV does not mediate K. pneumoniae adherence, potentially reflecting the rare association of this bacterium with coinfection.
Fig. 4.

IAV-mediated increase in bacterial adherence is dependent on Fn-binding protein. Adhesion assays were performed at an MOI of 10 with GAS and K. pneumonia (A) or with L. lactis M1+ and L. lactis M1− (B) after A549 cells were infected with PR8. The results are expressed as a percentage of the inoculum. Data are presented as means ± SEM of two independent experiments (n = 4). *P ≤ 0.05.
The difference in the adherence of GAS and K. pneumoniae to PR8-infected cells suggests that IAV promotes GAS adherence through the ECM and integrins. GAS expresses multiple adhesins, which bind to different ECM molecules. One of these adhesins is the M1 protein, which contributes significantly to the adherence and invasion of GAS (20, 21). The deletion of M1 significantly reduced GAS adherence to cells. Lactococcus lactis does not express adhesins and is considered nonadherent. The expression of M1 protein on the surface of L. lactis (M1+) confers the ability to adhere efficiently (22). This system provides a strategy for characterizing the function of bacterial Fn-binding protein in coinfection. Without PR8 infection (Fig. 4B, PBS), the adherence of L. lactis (M1−) to A549 cells was less than 0.7% of the inoculum and increased significantly to 8% of the inoculum when the strain expressed the M1 protein of GAS (L. lactis M1+) (Fig. 4B). Notably, the adherence of L. lactis (M1−) was not affected after PR8 infection (Fig. 4B, Ll M1−, PR8), whereas the adherence of the M1-expressing L. lactis (M1+) strain was increased significantly (12% of the inoculum) (Fig. 4B, Ll M1+, PR8). These results indicate that PR8-promoted bacterial adherence is facilitated through Fn binding.
The Inhibition of TGF-β Signaling Prevented the Adherence of Other Common Coinfecting Bacteria.
S. pneumoniae, S. aureus, and H. influenza frequently have been isolated from patients with coinfection (23). For adherence or invasion, these bacteria interact with ECM components and integrin receptors through Fn-binding molecules, such as PfbA, an Fn-binding protein of S. pneumoniae (24); FnBPA and FnBPB, the Fn-binding proteins of S. aureus (15, 16); and Hap, an adhesin of H. influenza that binds to Fn, laminin, and collagen IV (17). The specific association of coinfection with these bacteria suggests that these microbes share a similar mechanism. Consistent with GAS, higher numbers of S. pneumoniae and S. aureus Fn-binding protein A and B (FnBPA+B+) bacteria were associated with PR8-infected cells than with PBS control cells, and the number of bacteria was reduced in the presence of TGF-β inhibitors during PR8 infection (Fig. 5 A and B), suggesting that TGF-β activity also is required for the adherence of these bacteria. The adherence of nontypeable H. influenza (NTHi, Hap+) to PR8-infected cells was much higher (up to 24-fold) than that of GAS. Interestingly, the adherence was prevented partially by the TGF-β receptor inhibitor SB 431542 and was not affected by the Smad3 (a molecule of TGF-β signal pathway) inhibitor SIS3 (Fig. 5C). Because TGF-β uses multiple intracellular signaling pathways after receptor activation (25), in contrast with GAS, an Smad-independent TGF-β pathway likely is used in NTHi adherence. In addition, the dramatic increase in NTHi adherence and partial inhibition by SB431542 suggest that mechanisms other than TGF-β are involved. Taken together, these results suggest that TGF-β activation in IAV infection plays an important role in coinfection and likely is a common mechanism among coinfecting pathogens.
Fig. 5.

TGF-β inhibitors prevented IAV-enhanced adherence of other coinfective bacterial pathogens. A549 cells were treated with PBS and DMSO, PR8 and DMSO, PR8 and the TGF-β inhibitor SB 431542 (SB), or PR8 and the TGF-β inhibitor SIS3. Adhesion assays then were performed with S. pneumonia (T19F) (A), S. aureus (RN 6390, FnBPA+B+) (B), or H. influenzae (C) at an MOI of 10. Data are presented as means ± SEM of two or three independent experiments (n = 4–12). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.0001.
IAV Infection Increased GAS Colonization in the Lungs of Wild-Type Mice but Not in Mice Deficient inTGF-β Signaling.
GAS coinfection was studied further in vivo. Wild-type mice were infected with PR8 at low or high doses (500 or 10,000 times the 50% tissue culture infective dose, TCID50), were challenged with GAS 3 d after PR8 infection, and were killed 24 h later. In the two groups of mice receiving high or low doses of PR8, 100% of the mice survived at the end of the experiment (day 5), and the mice in the two groups showed similar weight loss (Fig. S3A). Single lung cell suspensions were plated to determine bacterial numbers in the lungs. Consistent with the results of the in vitro assays, higher cfus of GAS were recovered from PR8-infected mice than from control mice (Fig. 6A). The expression of α5 integrin/Fn in lung cells was examined by flow cytometric analysis. Similar to PR8-infected A549 cells, viral-infected mice had an increased number of lung cells with higher expression of α5 integrin (Fig. 6B, Upper) or Fn (Fig. 6B, Lower). Pathology of the lung tissue showed that, compared with PBS-treated control mice (Fig. S3 B, a), thickening of alveolar septa and leukocyte infiltration were found in lung sections of GAS-infected or PR8-infected mice (Fig. S3 B, b, c, and e). In coinfected mice lung consolidation, thickening of alveolar septa, and liquid in alveolar space were more obvious (indicated by arrow, Fig. S3 B, d, and f). In addition, no necrosis or apoptosis was found in PR8-infected lungs, suggesting that increased bacterial colonization is unlikely to be caused by a greater number of dead cells in PR8-infected lungs.
Fig. 6.

(A) Mice deficient in TGF-β signaling were resistant to GAS after PR8 infection. Wild-type C57BL/6 (B6) mice were infected intranasally with PR8 at 500 TCID50 or 10,000 TCID50 or received PBS as control. Three days later the mice were inoculated intranasally with 1 × 107 cfu of GAS. Mice were killed 24 h after the inoculation. Aliquots of lung homogenate were diluted to quantify cfus. Data are presented as means ± SEM of two independent experiments. ***P ≤ 0.0001. (B) Lung homogenate from four randomly selected mice was stained, and 20,000 cells were acquired and analyzed for α5 integrin (Upper) and Fn (Lower) by flow cytometry. Gray areas, black lines, and dashed lines in histograms represent lung cells from mice that received PBS or PR8 at 500 TCID50 or 10,000 TCID50, respectively. Relative numbers of lung cells expressing high levels of α5 integrin or Fn are summarized in the bar graphs (n = 4, **P ≤ 0.01, ***P ≤ 0.0001). (C and D) Smad3−/− mice and Smad3+/+ littermates were infected with PR8 at 10,000 TCID50 and were coinfected with GAS as in A. Cfus (*P ≤ 0.05) (C) and the expression of α5 integrin (Left) or Fn (Right) (D) in the lungs were determined as in A and B.
As shown in the lung sections, inflammatory cells such as neutrophils are infiltrated into the lungs in response to IAV infection. To determine if overexpressed α5 integrin/Fn in lung epithelial cells is related to the increased bacteria, lung epithelial cells were enriched by Percoll density gradient and examined for α5 integrin/Fn. Similar to the results from total lung cells (Fig. 6B), significantly higher levels of these adhesin molecules were expressed on the lung epithelial cells (Fig. S4).
The prevention of GAS adherence to IAV-infected cells through the inhibition of Smad3 signaling in vitro suggested that an Smad-signaling deficiency would disrupt the TGF-β–mediated overexpression of host adhesins and would impede IAV-facilitated GAS colonization. This hypothesis was tested using Smad3-knockout (Smad3−/−) mice (26). The cfus recovered from the lungs of PR8-infected Smad3−/− mice were significantly lower than those obtained from PR8-infected Smad3+/+ littermates but were similar to the cfus from Smad3+/+ littermates without PR8 infection (Fig. 6C). Flow cytometric analysis showed that the expression of α5 integrin/Fn in lung cells was similar in Smad3−/− mice and Smad3+/+ mice without PR8 infection (Fig. 6D, Upper). After PR8 infection, a substantial increase in the number of lung cells with higher levels of α5 integrin/Fn expression was observed in Smad3+/+ mice (Fig. 6D, Middle), whereas the number of these cells remained similar in Smad3−/− mice before and after PR8 infection (Fig. 6D, Lower). These results indicate that the TGF-β–signaling deficiency prevents the up-regulation of α5 integrin/Fn expression in PR8-infected Smad3−/− mice. Consistently, the in vivo experiments demonstrated that the TGF-β–mediated up-regulation of cellular adhesins in IAV infection facilitates bacterial colonization in the lungs.
Discussion
Bacterial colonization is a crucial step in the establishment of infection. Coinfection during influenza epidemics often is preceded by excess bacterial density within the upper respiratory tract (27), indicating that coinfection is initiated by increased bacterial loading. IAV NA helps release progeny virions from infected cells by stripping sialic acids from cells, likely exposing tissue receptors, and resulting in increased bacterial attachment (3). However, chemically induced lung damage did not promote secondary bacterial infection, suggesting that other mechanisms are involved (3). We observed that the inhibition of TGF-β signaling prevented the NA-mediated up-regulation of α5 integrin/Fn and excess bacterial adherence. The study demonstrated that viral NA activates TGF-β, which promotes the expression of cellular adhesins, leading to increased bacterial colonization (Fig. 7).
Fig. 7.

IAV facilitates bacterial adherence through the activation of the TGF-β signaling pathway. IAV NA activates latent TGF-β, which turns on the Smad signaling pathway, resulting in the up-regulation of integrins and Fn expression in cells. As bacterial receptors, the overexpression of integrins and Fn increases host susceptibility to bacterial infection.
Other respiratory viruses, such as influenza B virus (IBV) and human parainfluenza viruses (HPIVs), also express NA. Both these viruses have been associated with bacterial pneumonia, although IBV has little impact on morbidity and mortality (28, 29). In addition, a specific inhibitor of HPIV NA reduces bacterial titers in the lungs and prevents the death of infected mice (29). Whether the NAs of these viruses play a similar role in coinfection is unknown. Bacterial coinfection often has been associated with other viruses without NAs, such as respiratory syncytial virus (RSV) and rhinovirus. Interestingly, both RSV and rhinovirus induce TGF-β expression in epithelial cells (30, 31). Thus, it would be interesting to determine the relevance of TGF-β to coinfection with these viruses.
TGF-β is an important regulator of immune cells and is known to inhibit natural killer (NK) cell infiltration and activation (32, 33). It has been reported that IAV infection impairs NK cell responses to S. aureus coinfection in mice (34). It is likely that NK cell function is less impaired in PR8-infected Smad3−/− mice and also contributes to the reduction of GAS colonization in these mice. In addition, TGF-β regulates the development of Th17 and Treg cells. A coinfection model in WT mouse showed that Th17 immunity to coinfection with S. aureus is inhibited, resulting in the inefficient clearance of S. aureus from the lungs (35). Whether TGF-β regulates adaptive immunity to coinfection is currently unknown.
The inhibition of TGF-β signaling prevented adherence of GAS and other coinfecting bacteria to PR8-infected cells, suggesting that a similar cellular mechanism is used. Many bacteria express Fn-binding proteins or MSCRAMM (4, 36), although not all these microbes have been associated with IAV. However, all identified coinfecting bacteria are found commonly in the upper respiratory tract and are able to express MSCRAMM. As commensals in the nasopharyngeal niche, it is likely that the expression of MSCRAMM confers an advantage to the preexistent bacteria in using IAV-induced cellular changes for the efficient colonization in the lungs.
Only PR8 and NA1 protein were tested in the study, although most IAV subtypes and NAs can activate TGF-β (10). Because viral NA activity has been associated with the levels of bacterial adherence and mortality of coinfected mice (5), different types of IAV NA might contribute differently to up-regulation of α5 integrin/Fn. We observed that within inhibitory ranges, the NA inhibitor zanamivir was less effective than the TGF-β inhibitors in suppressing Fn expression mediated by NA (Fig. 3D) or by PR8 (Fig. S2A). Because other IAV-associated proteases (5) or cellular mechanisms (9) also activate TGF-β, it is likely that the NA-independent activation of TGF-β is responsible for the residual expression of Fn, which is prevented by TGF-β inhibitors but not by NA inhibitors. Interestingly, inhibition of either TGF-β (Fig. 2C) or NA activity (Fig. 3E and Fig. S2B) efficiently prevented GAS adherence, and currently there is no explanation for this phenomena.
We also observed an increased expression of α5 integrin/Fn in lung leukocytes after PR8 infection. Because inflammatory cells have been implicated in both protective and pathological responses following influenza virus infections, it is not known whether these cells promote bacterial killing or contribute to bacterial binding. It is likely that up-regulated adhesin molecules on the inflammatory cells also contribute to bacterial binding at the time point of observation, regardless the outcome of the bound bacteria later.
In this study we demonstrate that the activation of TGF-β in IAV infection plays an important role in the pathogenesis of coinfection. The results suggest that TGF-β and cellular adhesion molecules are attractive targets for the prevention of coinfection and have an advantage in infections involving IAV strains that are resistant to NA inhibitors.
Materials and Methods
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Institute of Microbiology, Chinese Academy of Sciences (IMCAS) Ethics Committee. The protocol was approved by the Committee on the Ethics of Animal Experiments of IMCAS (permit no. PZIMCAS2011002). The mice were bred under specific pathogen-free conditions in the laboratory animal facility at IMCAS. All animal experiments were conducted under isoflurane anesthesia, and all efforts were made to minimize suffering.
Details of cell-culture conditions, the TGF-β1 bioactivity assay, and the bacterial adhesion assay are given in SI Materials and Methods.
Reagents.
SB431542 was purchased from Cayman Chemical. SIS3 and zanamivir were obtained from Sigma. Antibodies directed against α5 integrin (ab55988) and Fn (S-9068) were obtained from R&D Systems and Santa Cruz Biotechnology, respectively. FITC-conjugated goat anti-rabbit IgG was purchased from Sigma. NA was purchased from Sino Biological Inc. [A/California/04/2009 (H1N1)] or was a kind gift from G.F.G. [PR8, A/Puerto Rico/08/1934]. NA activity was determined using methylumbelliferyl-N-acetylneuraminic acid (J&K Scientific, Ltd.).
Virus and Bacterial Strains.
PR8 was obtained from B. Gao (IMCAS), cultured in the allantoic cavities of 9-d-old specific pathogen-free embryonated hen eggs, and incubated for 2 d at 35 °C. The allantoic fluid was collected and stored at −80 °C. The viruses were quantified in MDCK cells and expressed as TCID50. GAS strain 90-226 is a serotype of the M1 strain. S. pneumoniae strain T19F was a clinical isolate kindly provided by X. Shen (Beijing Children's Hospital, Beijing). S. aureus strain RN 6390 contains FnBPA+B+. These bacteria were maintained on sheep blood agar and grown in THY-Neo medium at 37 °C with 5% CO2. K. pneumoniae (strain 10617) was purchased from China General Microbiological Culture Collection Center and was grown in tryptic soy broth (Difco) for 18 h at 37 °C. M1+ L. lactis (pLM1) contains the plasmid pLM1, which encodes the full-length M1 protein. L. lactis M− (pP59) carries the empty vector. L. lactis strains were grown in M17 medium (Difco) containing erythromycin (5 μg/mL) at 30 °C. H. influenzae strain NTHi Sc70040 was obtained from Z. J. Shao, Chinese Center for Disease Control and Prevention, Beijing, and grown on chocolate agar at 37 °C with 5% CO2.
Flow Cytometry Analysis of α5 Integrin and Fn Expression.
A549 cells were infected with PR8 or were treated with 10 U of recombinant NA. SB431542, SIS3, or zanamivir was added as described above. After incubation at 37 °C in 5% CO2 for 18 h, the cells were collected using 0.25% trypsin and were resuspended in cold PBA (1.0% BSA and 0.1% sodium azide in PBS) to analyze α5 integrin and Fn expression by flow cytometry. Briefly, the cells were stained with either anti-human α5 integrin or Fn antibodies and were washed with PBA. Fluorescent-conjugated secondary antibody was added, and the cells were incubated according to the manufacturer's instructions. The cells were fixed in 4% (wt/vol) paraformaldehyde for 10 min on ice, resuspended in PBA, and analyzed using a FACScan (Becton-Dickinson) flow cytometry analyzer. The percentage of cells with high α5 integrin or Fn expression was determined using FlowJo software.
Mice and Infection Models.
Female C57BL/6 (B6) mice (6–8 wk old) were purchased from Vital River Laboratory Animal Technology, whose colonies were all introduced from Charles River Laboratories. Smad3+/− mice were obtained from Y.Z. and were backcrossed for at least six generations to B6 mice in the animal facility of the Institute of Microbiology, followed by intercrossing to produce homozygous offspring. The resulting progeny were screened using PCR to identify Smad3−/− mice and Smad3+/+ littermates; the latter were used as wild-type control mice. The mice were anesthetized with inhaled 2.5% isoflurane (Baxter Healthcare). The anesthetized mice were inoculated intranasally with 500 or 10,000 TCID50 of PR8 suspended in 30 μL PBS. After 3 d, the mice were inoculated intranasally with 1 × 107 cfus of GAS. The mice were killed 24 h after inoculation; the lung homogenates were diluted to quantify the cfus and were analyzed by flow cytometry.
Statistical Analysis.
Statistical analyses were performed with two-tailed unpaired Mann–Whitney U nonparametric t tests using the GraphPad Prism version 5.01 for Windows (GraphPad Software). Data were considered significantly different at P ≤ 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Bin Gao of the Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, and Dr. Zhujun Shao of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, for providing the PR8 virus and NTHi strains; Dr. X. Shen of Beijing Children's Hospital for providing S. pneumoniae strain T19F; and Dr. P. Schlievert of the Department of Microbiology, University of Iowa, for providing S. aureus strain RN 6390. This research was supported by National Natural Science Foundation of China Grant 31170845 (to B.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414422112/-/DCSupplemental.
References
- 1.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J Infect Dis. 2008;198(7):962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anonymous Centers for Disease Control and Prevention (CDC) Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1) - United States, May-August 2009. MMWR Morb Mortal Wkly Rep. 2009;58(38):1071–1074. [PubMed] [Google Scholar]
- 3.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19(3):571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. 2014;12(4):252–262. doi: 10.1038/nrmicro3231. [DOI] [PubMed] [Google Scholar]
- 5.Peltola VT, Murti KG, McCullers JA. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J Infect Dis. 2005;192(2):249–257. doi: 10.1086/430954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J Infect Dis. 2003;187(6):1000–1009. doi: 10.1086/368163. [DOI] [PubMed] [Google Scholar]
- 7.Patti JM, Allen BL, McGavin MJ, Höök M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol. 1994;48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Li S, Southern PJ, Cleary PP. Streptococcal modulation of cellular invasion via TGF-beta1 signaling. Proc Natl Acad Sci USA. 2006;103(7):2380–2385. doi: 10.1073/pnas.0506668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li MO, Flavell RA. TGF-beta: A master of all T cell trades. Cell. 2008;134(3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol. 1996;70(12):8624–8629. doi: 10.1128/jvi.70.12.8624-8629.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson CM, et al. Transforming growth factor-β: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010;6(10):e1001136. doi: 10.1371/journal.ppat.1001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brundage JF. Interactions between influenza and bacterial respiratory pathogens: Implications for pandemic preparedness. Lancet Infect Dis. 2006;6(5):303–312. doi: 10.1016/S1473-3099(06)70466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry-Acosta M, Puius YA. Post-influenza pneumonia: Everything old is new again. Med Health R I. 2010;93(7):204–207, 211. [PubMed] [Google Scholar]
- 14.Pracht D, et al. PavA of Streptococcus pneumoniae modulates adherence, invasion, and meningeal inflammation. Infect Immun. 2005;73(5):2680–2689. doi: 10.1128/IAI.73.5.2680-2689.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B, Yurecko RS, Dedhar S, Cleary PP. Integrin-linked kinase is an essential link between integrins and uptake of bacterial pathogens by epithelial cells. Cell Microbiol. 2006;8(2):257–266. doi: 10.1111/j.1462-5822.2005.00618.x. [DOI] [PubMed] [Google Scholar]
- 16.Sinha B, et al. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol. 1999;1(2):101–117. doi: 10.1046/j.1462-5822.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 17.Fink DL, Green BA, St Geme JW., 3rd The Haemophilus influenzae Hap autotransporter binds to fibronectin, laminin, and collagen IV. Infect Immun. 2002;70(9):4902–4907. doi: 10.1128/IAI.70.9.4902-4907.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fader RC, Gondesen K, Tolley B, Ritchie DG, Moller P. Evidence that in vitro adherence of Klebsiella pneumoniae to ciliated hamster tracheal cells is mediated by type 1 fimbriae. Infect Immun. 1988;56(11):3011–3013. doi: 10.1128/iai.56.11.3011-3013.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cano V, Moranta D, Llobet-Brossa E, Bengoechea JA, Garmendia J. Klebsiella pneumoniae triggers a cytotoxic effect on airway epithelial cells. BMC Microbiol. 2009;9:156–164. doi: 10.1186/1471-2180-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, et al. 2006. Intracellular invasion by Streptococcus pyogenes: Invasins, host receptors, and relevance to human disease. Gram-Positive Pathogens, eds Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA (ASM Press, Washington, DC), pp 29–36.
- 21.Cue D, et al. A nonpeptide integrin antagonist can inhibit epithelial cell ingestion of Streptococcus pyogenes by blocking formation of integrin alpha 5beta 1-fibronectin-M1 protein complexes. Proc Natl Acad Sci USA. 2000;97(6):2858–2863. doi: 10.1073/pnas.050587897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cue D, Lam H, Cleary PP. Genetic dissection of the Streptococcus pyogenes M1 protein: Regions involved in fibronectin binding and intracellular invasion. Microb Pathog. 2001;31(5):231–242. doi: 10.1006/mpat.2001.0467. [DOI] [PubMed] [Google Scholar]
- 23.Chertow DS, Memoli MJ. Bacterial coinfection in influenza: A grand rounds review. JAMA. 2013;309(3):275–282. doi: 10.1001/jama.2012.194139. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi M, Terao Y, Mori Y, Hamada S, Kawabata S. PfbA, a novel plasmin- and fibronectin-binding protein of Streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J Biol Chem. 2008;283(52):36272–36279. doi: 10.1074/jbc.M807087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, et al. Smad3-deficient CD11b(+)Gr1(+) myeloid-derived suppressor cells prevent allograft rejection via the nitric oxide pathway. J Immunol. 2012;189(10):4989–5000. doi: 10.4049/jimmunol.1200068. [DOI] [PubMed] [Google Scholar]
- 27.Mina MJ, Klugman KP, McCullers JA. Live attenuated influenza vaccine, but not pneumococcal conjugate vaccine, protects against increased density and duration of pneumococcal carriage after influenza infection in pneumococcal colonized mice. J Infect Dis. 2013;208(8):1281–1285. doi: 10.1093/infdis/jit317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aebi T, et al. Co-infection of Influenza B and Streptococci causing severe pneumonia and septic shock in healthy women. BMC Infect Dis. 2010;10:308–317. doi: 10.1186/1471-2334-10-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alymova IV, et al. The novel parainfluenza virus hemagglutinin-neuraminidase inhibitor BCX 2798 prevents lethal synergism between a paramyxovirus and Streptococcus pneumoniae. Antimicrob Agents Chemother. 2005;49(1):398–405. doi: 10.1128/AAC.49.1.398-405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibbs JD, Ornoff DM, Igo HA, Zeng JY, Imani F. Cell cycle arrest by transforming growth factor beta1 enhances replication of respiratory syncytial virus in lung epithelial cells. J Virol. 2009;83(23):12424–12431. doi: 10.1128/JVI.00806-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dosanjh A. Transforming growth factor-beta expression induced by rhinovirus infection in respiratory epithelial cells. Acta Biochim Biophys Sin (Shanghai) 2006;38(12):911–914. doi: 10.1111/j.1745-7270.2006.00234.x. [DOI] [PubMed] [Google Scholar]
- 32.Meadows SK, Eriksson M, Barber A, Sentman CL. Human NK cell IFN-gamma production is regulated by endogenous TGF-beta. Int Immunopharmacol. 2006;6(6):1020–1028. doi: 10.1016/j.intimp.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 33.Marcoe JP, et al. TGF-β is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat Immunol. 2012;13(9):843–850. doi: 10.1038/ni.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Small CL, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. 2010;184(4):2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
- 35.Kudva A, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186(3):1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patti JM, Höök M. Microbial adhesins recognizing extracellular matrix macromolecules. Curr Opin Cell Biol. 1994;6(5):752–758. doi: 10.1016/0955-0674(94)90104-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.