

Significance
Fusion of intracellular membranes is involved in many critical cellular processes, such as neurotransmission, protein trafficking, and in the lysosomal degradation of invading bacterial pathogens. Accordingly, some intracellular bacterial pathogens use protein effectors to alter host membrane fusion directly as a survival mechanism. In this study, we show that the Vibrio secreted effector, VopQ, is a potent inhibitor of yeast homotypic vacuole fusion in vitro. Although VopQ was shown to deacidify yeast vacuoles via its known V-type H+-ATPase (V-ATPase)-binding and channel-forming activities, its ability to inhibit vacuole fusion does not depend on channel-forming activity. Our studies suggest that yeast vacuole fusion is not regulated by lumenal acidification and identify a reagent to study the V-ATPase role in some membrane fusion events.
Keywords: Vibrio parahaemolyticus, SNARE, vesicle fusion, yeast vacuole, vp1680
Abstract
Vesicle fusion governs many important biological processes, and imbalances in the regulation of membrane fusion can lead to a variety of diseases such as diabetes and neurological disorders. Here we show that the Vibrio parahaemolyticus effector protein VopQ is a potent inhibitor of membrane fusion based on an in vitro yeast vacuole fusion model. Previously, we demonstrated that VopQ binds to the Vo domain of the conserved V-type H+-ATPase (V-ATPase) found on acidic compartments such as the yeast vacuole. VopQ forms a nonspecific, voltage-gated membrane channel of 18 Å resulting in neutralization of these compartments. We now present data showing that VopQ inhibits yeast vacuole fusion. Furthermore, we identified a unique mutation in VopQ that delineates its two functions, deacidification and inhibition of membrane fusion. The use of VopQ as a membrane fusion inhibitor in this manner now provides convincing evidence that vacuole fusion occurs independently of luminal acidification in vitro.
Vesicle fusion governs many important physiological processes including neurotransmitter release and exocytosis. As such, many studies have focused on understanding this process and the proteins involved in fusion using various models such as yeast vacuoles and Drosophila synaptic vesicles (1, 2). Yeast vacuoles are an established and elegant model to study eukaryotic membrane fusion because of the ease of their isolation and the conserved nature of the fusion machinery required for their homotypic fusion (3). Although the core SNARE and Rab GTPase fusion machinery alone can drive the physiologically relevant fusion of liposomes in vitro (2), genetic and biochemical experiments have identified a number of additional regulators of vacuole fusion, including the membrane sector of the highly conserved V-type H+-ATPase (V-ATPase) (4, 5).
The eukaryotic V-ATPase is the main electrogenic proton pump involved in the acidification of many intracellular organelles such as endosomes, lysosomes, and the yeast vacuole (6). The V-ATPase consists of two conserved, multisubunit domains: the cytoplasmic V1 domain and the membrane bound Vo domain. The V1 domain hydrolyzes ATP, providing the energy for proton translocation through the membrane-bound Vo proteolipid proton channel, thus acidifying the lumen of the vesicle. The loss of V-ATPase subunits is lethal in higher eukaryotes, highlighting the importance of this vital protein complex for normal eukaryotic physiology. However, yeast that lack subunits of the V-ATPase exhibit conditional lethality that is rescued by growth on acidic media, thus providing a unique and powerful system for the study of V-ATPase functions in vivo. In addition to its acidification function, the V-ATPase has been implicated in a broad range of biological processes, including the proper trafficking of secreted and endocytosed cargos (7), viral fusion (8), exocytosis (1, 9, 10), and the SNARE-dependent membrane fusion of yeast vacuoles (4, 5, 11, 12). Even though the role of V-ATPase in fusion has been demonstrated in various model organisms, its role in this process remains controversial (4, 13–16).
Recent work has shown that a bacterial protein VopQ (also known as VP1680 or VepA) forms an outward rectifying, gated channel in membranes that contain the V-ATPase, resulting in the collapse of ion gradients and the disruption of autophagic flux (17). VopQ is a type III effector protein from the marine bacterium Vibrio parahaemolyticus that strongly associates with the Vo domain of the eukaryotic V-ATPase (17, 18). Given the proposed role of the Vo domain in fusion, we sought to examine the effect of VopQ on fusion using the well-defined biochemical model of eukaryotic membrane fusion from the budding yeast Saccharomyces cerevisiae. In this study, we demonstrate that expression of VopQ causes extensive yeast vacuolar fragmentation indicative of a defect in homotypic vacuole fusion. In vitro vacuole fusion assays confirmed VopQ is a potent inhibitor of a conserved Rab GTPase- and SNARE-dependent membrane fusion. Furthermore, we identify a mutant of VopQ, VopQS200P, that deacidifies the vacuole via its known channel-forming activity but no longer inhibits vacuole fusion in vitro because of its reduced affinity for the V-ATPase Vo domain. Therefore, by using a bacterial effector protein with yeast vacuoles containing wild-type V-ATPase machinery, we show that yeast vacuole fusion does not require luminal acidification in vitro.
Results
VopQ Inhibits Yeast Homotypic Vacuole Fusion.
VopQ interacts directly with the Vo domain of the V-ATPase on the yeast vacuole. To assess whether this interaction alters vacuole morphology in vivo, we stained and visualized the vacuolar membrane of VopQ-expressing yeast. Normally, yeast cells contain one to three large vacuoles (Fig. S1A, WT). The Vps33p protein, a component of the endosomal (class C core vacuole/endosome tethering, CORVET) and vacuolar (homotypic fusion and vacuole protein sorting, HOPS) membrane-tethering complexes, is required for normal vacuolar morphology and fusion (19). Yeast strains lacking Vps33p display highly fragmented vacuoles typical of strains with defects in homotypic vacuole fusion (Fig. S1A, vps33∆). When VopQ is expressed in yeast under a galactose-inducible promoter, the yeast are viable (Fig. S1B) but have fragmented vacuoles similar to those seen in vps33∆ strains (Fig. S1A, VopQ+). Therefore, VopQ appears to modulate vacuolar membrane dynamics in vivo.
To assay whether this fragmentation is mediated through inhibition of the known yeast vacuole fusion machinery, we measured the effect of VopQ on a purified, well-studied homotypic vacuole fusion system. Because this system is known to be sensitive to a full complement of Rab GTPase-, SNARE-, and lipid-directed reagents, observations made in the context of this system often are found to be conserved in lysosomal fusion events in higher organisms (2). Yeast vacuoles were isolated from two strains of yeast: one lacking Pho8p, the normal vacuolar alkaline phosphatase (ALP), and one lacking Pep4p, required for the activation of the luminal vacuolar protease (Prb1p) responsible for activating Pho8p in vivo. Although neither of these purified vacuoles has ALP activity alone, upon successful fusion active proteases gain access to the catalytically inactive Pho8p, thereby activating Pho8p phosphatase activity to be measured colorimetrically (20). Recombinant VopQ protein was a powerful inhibitor of vacuole fusion in a dose-dependent manner (Fig. 1A, closed squares), on par with standard inhibitors of the fusion reaction including antisera against the Sec17p [α-soluble N-ethylmaleimide–sensitive fusion (NSF) attachment protein, α-SNAP) SNARE chaperone and the Vam3p syntaxin (21). Therefore, VopQ is capable of directly inhibiting homotypic vacuole fusion in vitro.
Fig. 1.

VopQ inhibits yeast homotypic vacuole fusion. (A) ALP vacuole fusion reactions in the absence or presence of increasing concentrations of recombinant His6-VopQ. Standard fusion inhibitors were α-Vam3p, α-Sec17p, 1 µM Gdi1p, Gyp1-46, or ice incubation. Error bars indicate SD from the mean; n = 4. (B and C) The β-lactamase content-mixing assay (B) was performed in parallel with the Rh-PE dequenching lipid-mixing assay (C) in the absence of ATP or including α-Vam3p, MBP-VopQ, or MBP. Data for the content-mixing assay are reported as mean ± SD, n = 3. The Rh-PE dequenching plot is a representative curve measured from the same three experiments used for the content-mixing assay.
Because VopQ is known to form channels in the vacuolar membrane, we wanted to confirm that VopQ does not disrupt the vacuolar membrane during fusion. We routinely included the ∼8-kDa inhibitor of Prb1p, Pbi2p, in these assays to prevent aberrant proPho8p activation; Pbi2p access to the vacuole lumen would be measured as fusion inhibition. ALP reactions performed in the absence of Pbi2p continue to display potent VopQ-dependent fusion inhibition (Fig. S2A), and vacuoles treated with VopQ do not release additional luminal GFP during fusion, unlike reactions treated with the soluble Vam7p SNARE and HOPS tethering complex (Fig. S2B), a treatment known to disrupt the integrity of yeast vacuoles (22). Therefore, VopQ does not disrupt vacuolar integrity during fusion.
VopQ Inhibits Vacuole Content Mixing.
The proteolytic processing and activation of the ALP can be used as a measure for successful yeast vacuolar fusion. However, luminal proteases that cleave the phosphatase require an acidic environment for proper trafficking and optimal activity in vitro (23). Previous work with the purified homotypic vacuole fusion system has shown that V-ATPase inhibitors that inhibit acidification of the vacuole, such as bafilomycin, also inhibit in vitro vacuolar fusion reaction as measured by ALP activity; therefore vacuolar acidification appears to be required for homotypic fusion (24). Because VopQ deacidifies yeast vacuoles, and our readout for fusion may be sensitive to luminal pH, we chose to revisit VopQ’s ability to inhibit membrane fusion with assays that do not depend on acidification.
Yeast homotypic vacuole fusion can be measured by reconstituting the activity of a soluble, vacuole lumen-directed bifurcated enzyme, β-lactamase (25). When vacuolar contents are mixed as a result of fusion, the domains of β-lactamase are brought together by interacting leucine zipper domains of the c-Jun and c-Fos proteins, resulting in an active β-lactamase enzyme that can be assayed via its hydrolytic activity (25). Fusion observed in the absence of inhibitors is used to standardize the effects of each biochemical inhibitor of the reaction (Fig. 1B). Vacuolar fusion is strongly inhibited in the presence of fusion inhibitors (no ATP, α-Vam3p; Fig. 1B). Addition of MBP-VopQ protein, but not MBP alone, inhibited vacuolar fusion, demonstrating that VopQ still inhibits homotypic vacuole fusion in an acidification-independent manner using an alternative ALP-independent fusion assay.
VopQ Inhibits Lipid Mixing.
We next measured the ability of VopQ to inhibit lipid bilayer mixing during in vitro vacuole fusion. Purified vacuolar membranes containing the components for the β-lactamase reconstitution fusion assay can be labeled with Rhodamine-PE and fused to unlabeled vacuoles to measure lipid bilayer mixing via fluorescence dequenching (26) and content mixing via nitrocefin hydrolysis (Fig. 1 B and C). Fusion inhibitors prevent both content and lipid mixing (Fig. 1 B and C, red and blue, respectively). Maltose-binding protein (MBP)-VopQ, but not MBP, inhibited both content and lipid mixing (Fig. 1 B and C, purple and green respectively). Coupled with its ability to inhibit Pho8p activation, β-lactamase reconstitution, and vacuolar lipid mixing, VopQ was validated as an authentic inhibitor of yeast vacuole fusion in vitro (Fig. 1).
VopQ Inhibits Docking and Trans-SNARE Complex Formation.
We next took advantage of the fact that fusion must occur in three biochemically distinct stages: (i) Priming, in which the SNARE chaperones Sec17p and Sec18p (α-SNAP and NSF protein, respectively) disassemble cis-SNARE complexes in an ATP-dependent manner (27); (ii) docking, which requires the activities of the Rab-GTPase Ypt7p and a multisubunit tethering complex, the HOPS complex, to tether vacuoles and enable the formation of the essential trans-SNARE complexes composed of Vam3p, Vti1p, Vam7p, and Nyv1p across opposing membranes (21, 28, 29); and (iii) fusion, in which trans-SNARE complexes catalyze the mixing of lipid bilayers, and luminal contents are exchanged (Fig. S2C) (30, 31). Fusion can be analyzed using a kinetic assay designed to determine the step at which the vacuole fusion reaction becomes resistant to particular subreaction inhibitors (32). The kinetic profiles for blocking the priming, docking, and fusion steps in vacuole fusion were exemplified by the addition of antibodies to Sec17p, Ypt7p, and Vam3p or ice, respectively, over the course of the Pho8p-dependent fusion reaction. The addition of the priming inhibitor α-Sec17p to the reaction after 20 min did not inhibit fusion, indicating that the priming step had passed (Fig. 2A, closed circles). Similarly, the addition of the docking inhibitors α-Ypt7p and α-Vam3p after 45 min did not inhibit fusion, indicating the completion of docking by this time (Fig. 2A, open diamond and black triangle, respectively). Placing reactions on ice inhibited the final stage of fusion, lipid mixing (Fig. 2A, open squares). We find that vacuoles become resistant to VopQ inhibition (Fig. 2A, closed squares) after resistance to the priming inhibitor α-Sec17p but before the final stage of lipid and content exchange, as measured by incubation on ice. Vacuoles display kinetic resistance to VopQ similar to that of the docking/tethering inhibitors α-Ypt7p and α-Vam3p, suggesting that VopQ may inhibit vacuole docking and the formation of trans-SNARE complexes.
Fig. 2.

VopQ inhibits trans-SNARE complex formation. (A) At 0-, 10-, 20-, and 45-min intervals, ALP vacuole fusion reaction was incubated on ice, or one of the following fusion inhibitors was added: α-Sec17p, α-Ypt7p, α-Vam3p, or 200 nM VopQ. After 90 min, fusion was measured and compared with the fusion of an uninhibited fusion reaction. Error bars indicate SD from the mean; n = 3. (B) The quantitative docking assay was performed in the presence of 500 nM MBP, 2.8 µM Gdi1p and 11.4 µM Gyp1-46, or 500 nM MBP-VopQ. At least 130 clusters from 10 random fields were scored for each condition. (C) Trans-SNARE assays were performed without or with the following inhibitors: ice incubation, 1 µM Gdi1p/1 µM Gyp1-46, or 200 nM VopQ. (D) ALP vacuole fusion reactions were performed in the absence or presence of 200 nM MBP-VopQ. Recombinant 50 nM Vam7p SNARE, 20 nM HOPS complex, or α-Vam3p was added where indicated.
To analyze further the effect of VopQ on the Rab GTPase- and SNARE-dependent tethering of yeast vacuoles, we used a visual “docking” assay that measures the number of vacuoles in clusters (33). The addition of MBP-VopQ or MBP does not disrupt the integrity of vacuoles (Fig. S2D). Incubation with MBP results in clusters of five or more vacuoles ∼30% of the time (Fig. 2B, black bars). Fewer vacuoles were observed in clusters upon treatment with a combination of GDP dissociation inhibitor (GDI) and Gyp1-46, which causes the release of Ypt7p and inhibits docking (Fig. 2B, white bars). Addition of MBP-VopQ closely mimicked GDI/Gyp1-46 activity (Fig. 2B, gray bars), indicating that VopQ inhibits the docking of yeast vacuoles.
To test the effect of VopQ on trans-SNARE complex formation during fusion, we measured the extent of trans-SNARE complex formation using an epitope-tagged Vam3p SNARE [calmodulin-binding protein (CBP)-Vam3p)] with its cognate vacuolar R-SNARE, Nyv1p (Fig. S2E) (21). Under normal fusion conditions, CBP-Vam3p and Nyv1p interact (Fig. 2C, lane 5). This SNARE interaction was strongly disrupted by incubation on ice (Fig. 2C, lane 6) and with GDI/Gyp1-46 (Fig. 2C, lane 7). Addition of VopQ prevented the formation of trans-SNARE complexes (Fig. 2C, lane 8), consistent with its distinct mechanism of vacuole fusion inhibition observed in the kinetic fusion assay (Fig. 2A).
The kinetic assay along with the docking and trans-SNARE assay suggests that VopQ inhibits fusion downstream of priming but upstream of trans-SNARE complex formation. In an attempt to reverse VopQ inhibition by increasing trans-SNARE complex formation, we provided an excess of the soluble SNARE Vam7p and the HOPS tethering complex; these proteins are known to bypass inhibitors that prevent SNARE complex assembly (29, 34). In the absence of HOPS and Vam7p, VopQ inhibits vacuole fusion similar to the addition of Vam3p antibody (Fig. 2D). Upon addition of HOPS or Vam7 alone, only modest effects on VopQ inhibition are observed. However, upon addition of HOPS and Vam7p, VopQ-mediated inhibition is reversed; this reversal remains sensitive to the fusion inhibitor α-Vam3p. Taken together, these data suggest that VopQ may hinder trans-SNARE complex assembly, thus inhibiting membrane fusion.
Vacuolar Acidification and Electrochemical Gradient Are Not Required for Fusion.
VopQ deacidifies the lumen of vacuoles through the rapid formation of an 18-Å channel in the membrane after direct interactions with Vo subunits of the V-ATPase (17). A recent publication by Coonrod et al. (13) shows that acidification of the yeast vacuole by the V-ATPase is absolutely required for vacuole fusion in vivo. Therefore, we tested if VopQ inhibits fusion simply through the collapse of the vacuole pH gradient.
Previously, the specific V-ATPase inhibitor bafilomycin was shown to be an inhibitor of yeast vacuole fusion in vitro using the ALP assay (24). We therefore compared VopQ and bafilomycin activities in ALP-dependent and -independent fusion assays and found that MBP-VopQ inhibited both the ALP (Fig. 3A, black bars) and β-lactamase fusion assays (Fig. 3A, white bars). In contrast, bafilomycin inhibited the ALP assay to ∼60% of the maximal fusion level (Fig. 3A, black bars) but had no effect on the activation of β-lactamase (Fig. 3A, white bars). Therefore, in contrast to VopQ, bafilomycin does inhibit vacuole fusion based on the ALP assay but does not inhibit vacuole fusion based on β-lactamase reconstitution.
Fig. 3.

A pH gradient is not required for vacuole fusion. (A) ALP and β-lactamase fusion reactions were performed in parallel, using standard inhibitors or 200 nM MBP, 200 nM MBP-VopQ, or 500 nM bafilomycin. Maximal fusion was defined as the extent of fusion of the no-inhibitor reaction; error bars indicate SD from the mean; n = 3. (B) Schematic diagram of the mechanism of action of the ionophores used. (C) Proton translocation activity of vacuoles was assayed in the absence or presence of ATP, α-Vam3p, 500 nM bafilomycin, 1 µM valinomycin, 1 µM nigericin, or combinations of ionophores and bafilomycin. Curves are representative of three independent experiments. (D) ALP and β-lactamase fusion reactions were performed in parallel, using a combination of ionophores at the concentrations used in C. Maximal fusion was defined as the no-inhibitor reaction. Error bars indicate SD from the mean; n = 3.
The possibility remained that the lumen of the vacuole retained acidification caused by V-ATPase activity before vacuole isolation and that this acidification was sufficient for driving homotypic fusion in vitro. Furthermore, the V-ATPase generates an electrochemical gradient (∆ψ) that also could be the driving force in vacuole fusion. To address the role of acidification and ∆ψ in yeast vacuole fusion, we used a pair of well-studied ionophores, valinomycin and nigericin, in combination with bafilomycin. Valinomycin is a neutral K+-specific ionophore, which transports K+ across the membrane down the concentration gradient (35, 36), and nigericin is an electroneutral K+/H+ antiporter (37). In the presence of V-ATPase activity, valinomycin and nigericin together equilibrate H+ and K+ ions to collapse both the electrochemical and pH gradients of the vacuole (Fig. 3B). Valinomycin and nigericin in combination with bafilomycin will collapse any preexisting electrochemical and pH gradient and inhibit any further V-ATPase–dependent translocation of H+.
First, we measured vacuole acidification by measuring the change in absorbance of acridine orange upon protonation (38). Similar to the no-inhibitor control, the α-Vam3p fusion inhibitor did not inhibit H+ translocation (Fig. 3C, black and blue curves). When added individually, bafilomycin and nigericin inhibited H+ translocation (Fig. 3C, orange and purple curves). Valinomycin did not completely inhibit proton translocation (Fig. 3C, pink curve), as expected for a K+-specific ionophore. However, valinomycin in combination with nigericin (Fig. 3C, green curve) inhibited luminal acidification. Likewise, no acidification was detected when a mixture of all three inhibitors was added (brown curve). These results indicate that these inhibitors are capable of neutralizing the yeast vacuole.
Next, we measured vacuole fusion in the presence of these inhibitors to study the role of electrochemical and pH gradients in fusion. As expected, bafilomycin inhibited the ALP but not the β-lactamase fusion assay (Fig. 3D). Combinations containing valinomycin and nigericin partially inhibit the ALP fusion assay (Fig. 3D, black bars) but do not inhibit the β-lactamase fusion assay (white bars). Vacuole fusion detected in the presence of these inhibitors is authentic, SNARE-dependent fusion, because the addition of α-Vam3p inhibited this fusion to baseline levels in both the ALP (Fig. S3A) and β-lactamase (Fig. S3B) assays. Therefore, vacuoles fuse normally in the absence of a pH gradient and ∆ψ in vitro. Furthermore, ALP-dependent fusion proceeds normally when KCl is completely replaced with the lysomotrophic, weakly basic NH4Cl (39) in the fusion reaction to neutralize the lumen of vacuoles (Fig. S3C); vacuoles do not acidify in the presence of NH4Cl, in contrast to KCl (Fig. S3 D and E).
To address the role of ∆ψ in fusion, we measured the ability of the vacuole to quench the potential-sensitive dye, Oxonol V. Dye quenching occurs as Oxonol V changes its membrane association in response to an inside-positive ∆ψ (40). In the absence of inhibitors, vacuoles rapidly quench Oxonol V fluorescence (Fig. S3F, black curve). Inhibiting V-ATPase activity eliminates most Oxonol V quenching (Fig. S3F, blue curve). Similar to bafilomycin, MBP-VopQ inhibits Oxonol V quenching (Fig. S3G, purple curve), showing that VopQ can inhibit the formation of ∆ψ during vacuole fusion. Finally, in a reconstituted proteoliposome fusion system that lacks the V-ATPase and depends solely on the activities of vacuolar SNAREs, the Rab GTPase Ypt7p, HOPS, and SNARE chaperones (Fig. S4A) (42), VopQ is completely unable to inhibit either content (Fig. S4B) or lipid (Fig. S4C) mixing. These data support the proposal that VopQ requires the V-ATPase to bind to the vacuole and inhibit fusion, because we observe that VopQ does not directly inhibit the “core” SNARE/Rab GTPase/HOPS membrane fusion machinery.
VopQ Does Not Inhibit Vacuole Fusion via Channel-Forming Activity.
With the knowledge that VopQ binds Vo subunits, we sought to identify mutants of VopQ that were defective in V-ATPase binding to characterize better the mechanism by which VopQ inhibits vacuole fusion. VopQ expression arrests yeast cell growth, and this effect appears to be mediated through the interaction of VopQ with Vma3p; vma3∆ strains are fully resistant to VopQ growth repression (Fig. S5A) (17, 18). Furthermore, VopQ-dependent vacuolar fragmentation in vivo also was completely dependent on the presence of Vma3p (Fig. S5B). Therefore, we hypothesized that VopQ mutants defective in V-ATPase binding would be less toxic upon expression in wild-type yeast. Using this reduced toxicity as a genetic selection, the galactose-inducible VopQ expression plasmid was transformed into an error-prone Escherichia coli mutator strain to create a library of random vopQ mutants. Sequencing of the mutants that did not arrest growth identified a plasmid containing a missense mutation in the vopQ coding sequence: a T-to-C transition at position 598. This mutation produced a mutant protein substituting a proline for serine at position 200 (VopQS200P). In contrast to wild-type VopQ, the mutant VopQS200P allowed yeast growth (Fig. 4A), despite being more highly expressed than the wild-type VopQ protein (Fig. S6A). Furthermore, the VopQS200P mutant, unlike the wild-type protein, did not cause extensive fragmentation of yeast vacuoles in vivo (Fig. 4B and Fig. S6B), suggesting that the mutant VopQS200P protein might be defective in its ability to inhibit homotypic vacuole fusion in vivo.
Fig. 4.
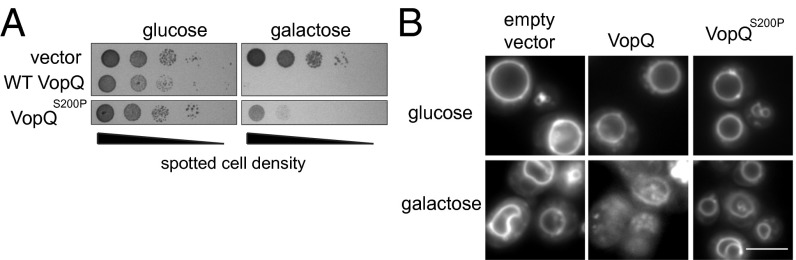
VopQS200P does not induce vacuole fragmentation. (A) Serial dilutions of yeast strain BY4742 harboring pRS413-Gal1, pRS413-Gal1-VopQ, or pRS413-Gal1-VopQS200P in Complete Supplement Mixture (CSM) medium lacking histidine, supplemented with either 2% glucose or 2% galactose. (B) Vacuolar morphology of the BY4742 strain harboring pRS413-Gal1, pRS413-Gal1-VopQ, or pRS413-Gal1-VopQS200P was visualized via FM4-64 staining. (Scale bar, 5 μm.)
To examine the activity of VopQS200P in comparison with wild type, recombinant proteins were purified (Fig. S7A) and assayed for both inhibition of vacuole fusion and vacuolar deacidification in vitro. Strikingly, recombinant VopQS200P protein did not inhibit the ALP (Fig. 5A) or β-lactamase (Fig. 5B) fusion assays at concentrations up to 1 µM. Even concentrations of VopQS200P up to 10 µM were no more effective at fusion inhibition than MBP alone (Fig. S7B). To study the S200P mutant for the known vacuolar channel-forming activity of VopQ (17), we assayed the ability of VopQS200P to inhibit vacuolar acidification in vitro. VopQS200P completely inhibited vacuolar acidification when added before the addition of ATP (Fig. 5C) and deacidified the lumen after the vacuoles had been fully acidified (Fig. S7C). To eliminate the possibility that MBP interfered with VopQS200P fusion inhibition, we used the channel-forming His-tagged version of VopQS200P (Fig. S7 D and E) and observed that it also is incapable of inhibiting vacuole fusion (Fig. S7F). Accordingly, VopQS200P still associates with yeast vacuoles in a V-ATPase–dependent manner, because vacuoles lacking the V-ATPase no longer bind VopQ or VopQS200P at pH 7.5 (Fig. S7G), confirming previous observations of VopQ–vacuole interactions (17). Therefore, VopQS200P still has some affinity for the V-ATPase, which is required for its channel-forming activity in vitro.
Fig. 5.

VopQS200P deacidifies the vacuole but does not inhibit fusion. (A) ALP vacuole fusion reactions or (B) β-lactamase fusion reactions were performed using standard inhibitors, MBP-VopQ, or MBP-VopQS200P. (C) Proton translocation activity was measured in the presence of MBP-VopQ or MBP-VopQS200P when proteins were added before the addition of ATP.
Although the rate of vacuolar deacidification induced by VopQS200P was slower than that induced by VopQ, the extents of deacidification were nearly identical over time and over a broad concentration range (Fig. S8). We used a dye-release assay that measures the release of a self-quenching concentration of carboxyfluorescein dye encapsulated in liposomes (41). Because these are protein-free liposomes, this assay tests for VopQ channel activity independent of its V-ATPase–dependent targeting activity (17). Interestingly, VopQS200P induced carboxyfluorescein release from protein-free liposomes (17) with rates identical to wild-type VopQ (Fig. S9A). Therefore, VopQS200P appears to retain the channel-forming and deacidification activities of VopQ on both vacuoles and liposomes but is unable to inhibit in vitro vacuole fusion.
Because we noticed a reduction in the ability of VopQS200P to induce the release of protons from yeast vacuoles, as compared with wild-type VopQ, but not in its ability to release carboxyfluorescein from protein-free liposomes, we hypothesized that VopQS200P has a lower affinity for the V-ATPase. Accordingly, VopQS200P-GFP does not localize to the yeast vacuole as was observed for VopQ-GFP (Fig. S9B). Furthermore, much lower levels of the Vo subunits Vph1p and Vma6p were found in coprecipitation with VopQS200P than with VopQ (Fig. S9C). Therefore, VopQS200P binds more weakly than VopQ to V-ATPase subunits but still retains its channel-forming activity on yeast vacuoles. The weak binding of VopQS200P is sufficient to neutralize the vacuole completely in vitro but is insufficient to inhibit vacuolar fusion.
Discussion
VopQ is a 53-kDa bacterial effector protein that binds to the conserved eukaryotic V-ATPase and forms a voltage-gated channel in the vacuolar membrane to disrupt ion homeostasis (17). We now show that nanomolar amounts of VopQ inhibit the homotypic fusion of yeast vacuoles, a biochemical model of Rab GTPase- and SNARE-dependent membrane fusion. Furthermore, we identified a VopQ mutant, VopQS200P, that retained V-ATPase binding and membrane channel-forming activities but no longer inhibited vacuole fusion in vitro. Coprecipitation assays between VopQS200P and Vo domain subunits show a weaker overall interaction than with the wild-type VopQ protein, and thus we speculate that the direct interaction of VopQ with the Vo membrane sector is required to inhibit vacuole fusion. Importantly, the use of VopQ as a channel-forming reagent for the study of membrane fusion regulation shows that yeast vacuoles do not require active acidification or ∆ψ for homotypic fusion in vitro.
This finding is in contrast to recent work showing that yeast vacuole fusion absolutely requires acidification in vivo, as was measured by protein maturation in an elegant in vivo protein-localization assay during haploid cell mating and subsequent vacuolar membrane fusion (13). However, both these assays were completely dependent upon the localization and maturation of the vacuolar ALP. The physiological readout of these assays may be sensitive to vesicular ion concentrations and therefore could be completely independent of physiological membrane fusion. Consistent with this theory, we show that the yeast ALP-dependent content-mixing assay is sensitive to a number of ionophores, although an alternative β-lactamase content-mixing assay is unaffected by these reagents. Furthermore, the previous study showed that a mutation in Vph1p (Vo, a subunit) that disrupts the ability of the V-ATPase to translocate protons, Vph1pR735Q, was unable to support the fusion of intracellular vacuoles (13). This finding also was in contrast to studies in Drosophila, which show that a mutant v100 a1 subunit with the corresponding arginine disrupted (v100R755A) restored vesicular trafficking and synaptic transmission in v100-null mutant flies, confirming an acidification-independent role of the a1 subunit in membrane fusion events (9). Further research demonstrated that Ca2+-calmodulin interactions with the a1 subunit positively regulate the formation of some SNARE complexes at the synapse in an acidification-independent fashion (14), thereby highlighting an important acidification-independent role of Vo upstream of SNARE complex formation.
Previous studies have indicated that the role of the Vo sector in vacuole fusion is required after the formation of the trans-SNARE complexes but before content mixing has occurred (4). In this study, when VopQ binds to Vo, we see inhibition of the trans-SNARE complex formation during homotypic vacuole fusion in vitro. These data could indicate two distinct roles for the Vo sector in membrane fusion, each implicating fusion-promoting activities upstream and downstream of SNARE pairing. Furthermore, by binding a large protein (VopQ) to the Vo channel, we may be imposing a fusion block distinct from other studies. It is important to note that VopQ does not inhibit V-ATPase activity (17); this lack of inhibition could account for some of the discrepancies in SNARE complex staging between this study and others.
Most published studies measuring the role of the V-ATPase in acidification and fusion events relied on mutant derivatives of the V-ATPase Vo domain, making a direct link of V-ATPase activities to membrane fusion regulation difficult. By using a specific Vo-binding, channel-forming protein from a pathogenic bacterium, we now separate the roles of acidification and ∆ψ formation in regulating SNARE-dependent membrane fusion. Yeast vacuoles unable to acidify because of inhibitors or the activity of VopQS200P protein are perfectly capable of fusing in vitro and in vivo, indicating that luminal acidification and membrane potential are not necessary for fusion. It remains possible, however, that additional unknown factors require acidification for efficient in vivo vacuolar fusion and that purification of vacuolar membranes removes this biochemical requirement. Many studies using reconstituted proteoliposomes containing cognate SNAREs, Rab GTPase, and tethering factors confirm that the V-ATPase (or acidification and ∆ψ) is not explicitly required to drive fusion (42–44); we see that VopQ is incapable of inhibiting fusion in these systems.
We therefore propose that the physical interaction of VopQ with the Vo domain is responsible for inhibiting membrane fusion in a V-ATPase–dependent manner, distinct from its deacidification activity. The Vo domain may simply serve as a receptor for VopQ, and VopQ could inhibit docking of vacuoles via several possible mechanisms including simple steric hindrance of vacuolar docking, uncoupling Vam3p SNARE–Vo interactions during fusion (12), or inhibiting a previously uncharacterized activity for Vo. Interestingly, it has been shown that the lack of the V-ATPase Vo a1 subunit induces an accumulation of autophagic vesicles without altering lysosomal acidity (45), and it has been proposed that functional V-ATPase may be required for membrane–membrane contacts between yeast autophagosomes and vacuoles (11). Given that VopQ also was shown to induce an accumulation of autophagic vesicles in HeLa cells (17), it is possible that this VopQ–V-ATPase interaction also may induce autophagosome accumulation by directly blocking autophagosome–lysosome fusion. Studies to address these possibilities are currently underway and may provide important insights into the mechanisms by which Vo domains or VopQ may regulate some intracellular membrane fusion events between V-ATPase–containing membranes. To the best of our knowledge, VopQ is the first bacterial V-ATPase–binding effector shown to inhibit membrane fusion and, as observed with other virulence factors, provides valuable insight into eukaryotic cellular signaling (46).
Materials and Methods
Detailed information on yeast strains, reagent preparation, yeast vacuole staining, vacuole isolation, vacuole fusion and docking assays, lipid-mixing assay, proton translocation assay, lysis assay, Oxonol V assay, liposome dye-release assay, trans-SNARE assay, and random mutagenesis is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank W. Wickner, C. Ungermann, R. Fratti, R. Hiesinger, and members of the K.O. laboratory for insightful discussions and supply of reagents. K.O. and A.S. are supported by National Institute of Allergy and Infectious Diseases (NIAID) Grants R01-AI056404 and R01-AI087808 and by Grant I-1561 from the Welch Foundation. A.S. is a Howard Hughes Medical Institute Med into Grad Scholar. K.O. is a Burroughs Wellcome Investigator in Pathogenesis of Infectious Disease and a W. W. Caruth Jr. Biomedical Scholar. V.J.S. is supported by University of Georgia Startup Funds and NIAID Grant R01-AI100913.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 8.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413764111/-/DCSupplemental.
References
- 1.Hiesinger PR, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121(4):607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wickner W. Membrane fusion: Five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol. 2010;26:115–136. doi: 10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong J. Yeast vacuoles: More than a model lysosome. Trends Cell Biol. 2010;20(10):580–585. doi: 10.1016/j.tcb.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Strasser B, Iwaszkiewicz J, Michielin O, Mayer A. The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. EMBO J. 2011;30(20):4126–4141. doi: 10.1038/emboj.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol. 2003;162(2):211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forgac M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8(11):917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 7.Nishi T, Forgac M. The vacuolar (H+)-ATPases—nature’s most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3(2):94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 8.van der Goot FG, Gruenberg J. Intra-endosomal membrane traffic. Trends Cell Biol. 2006;16(10):514–521. doi: 10.1016/j.tcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Williamson WR, Wang D, Haberman AS, Hiesinger PR. A dual function of V0-ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J Cell Biol. 2010;189(5):885–899. doi: 10.1083/jcb.201003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Giovanni J, et al. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron. 2010;67(2):268–279. doi: 10.1016/j.neuron.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 11.Mijaljica D, Prescott M, Devenish RJ. V-ATPase engagement in autophagic processes. Autophagy. 2011;7(6):666–668. doi: 10.4161/auto.7.6.15812. [DOI] [PubMed] [Google Scholar]
- 12.Peters C, et al. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409(6820):581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 13.Coonrod EM, et al. Homotypic vacuole fusion in yeast requires organelle acidification and not the V-ATPase membrane domain. Dev Cell. 2013;27(4):462–468. doi: 10.1016/j.devcel.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, et al. Ca2+-Calmodulin regulates SNARE assembly and spontaneous neurotransmitter release via v-ATPase subunit V0a1. J Cell Biol. 2014;205(1):21–31. doi: 10.1083/jcb.201312109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Far O, Seagar M. A role for V-ATPase subunits in synaptic vesicle fusion? J Neurochem. 2011;117(4):603–612. doi: 10.1111/j.1471-4159.2011.07234.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Hiesinger PR. The vesicular ATPase: A missing link between acidification and exocytosis. J Cell Biol. 2013;203(2):171–173. doi: 10.1083/jcb.201309130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sreelatha A, et al. Vibrio effector protein, VopQ, forms a lysosomal gated channel that disrupts host ion homeostasis and autophagic flux. Proc Natl Acad Sci USA. 2013;110(28):11559–11564. doi: 10.1073/pnas.1307032110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuda S, Okada N, Kodama T, Honda T, Iida T. A cytotoxic type III secretion effector of Vibrio parahaemolyticus targets vacuolar H+-ATPase subunit c and ruptures host cell lysosomes. PLoS Pathog. 2012;8(7):e1002803. doi: 10.1371/journal.ppat.1002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nickerson DP, Brett CL, Merz AJ. Vps-C complexes: Gatekeepers of endolysosomal traffic. Curr Opin Cell Biol. 2009;21(4):543–551. doi: 10.1016/j.ceb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J. 1995;14(21):5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins KM, Wickner WT. Trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc Natl Acad Sci USA. 2007;104(21):8755–8760. doi: 10.1073/pnas.0702290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starai VJ, Jun Y, Wickner W. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc Natl Acad Sci USA. 2007;104(34):13551–13558. doi: 10.1073/pnas.0704741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorensen SO, van den Hazel HB, Kielland-Brandt MC, Winther JR. pH-dependent processing of yeast procarboxypeptidase Y by proteinase A in vivo and in vitro. Eur J Biochem. 1994;220(1):19–27. doi: 10.1111/j.1432-1033.1994.tb18594.x. [DOI] [PubMed] [Google Scholar]
- 24.Ungermann C, Wickner W, Xu Z. Vacuole acidification is required for trans-SNARE pairing, LMA1 release, and homotypic fusion. Proc Natl Acad Sci USA. 1999;96(20):11194–11199. doi: 10.1073/pnas.96.20.11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jun Y, Wickner W. Assays of vacuole fusion resolve the stages of docking, lipid mixing, and content mixing. Proc Natl Acad Sci USA. 2007;104(32):13010–13015. doi: 10.1073/pnas.0700970104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reese C, Heise F, Mayer A. Trans-SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature. 2005;436(7049):410–414. doi: 10.1038/nature03722. [DOI] [PubMed] [Google Scholar]
- 27.Ungermann C, Nichols BJ, Pelham HR, Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol. 1998;140(1):61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer A, Wickner W. Docking of yeast vacuoles is catalyzed by the Ras-like GTPase Ypt7p after symmetric priming by Sec18p (NSF) J Cell Biol. 1997;136(2):307–317. doi: 10.1083/jcb.136.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25(8):1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merz AJ, Wickner WT. Trans-SNARE interactions elicit Ca2+ efflux from the yeast vacuole lumen. J Cell Biol. 2004;164(2):195–206. doi: 10.1083/jcb.200310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters C, Mayer A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature. 1998;396(6711):575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 32.Ungermann C, Wickner W. Vam7p, a vacuolar SNAP-25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO J. 1998;17(12):3269–3276. doi: 10.1093/emboj/17.12.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol. 2004;167(6):1087–1098. doi: 10.1083/jcb.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorngren N, Collins KM, Fratti RA, Wickner W, Merz AJ. A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 2004;23(14):2765–2776. doi: 10.1038/sj.emboj.7600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreoli TE, Tieffenberg M, Tosteson DC. The effect of valinomycin on the ionic permeability of thin lipid membranes. J Gen Physiol. 1967;50(11):2527–2545. doi: 10.1085/jgp.50.11.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tosteson DC, Cook P, Andreoli T, Tieffenberg M. The effect of valinomycin on potassium and sodium permeability of HK and LK sheep red cells. J Gen Physiol. 1967;50(11):2513–2525. doi: 10.1085/jgp.50.11.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pressman BC. Biological applications of ionophores. Annu Rev Biochem. 1976;45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- 38.Perzov N, Padler-Karavani V, Nelson H, Nelson N. Characterization of yeast V-ATPase mutants lacking Vph1p or Stv1p and the effect on endocytosis. J Exp Biol. 2002;205(Pt 9):1209–1219. doi: 10.1242/jeb.205.9.1209. [DOI] [PubMed] [Google Scholar]
- 39.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 40.Cooper CE, Bruce D, Nicholls P. Use of oxonol V as a probe of membrane potential in proteoliposomes containing cytochrome oxidase in the submitochondrial orientation. Biochemistry. 1990;29(16):3859–3865. doi: 10.1021/bi00468a009. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, et al. Requirements for the formation of membrane pores by the reovirus myristoylated μ1N peptide. J Virol. 2009;83(14):7004–7014. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zucchi PC, Zick M. Membrane fusion catalyzed by a Rab, SNAREs, and SNARE chaperones is accompanied by enhanced permeability to small molecules and by lysis. Mol Biol Cell. 2011;22(23):4635–4646. doi: 10.1091/mbc.E11-08-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mima J, Wickner W. Phosphoinositides and SNARE chaperones synergistically assemble and remodel SNARE complexes for membrane fusion. Proc Natl Acad Sci USA. 2009;106(38):16191–16196. doi: 10.1073/pnas.0908694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zick M, Stroupe C, Orr A, Douville D, Wickner WT. Membranes linked by trans-SNARE complexes require lipids prone to non-bilayer structure for progression to fusion. eLife. 2014;3:e01879. doi: 10.7554/eLife.01879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsin IL, et al. Inhibition of lysosome degradation on autophagosome formation and responses to GMI, an immunomodulatory protein from Ganoderma microsporum. Br J Pharmacol. 2012;167(6):1287–1300. doi: 10.1111/j.1476-5381.2012.02073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alto NM, Orth K. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol. 2012;4(9):a006114. doi: 10.1101/cshperspect.a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.