

Significance
We demonstrate that histone deacetylase (HDAC) inhibition can elicit changes in DNA methylation in Huntington’s disease (HD) human fibroblasts, as well as in sperm from HD transgenic mice, in association with DNA methylation-related gene expression changes. We suggest that alterations in sperm DNA methylation lead to transgenerational effects, and, accordingly, we show that first filial generation (F1) offspring of HDAC inhibitor-treated male HD transgenic mice show improved HD disease phenotypes compared with F1 offspring from vehicle-treated male HD transgenic mice. These findings have significant implications for human health because they enforce the concept that ancestral drug exposure may be a major molecular factor that can affect disease phenotypes, yet in a positive manner. Further, we implicate Lys (K)-specific demethylase 5d expression in this phenomenon.
Keywords: epigenetic, transgenerational, histone, neurodegenerative, therapeutic
Abstract
Increasing evidence has demonstrated that epigenetic factors can profoundly influence gene expression and, in turn, influence resistance or susceptibility to disease. Epigenetic drugs, such as histone deacetylase (HDAC) inhibitors, are finding their way into clinical practice, although their exact mechanisms of action are unclear. To identify mechanisms associated with HDAC inhibition, we performed microarray analysis on brain and muscle samples treated with the HDAC1/3-targeting inhibitor, HDACi 4b. Pathways analyses of microarray datasets implicate DNA methylation as significantly associated with HDAC inhibition. Further assessment of DNA methylation changes elicited by HDACi 4b in human fibroblasts from normal controls and patients with Huntington’s disease (HD) using the Infinium HumanMethylation450 BeadChip revealed a limited, but overlapping, subset of methylated CpG sites that were altered by HDAC inhibition in both normal and HD cells. Among the altered loci of Y chromosome-linked genes, KDM5D, which encodes Lys (K)-specific demethylase 5D, showed increased methylation at several CpG sites in both normal and HD cells, as well as in DNA isolated from sperm from drug-treated male mice. Further, we demonstrate that first filial generation (F1) offspring from drug-treated male HD transgenic mice show significantly improved HD disease phenotypes compared with F1 offspring from vehicle-treated male HD transgenic mice, in association with increased Kdm5d expression, and decreased histone H3 Lys4 (K4) (H3K4) methylation in the CNS of male offspring. Additionally, we show that overexpression of Kdm5d in mutant HD striatal cells significantly improves metabolic deficits. These findings indicate that HDAC inhibitors can elicit transgenerational effects, via cross-talk between different epigenetic mechanisms, to have an impact on disease phenotypes in a beneficial manner.
Epigenetic alterations and transcriptional dysregulation have emerged as common pathological mechanisms in many neurological disorders (1, 2). Accordingly, therapeutic approaches that target gene expression represent an encouraging new avenue of exploration for these disorders. Numerous phase I and II clinical trials using histone deacetylase (HDAC) inhibitors are ongoing. Novel HDAC inhibitors with improved safety, brain penetration, potency, and selectivity have greatly advanced the therapeutic potential of these agents for a wide range of noncancer indications, including neurodegenerative disease, psychiatric disorders, addiction, and inflammatory disease states (3–6).
Class I-specific, benzamide-type HDAC inhibitors have been developed as a therapeutic approach for Friedreich’s ataxia (7, 8), and we have previously tested these inhibitors for their potential benefit in Huntington’s disease (HD). Our studies have shown that inhibitors selectively targeting HDAC1 and HDAC3 are beneficial in several different HD model systems (9–11). In particular, in vivo studies have shown that pharmacological inhibition of HDAC1 and HDAC3 led to improved disease-associated body weight loss, motor dysfunction, and cognitive decline in two different HD mouse models (9, 11). These findings are consistent with other studies showing beneficial effects of broadly acting HDAC inhibitors in HD mouse models (12–14). However, the mechanisms by which these compounds are imparting benefit in disease models are unclear.
In general, HDAC inhibitors are known to elevate acetylation of histones, both globally and at specific loci, resulting in a more relaxed chromatin structure that facilitates gene transcription. However, several microarray studies have demonstrated that HDAC inhibitors can cause both up- and down-regulation of gene expression patterns (11, 15), suggesting that HDAC inhibition may alter the expression of other regulatory enzymes and/or cofactors, which subsequently act as activators or repressors of gene activity. Accumulating evidence suggests that many HDACs, including class I HDACs, can also deacetylate nonhistone proteins (16), which may contribute to their beneficial properties.
In this study, we used gene expression microarrays to identify potential mechanisms of action of the HDAC1/3-targeting inhibitor, HDACi 4b. We find that HDAC inhibition alters the expression of several DNA methylation-related genes and, further, that HDAC inhibition could induce DNA methylation changes on a genome-wide scale. The potential for HDAC inhibitors to elicit alterations in DNA methylation is highly relevant because DNA methylation is an epigenetic modification that can be inherited in the germ line and can lead to transgenerational effects in subsequent progeny; accordingly, we find that HD transgenic offspring of HDACi 4b-treated male HD transgenic mice exhibit improved disease phenotypes compared with HD transgenic offspring from vehicle-treated male HD transgenic mice. In addition, we demonstrate a role for the gene lysine (K)-specific demethylase 5d (Kdm5d) in this phenomenon, further implicating cross-talk among different epigenetic mechanisms. Identifying the relationship(s) between chromatin and DNA methylation is imperative for understanding the programming of gene expression within the epigenome and mechanisms of epigenetic transgenerational effects, as well as for revealing the potential impact of the clinical use of HDAC inhibitors.
Results
We have previously shown that HDAC1/3-targeting HDAC inhibitors show therapeutic efficacy in HD mouse models, in association with elevated histone acetylation levels (9–11). To identify gene expression changes associated with altered histone acetylation levels, we performed microarray analysis on skeletal muscle samples from N171-82Q HD transgenic mice treated with the HDAC1/3-targeting compound HDACi 4b and reanalyzed microarray data from brain samples of HDACi 4b-treated R6/2 transgenic mice from our earlier studies (11). We used skeletal muscle in light of the fact that many reports have implicated skeletal muscle dysfunction as an important factor in motor dysfunction, both in human and animal HD models (17–20). Using two pathway analysis programs, Ingenuity Systems Pathway Analysis (Ingenuity Systems) and Database for Annotation, Visualization, and Integrated Discovery (DAVID), we found that gene expression alterations in response to HDACi 4b treatment were significantly associated with DNA methylation and chromatin organization (Tables S1 and S2). In fact, the category “DNA methylation and transcriptional repression signaling” was the third most significantly associated canonical pathway related to drug treatment, with 15 genes showing altered expression related to this pathway (Table S1). These 15 genes include two genes encoding major DNA methyltransferase enzymes: DNA methyltransferase 1 (Dnmt1) and DNA methyltransferase 3a (Dnmt3a); several genes encoding putative DNA demethylase enzymes: methyl-CpG binding domain proteins 3–5 (Mbds3–5), poly (ADP ribose) polymerase 1 (Parp1), and growth arrest and DNA damage-inducible 45 beta (Gadd45b) (21–24); and genes encoding proteins comprising two well-characterized class I HDAC-containing corepressor complexes: the nucleosome remodeling and histone deacetylase (NuRD) and Sin3 complexes (25). The fact that so many genes encoding proteins in these complexes were altered by HDACi 4b treatment strongly suggests a relationship between HDAC inhibition and alterations in DNA methylation and chromatin structure. The changes induced by HDACi 4b include both up-regulated and down-regulated genes, suggesting an overall multifaceted regulation of these complexes. We validated the expression differences for some of these genes in muscle and brain samples from HDACi 4b-treated WT and N171-82Q transgenic mice using real-time PCR analysis (Table 1). The genes most significantly altered in expression were those encoding enzymes related to the regulation of DNA methylation: Dnmt1, Dnm3a, Gadd45b, Parp1, and RING finger protein 4 (Rnf4) (Table 1). Increased expression of these genes could lead to either increased or decreased methylation of DNA.
Table 1.
qPCR validation of DNA methylation-related genes altered by HDACi 4b treatment in cortex, striatum, and muscle
| Symbol | Entrez gene name | Cortex | Striatum | Muscle | |||
| Fold change | P value | Fold change | P value | Fold change | P value | ||
| N171-82Q transgenic mice | |||||||
| Dnmt1 | DNA (cytosine-5-)-methyltransferase 1 | 0.91 | 0.140 | 0.90 | 0.143 | 1.61* | 0.025 |
| Dnmt3a | DNA (cytosine-5-)-methyltransferase 3a | 0.94 | 0.330 | 1.40* | 0.032 | 1.23* | 0.011 |
| Gadd45b | Growth arrest and DNA-damage–inducible 45 beta | 1.44* | 0.049 | 1.01 | 0.460 | 0.96 | 0.338 |
| Hdac1 | Histone deacetylase 1 | 0.81* | 0.040 | 0.57* | 0.011 | 0.81* | 0.049 |
| Hdac2 | Histone deacetylase 2 | 0.80 | 0.210 | 0.97 | 0.441 | 1.11 | 0.160 |
| Hdac3 | Histone deacetylase 3 | 0.88 | 0.310 | 0.44* | 0.031 | 0.98 | 0.437 |
| Mbd3 | Methyl-CpG binding domain protein 3 | 1.57** | 0.006 | 0.90 | 0.135 | 1.11 | 0.110 |
| Mecp2 | Methyl CpG binding protein 2 | 0.97 | 0.890 | 1.05 | 0.360 | 1.21* | 0.047 |
| Parp1 | Poly (ADP ribose) polymerase family member 1 | 1.41* | 0.007 | 1.22 | 0.107 | 0.92 | 0.186 |
| Rnf4 | RING finger protein 4 | 1.11* | 0.030 | 1.24* | 0.035 | 0.87* | 0.049 |
| WT mice | |||||||
| Dnmt1 | DNA (cytosine-5-)-methyltransferase 1 | 1.00 | 0.48 | 0.98 | 0.43 | 0.79 | 0.13 |
| Dnmt3a | DNA (cytosine-5-)-methyltransferase 3a | 0.78 | 0.16 | 1.37* | 0.03 | 0.90 | 0.21 |
| Gadd45b | Growth arrest and DNA-damage–inducible 45 beta | 1.11 | 0.12 | 1.32* | 0.03 | 0.92 | 0.31 |
| Hdac1 | Histone deacetylase 1 | 0.90 | 0.29 | 0.97 | 0.40 | 0.98 | 0.45 |
| Hdac2 | Histone deacetylase 2 | 0.98 | 0.46 | 1.09 | 0.26 | 1.12 | 0.12 |
| Hdac3 | Histone deacetylase 3 | 0.90 | 0.31 | 1.26* | 0.04 | 1.10 | 0.25 |
| Mbd3 | Methyl-CpG binding domain protein 3 | 0.74** | 0.01 | 0.94 | 0.31 | 0.91 | 0.33 |
| Mecp2 | Methyl CpG binding protein 2 | 0.85 | 0.17 | 1.08 | 0.33 | 0.78 | 0.08 |
| Parp1 | Poly (ADP ribose) polymerase family, member 1 | 1.25* | 0.03 | 0.99 | 0.48 | 0.95 | 0.35 |
| Rnf4 | RING finger protein 4 | 1.07 | 0.20 | 1.11 | 0.28 | 0.86 | 0.13 |
Asterisks indicate fold change that was significantly different, as determined by Student t tests: *P < 0.05, **P < 0.01.
To test the direct effects of HDAC inhibition on DNA methylation genome-wide, we measured the locus-specific DNA methylation patterns in cultured human fibroblasts from normal controls and patients with HD using the Infinium HumanMethylation450 BeadChip (Illumina, Inc.), which interrogates DNA methylation at >450,000 CpG sites associated with both coding and noncoding genes. First, comparing the HD vs. control condition, we found 16,431 positions increased in methylation in HD cells relative to control cells and 8,118 positions decreased in methylation, using a multiple hypothesis corrected P value of 0.001 and a difference in methylation cutoff of beta change of >|0.2|. These findings are similar to recently published work on striatal cells carrying polyglutamine-expanded HTT (STHdh) that showed extensive DNA methylation changes in mutant STHdhQ111 cells compared with normal STHdhQ7 striatal cells (26). Also consistent with these previous studies was that a large majority of the DNA methylation changes occurred in CpG-poor regions of the genome, compared with CpG-rich regions (Fig. S1). Using the program Genomic Regions Enrichment of Annotations Tool (GREAT), which associates input genomic regions with their putative target genes and then assigns significant functional annotations (27), we found that genes associated with methylation changes at either CpG-poor or CpG-rich regions were enriched for developmental processes, cell differentiation, gene transcription, chromatin binding, and immune signaling (Table S3), similar to previous reports (26).
We next identified methylated positions that were regulated by HDACi 4b exposure in human fibroblast cells from both normal controls and patients with HD. All differentially methylated positions (DMPs) were associated with one or two annotated genes, and 87% of the DMPs were within 500 kb of an annotated transcription start site. A greater proportion of genomic sites were found to be decreased in methylation by HDACi 4b treatment in HD cells compared with normal cells (62% and 48% for HD and normal cells, respectively) (Fig. 1). GREAT analysis found that HDACi 4b-induced DMPs were significantly associated with the biological processes of spermatogenesis, the multicellular organismal reproductive process, and antigen presentation and processing (Table 2). In addition, both sets returned a significant relationship to the human phenotype ontology enrichment category of Y-linked inheritance and gonosomal inheritance (Table 2). From the human dataset of DMPs, 35.2% and 42.3% of the human genomic coordinates (hg19) in the WT and HD cells, respectively, had a corresponding region in the mouse genome (mm10). Among the differentially methylated genes that showed a corresponding lift-over to mouse were Kdm5d (also known as Jarid1d or Smcy), sex-determining region of Chr Y (Sry), eukaryotic translation initiation factor 1A, Y-linked (Eif1ay), and synaptoporin (Synpr). HDACi 4b did not alter the expression of any known imprinted genes, as determined by cross-referencing the Geneimprint database (geneimprint.com/).
Fig. 1.
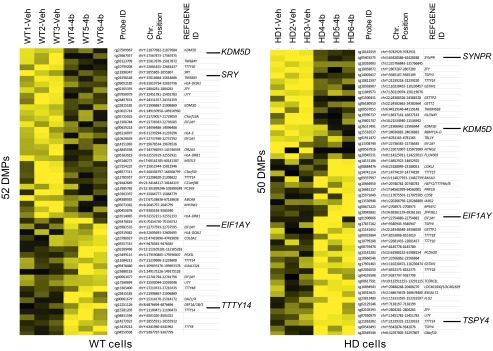
Differential DNA methylation due to HDAC inhibitor treatment. Data for CpG sites differentially methylated between HDACi 4b-treated and vehicle-treated (delta beta > 0.2) in normal and HD human fibroblasts on the 27K DNA methylation array. Yellow denotes a decrease in methylation due to HDACi 4b treatment; black denotes an increase in methylation. The total number of DMPs in each cell type is shown along the y axis. Chr., chromosome, SRY, sex-determining region of Chr Y; SYNPR, synaptoporin; TSPY4, testis-specific protein, Y-linked 1; TTTY14, testis-specific transcript, Y-linked 14.
Table 2.
GREAT analysis of HDACi 4b-induced DNA methylation differences in human HD fibroblasts
| Term | Binomial P value | Binomial FDR Q value | Binomial FE |
| Top enriched GO biological process terms | |||
| Spermatogenesis | 1.43E-11 | 3.14E-08 | 8.84889 |
| Sexual reproduction | 2.29E-10 | 4.02E-07 | 6.123311 |
| Gamete generation | 2.56E-09 | 2.49E-06 | 6.19334 |
| Gonadal mesoderm development | 6.54E-09 | 5.73E-06 | 79.2003 |
| Multicellular organismal reproductive process | 2.29E-07 | 1.18E-04 | 4.483269 |
| Antigen presentation via MHC class II | 2.53E-07 | 1.23E-04 | 77.53082 |
| Reproductive process | 1.37E-06 | 5.46E-04 | 3.301294 |
| Reproduction | 1.43E-06 | 5.43E-04 | 3.292752 |
| Antigen processing and presentation | 1.22E-05 | 3.81E-03 | 16.96118 |
| Mesoderm development | 7.11E-04 | 1.00E-01 | 7.069603 |
| Sex differentiation | 3.28E-03 | 3.55E-01 | 3.567114 |
| Gonad development | 4.51E-03 | 4.44E-01 | 3.853899 |
| Development of primary sexual characteristics | 8.14E-03 | 6.99E-01 | 3.402624 |
| Reproductive structure development | 9.96E-03 | 8.16E-01 | 3.257634 |
| Developmental process involved in reproduction | 1.80E-02 | 1.00E+00 | 2.577171 |
| Immune system process | 8.69E-02 | 1.00E+00 | 1.6056 |
| Top enriched human phenotype terms | |||
| Y-linked inheritance | 2.90E-19 | 1.74E-15 | 143.0223 |
| Gonosomal inheritance | 4.64E-08 | 1.39E-04 | 10.1296 |
FDR, false discovery rate; FE, fold enrichment.
We focused on one of these Y-linked genes, Kdm5d, further testing whether HDACi 4b could alter its methylation status in mouse sperm DNA from N171-82Q transgenic mice. After treatment of male mice for 1 mo with HDACi 4b, using methylated DNA immunoprecipitation in combination with real-time quantitative PCR (qPCR), we demonstrated increases in methylation elicited by HDACi 4b treatment at three different Kdm5d loci in DNA from mouse sperm (Fig. 2).
Fig. 2.
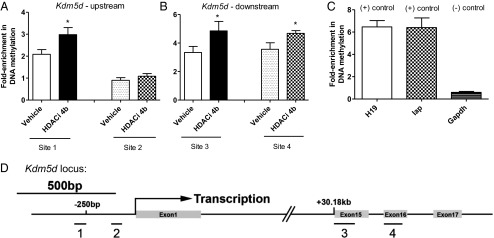
Effect of HDACi 4b on DNA methylation at the Kdm5d locus in mouse sperm DNA. The DNA methylation array revealed two regions increased in methylation in the Kdm5d locus with HDACi 4b treatment. Two pairs of primers were designed to verify each region in ±100 bp of the methyl-array probe site. Sites 1 and 2 are designed to detect a promoter region of 250 bp upstream of the Kdm5d transcription start site. Sites 3 and 4 are designed to detect a region 30.18 kb downstream of the Kdm5d transcription start site. (A) HDACi 4b treatment significantly increased the methylation signals in the promoter “site 1” region of Kdm5d but not in the “site 2” region. (B) Both “sites 3 and 4” increased the methylation signal with HDACi 4b treatment. (C) Promoter CpG region of glyceraldehyde-3-phosphate dehydrogenase (Gapdh) is a negative control, and Intracisternal A-particle (IAP) and H19 imprinted maternally expressed transcript (H19) are maternally imprinted positive controls for DNA methylation. All relative methylation was normalized to the methylation level of the Gapdh promoter region. Statistical differences were determined by a two-tailed Student t test: *P < 0.05 vs. vehicle group (n = 6).
DNA methylation is an epigenetic modification that can be inherited in the germ line and can lead to transgenerational effects on subsequent progeny. Therefore, we compared behavioral phenotypes in offspring from HDACi 4b- and vehicle-treated male HD mice between 14 and 16 wk of age. We found that first filial generation (F1) transgenic offspring from HDACi 4b-treated male transgenic F0 mice show significantly improved motor function in the rotarod test, improved cognition in the alternating T-maze test, and increased motor function in the open-field activity test (Fig. 3 B–E), compared with F1 transgenic offspring from vehicle-treated male transgenic F0 mice. Interestingly, these improvements were more pronounced in male transgenic mice compared with female transgenic mice (Fig. 3 C–E). No difference in the body weight of either sex was observed (Fig. 3A). No differences in CAG repeat length were observed in HDACi 4b-exposed offspring compared with their parents (82.4 ± 1.1 vs. 82.0 ± 1 CAG repeats). We further tested for transgenerational effects of another inhibitor, RGFP966 (Repligen Corporation), that is structurally related to HDACi 4b but selectively targets the HDAC3 subtype (6). RGFP966 treatment beginning at 8 wk of age showed direct beneficial effects on HD disease phenotypes in N171-82Q transgenic mice (F0 generation measured at 12–16 wk of age) (Fig. S2). These results are highly similar to our previous studies using HDACi 4b (9). However, we did not observe any significant transgenerational effects of RGFP966 on the F1 generation of N171-82Q transgenic mice (i.e., the offspring of RGFP966-treated fathers) (Fig. S3).
Fig. 3.
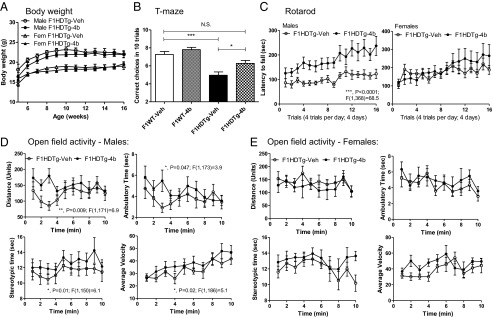
Comparisons of disease phenotypes in offspring of male N171-82Q transgenic mice treated with HDACi 4b (F1HDTg-4b) relative to offspring of N171-82Q transgenic mice treated with vehicle (F1HDTg-veh). (A) Effects of HDACi 4b exposure on the body weights of N171-82Q transgenic mice. (B) Rotarod performance of offspring of vehicle- and HDACi 4b-exposed N171-82Q transgenic mice. Males and females are shown separately. Two-way ANOVA revealed significant differences between male F1HDTg-4b mice compared with male F1HDTg-veh mice, whereas female mice did not show statistically significant differences. N.S., not significant. (C) Number of correct choices of first 10 trials in the alternating T-maze test. One-way ANOVA revealed significant differences between vehicle-exposed WT and N171-82Q transgenic mice (**P < 0.001) and a significant difference between vehicle-exposed and HDACi 4b-exposed N171-82Q mice (*P < 0.05). Both male and female mice were combined in this assay. (D and E) Effects of HDACi 4b exposure on open-field activity of N171-82Q transgenic mice over a 10-min test period. Two-way ANOVA revealed significant differences between HDACi 4b-exposed and vehicle-exposed N171-82Q transgenic mice for distance, ambulatory time, stereotypic time, and average velocity only in male mice, as indicated.
We next tested Kdm5d expression levels in these same offspring at 16 wk of age. F1 transgenic offspring from HDACi 4b-treated male transgenic mice showed a 7.8-fold increase in the cortical expression of Kdm5d compared with F1 transgenic offspring from vehicle-treated male transgenic mice (Fig. 4A). Additional genes from our DNA methylome analysis were tested for gene expression difference in the cortex of male offspring, and only Eif1ay showed a small but significant difference (Fig. S4). Consistent with the Kdm5d mRNA findings, we detected an increase in Kdm5d protein expression in the cortex of F1 transgenic offspring from HDACi 4b-treated males by Western blot analysis (Fig. 4B). Because Kdm5d is a Lys-specific demethylase, which specifically acts at H3K4, we also measured histone methylation at H3K4, and found lower levels in the brains of F1 transgenic offspring from HDACi 4b-treated males vs. F1 transgenic offspring from vehicle-treated males (Fig. 4B). We found that Kdm5d expression levels were negatively correlated with H3K4 methylation (H3K4me) levels (Fig. 4C), as would be predicted.
Fig. 4.

(A) Real-time qPCR results showing altered expression of Kdm5d in cortex of male offspring of vehicle-treated or HDACi 4b-treated male mice. Values shown are the mean ± SEM expression value from n = 4–5 mice per group. One-way ANOVA was used to determine significant differences, and these are indicated by asterisks: **P < 0.001; ***P < 0.0001. (B) Western blot analysis showing elevated Kdm5d protein and reduced H3K4me expression in cortex of male offspring of vehicle-treated (F1HD-veh) and HDACi 4b-treated (F1HD-4b) male HD transgenic mice. Representative blots are shown, although n = 8 F1HD-veh and n = 9 F1HD-4b samples were analyzed in total. (Right) Bar graph quantification of band signals is shown. Student t test was used to determine significance, as indicated by asterisks: *P < 0.05. (C) Correlation between Kdm5d expression levels and H3K4me levels. Pearson correlation analysis of Western blot data for Kdm5d and H3K4me was used, with each point representing a different sample (R = −0.502, P = 0.045). (D) Improvement of the Htt-elicited metabolic deficits in STHdhQ111 striatal cells elicited by KDM5D overexpression. Cells were transfected with human KDM5D cDNA using FuGENE transfection reagent and XTT assays performed after 24 h. Significant differences were determined by one-way ANOVA, followed by a Dunnett’s posttest comparing to the mock-transfected control: *P < 0.05; ***P < 0.0001.
We further tested the direct role of elevating Kdm5d expression in the mechanism for improving HD phenotypes using conditionally immortalized homozygous mutant STHdhQ111 striatal neuronal progenitor cells. These cells express full-length endogenous huntingtin (Htt) protein containing 111 glutamines and display a phenotype of decreased mitochondrial function and energy metabolism compared with WT cells expressing Htt with seven glutamines (28, 29). Overexpression of Kdm5d in STHdhQ111 striatal cells resulted in a significant improvement in the Htt-induced metabolic deficit (Fig. 4D).
Discussion
In summary, we demonstrated that HDAC1/3 inhibition can elicit changes in the expression of genes related to DNA methylation in mouse brain and cause selected alterations in CpG methylation in DNA from control and HD human fibroblasts. Methylation of CpG sites induced by HDACi 4b was significantly enriched at Y chromosome genes, including EIF1AY, TSPY4, and KDM5D. KDM5D/Kdm5d showed increased methylation at several CpG loci not only in human cells, but also in mouse sperm DNA from HDACi 4b-treated male mice. HD transgenic F1 offspring from drug-treated male HD transgenic mice exhibited improved disease phenotypes compared with transgenic offspring from vehicle-treated HD transgenic mice. The behavioral differences were sexually dimorphic, with males showing more pronounced improvements compared with females; this effect is consistent with the high proportion of Y chromosome-linked genes showing differential methylation in response to HDACi 4b.
Elevation in the expression of Dnmt3a, which establishes new DNA methylation patterns, or Dnmt1, a maintenance DNA methyltransferase, could account for increases in DNA methylation, whereas elevated expression of genes encoding putative DNA demethylase enzymes, such as Gadd45b and Parp1, would lead to decreases in DNA methylation. From our global methylation profiling of human fibroblasts, we found that HDACi 4b elicited both increases and decreases in CpG methylation, although a greater number of sites were found to be decreased in methylation in HD cells compared with WT cells. These effects might explain, in part, why many microarray studies have revealed that HDAC inhibitors can cause both increases and decreases in gene expression (11, 15).
The effect of HDAC inhibitors to alter DNA methylation is highly relevant because DNA methylation is an epigenetic modification that can be inherited in the germ line and can lead to transgenerational effects on subsequent progeny. The ability of environmental factors to promote a phenotype or disease state not only in the individual exposed but also in offspring is termed “epigenetic transgenerational inheritance.” Epigenetic transgenerational inheritance of altered phenotypes has been observed in many species, such as worms, flies, plants, rodents, and humans (30–34), suggesting that ancestral environmental exposure can influence disease epidemiology. Importantly, studies have demonstrated transgenerational effects via paternal transmission (35, 36), even in humans (33, 34), and these effects can be detected in the F1 generation (compared with maternal transmission, which requires testing of the F2 and F3 generations). This effect most likely involves alterations in the sperm DNA methylome. After fertilization, the paternal and maternal alleles are demethylated, in part, to develop the pluripotent state of the ES cells, although it is now known that methylation clearing is not complete after fertilization (37–39). We propose that HDAC inhibitors can induce germ-line epigenetic modifications and become permanently programmed similar to the DNA methylation of an imprinted gene (38, 39). Accordingly, we show increased methylation at several CpG loci of the Kdm5d gene in mouse sperm DNA from HDACi 4b-treated male mice.
The transgenerational effects elicited by exposure to HDACi 4b were not mimicked by a related HDAC inhibitor, RGFP966, which selectively targets the HDAC3 subtype. HDACi 4b potently inhibits HDAC3 and HDAC1, but it also inhibits HDAC2 and HDAC8 at higher concentrations (micromolar) (10). Hence, inhibition of other class I HDACs, particularly HDAC1, could be responsible for the DNA methylation changes we observed, as well as in the ensuing downstream effects.
One important candidate gene we have implicated in this study is KDM5D/Kdm5d, which encodes a histone Lys (K)-specific demethylase specific for H3K4. We found that Kdm5d expression was elevated in the brains of F1 offspring of HDACi 4b-exposed fathers, in correlation with reduced levels of H3K4me. H3K4me is an important epigenetic mark that has been implicated in diverse biological functions, such as gene activation and repression, the DNA damage response, and genomic imprinting and development (40). Proteins that regulate the methylation of H3K4 are known to be involved in neurological and developmental disorders, and mutations in genes encoding histone demethylase enzymes have been identified in autism, Rett syndrome, and X-linked mental retardation (XLMR) (41). For example, mutations in the gene KDM5C, a closely related family member to KDM5D but expressed on the X chromosome (40), are associated with XLMR and result in reduced demethylase activity, which is detrimental to neuronal function (41). Microarray studies have shown that Kdm5c expression is decreased in human HD caudate (42) and in the striatum of R6/2 mice (43), which is consistent with our current studies on Kdm5d in N171-82Q mice, implicating reduced demethylase activity in HD pathology. It should be noted that another study reported an increase in KDM5C expression in human HD caudate (44); however, the discrepancy could be due to the X-linked expression of KDM5C and the unknown sexes of these patients in that study.
Of particular relevance to our work is a recent study implicating a related H3K4 dimethyl demethylase, Spr-5, in transgenerational epigenetic effects observed in Caenorhabditis elegans (41). These researchers showed that Spr-5 mutations resulted in reduced fertility in C. elegans across successive generations (41). These findings further support a role for the mammalian H3K4 demethylase, Kdm5d, in the transgenerational effects we observed in this study.
Previous work studying epigenetic transgenerational inheritance has shown that diet, endocrine disruptors, and toxic compounds can lead to heritable epigenetic alterations of the germ line, and can subsequently significantly affect the transcriptomes and phenotypes of following generations (42, 43). Typically, the resulting phenotypic effects are unwanted and/or detrimental (i.e., infertility or toxic effects in the offspring). In stark contrast, our results show that HDAC inhibitors can impart beneficial effects to offspring in a transgenerational manner. These findings will have significant implications for human health because they enforce the concept that ancestral drug exposure may be a major molecular factor that can affect disease phenotypes, yet in a positive manner. This phenomenon is especially relevant for HDAC inhibitors, which are increasingly being used to treat a broad range of human diseases.
Materials and Methods
Mice and Drug Treatments.
An B6C3-Tg(HD82Gln)81Dbo/J (N171-HD82Q) line (Jackson Laboratories) has been maintained at The Scripps Research Institute by breeding male heterozygous N171-HD82Q mice with F1 hybrids of the same background. The CAG repeat lengths in these mice were verified by commercial genotyping (Laragen) and found to be 82 ± 1 CAGs. The lifespan of the N171-82Q HD mouse is ∼17–20 wk, with HD-like symptoms beginning at 10–12 wk of age. HDACi 4b [N1-(2-aminophenyl)-N7-phenylheptanediamide] was synthesized by Scynexis Corporaton as described previously (7). N171-82Q transgenic mice were housed and maintained on a normal 12-h light/dark cycle with lights on at 6:00 AM and free access to food and water. For microarray and gene expression studies, groups of male mice (n = 6–8) were administered HDACi 4b (50 mg/kg) or vehicle for 10–12 wk by s.c. injection (three injections per week) beginning at 8 wk of age. For sperm DNA methylation studies and breeding, groups of male mice (n = 6–8) were administered HDACi 4b (50 mg/kg) or vehicle for 4 wk by s.c. injection (three injections per week) beginning at 8 wk of age. In separate studies, RGFP966 was administered in a similar manner (SI Materials and Methods). HDACi 4b was dissolved with 75% (vol/vol) polyethylene glycol 200; control mice received double-distilled H2O containing an equal volume of drug vehicle. Our previous studies have found that the s.c. injection paradigm for HDACi 4b is optimal for improving disease phenotypes in N171-82Q transgenic mice, given the poor solubility of HDACi 4b for drinking water paradigms (44, 45).
Body weights were recorded two times per week. Mice were killed 6 h after the final injection. Brain regions (striatum and cortex) and gastrocnemius and soleus skeletal muscle samples were dissected out for gene expression assays. All procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals (46). All experimental protocols were approved by the Animal Care and Use Committee at the Scripps Research Institute.
Cell Culture of Human Fibroblasts.
HD patient and control fibroblast cells were purchased from the Coriell Institute. HD and control fibroblast cells were cultured to 80% confluence in DMEM with 10% (wt/vol) FBS and 1% antibiotic penicillin-streptomycin according to the instructions of the supplier. Cells were plated in 12-well plates, treated with HDACi 4b at 500 nM for 48 h, and then harvested to extract genomic DNA (gDNA) using a QIAamp DNA Mini Kit (Qiagen) for methyl-array analysis.
Microarray Gene Expression Analysis.
RNA was extracted from gastrocnemius muscle samples dissected from vehicle- and drug-treated mice and converted to cDNA by standard protocols. RNA (200 ng) was amplified, biotinylated, and hybridized to Illumina MouseRef-8 v2.0 Expression BeadChips per the manufacturer’s protocol. Slides were scanned using Illumina BeadStation, and signals were extracted using Illumina BeadStudio software. Gene expression profiles were normalized using robust multiarray average and log2-transformed using the Bioconductor package “beadarray.” Differential expression was performed using the Bioconductor package “Limma” and adjusted P values for multiple testing, as described previously (43). The full microarray dataset can be found on the NCBI Gene Expression Omnibus (GEO) website, under accession no. GSE56963.
Functional Pathways Analysis.
The functional relevance of microarray data generated from different brain regions from HDACi 4b-treated HD mice from our previous studies (GEO accession no. GSE26317) and from muscle samples in the current study (GEO accession no. GSE56963) were analyzed using Ingenuity Pathways Analysis (Ingenuity Systems, www.ingenuity.com) and DAVID.
Real-Time qPCR Analysis.
The real-time qPCR experiments were performed using the ABI StepOne Detection System (Applied Biosystems) as described previously (43). Specific primers for each sequence were designed using Primer Express 1.5 software (Applied Biosystems), and their specificity for binding to the desired sequences was searched against the National Center for Biotechnology Information database (Tables S4 and S5). The amount of cDNA in each sample was calculated using SDS2.1 software (Applied Biosystems) by the comparative threshold cycle (Ct) method and expressed as 2exp(Ct) using hypoxanthine guanine phosphoribosyl transferase (Hprt) as an internal control. Statistical significance was determined using Student t tests (unpaired and one- or two-tailed). All statistical tests were performed using GraphPad software (GraphPad Prism).
DNA Methylation Profiling.
DNA from normal and HD human fibroblasts was quality controlled (DNA1000 Kit and BioAnalyzer 2100; Zymo Research) and bisulfate-converted (EZ DNA Methylation Kit; Zymo Research) according to each manufacturer’s protocol. Bisulfite-converted DNA was hybridized to Infinium HumanMethylation27K BeadChips (Illumina, Inc.), scanned with an iScan (Illumina, Inc), and quality-controlled using GenomeStudio. For 27K data, β values for each probe were range-scaled with data collected from DNA controls that were fully methylated [DNA methyltransferase-treated (New England Biolabs), bisulfite-converted DNA], unmethylated (untreated gDNA), and half-methylated (50:50 mix of methylated and unmethylated controls). For HD vs. normal comparisons, we used a multiple hypothesis corrected P value of 0.001 and a difference in methylation cutoff of beta change of >|0.2|. Due to the lower number of methylation changes induced by HDACi 4b treatment, a statistical cutoff of P < 0.1 and a beta change of |0.2| were used.
Methylated DNA Immunoprecipitation Real-Time qPCR.
Male transgenic N171-HD82Q mice were treated daily with HDACi 4b (50 mg/kg) for 2 wk. The sperm was released from the cauda epididymis, and the gDNA was extracted and purified using a Qiagen genome DNA purification kit as described previously (47). The purified gDNA was immunoprecipitated and subjected to real-time qPCR analysis according to previous protocols (48). Briefly, gDNA was sonicated with an S220 Focused-Ultrasonicator (Covaris, Inc.) and verified for an average size of 250–350 bp. Two micrograms of sonicated gDNA was immunoprecipitated with 1 μg of anti-5mC antibody (catalog no. BI-MECY-0100; Eurogentec) by incubation for 2 h at 4 °C with shaking. Sheep-M280 anti-mouse Dynabeads (Life Technologies) were incubated with the sample for another 2 h at 4 °C with shaking, followed by collection of the Dynabeads using a magnetic rack. The DNA binding magnetic beads were resuspended in proteinase K digestion buffer and incubated for 3 h at 50 °C to release gDNA. The immunoprecipitated gDNA was purified using a Qiagen MiniElute PCR Purification Kit followed by qPCR analysis to verify the target region.
Motor Behavioral Assessments of N171-82Q Offspring.
In total, n = 55 F1 offspring from F0 vehicle-treated male mice and n = 54 F1 offspring from F0 HDACi 4b-treated male mice (50-mg/kg dose) from >25 different parental matings were tested.
Rotarod.
An AccuRotor rotarod (AccuScan Instruments) was used to measure motor coordination and balance. Mice were tested during the light phase of the 12-h light/dark cycle by using an accelerating rotation paradigm. Mice were placed on a rotarod accelerating from 0 to 20 rpm over 600 s, and latency to fall was recorded.
Open-field test.
Open field exploration was measured in a square Plexiglas chamber (27.3 cm × 27.3 cm × 20.3 cm; Med Associates, Inc.). Several behavioral parameters (ambulatory distance, ambulatory time, stereotypic time, vertical time, vertical counts, and mean velocity) were recorded during a 10-min observation period.
Alternating T-maze.
The alternating T-maze test was performed using methods similar to those described previously (49). The T-maze is made of transparent Plexiglas with a central arm (75 cm long × 12 cm wide × 20 cm high) and two lateral arms (32 cm long × 12 cm wide × 20 cm high) positioned at a 90° angle relative to the central arm. Forced alternation was used for the T-maze training. For T-maze testing, mice were provided an initial free choice for either arm of the maze, and the percentage of alternation over the next nine trials was determined. This percentage can be used as an index of working memory performance (50). Statistical analyses for all behavioral tests were performed using one-way or two-way ANOVA (GraphPad Prism).
Striatal Cell Culture.
Conditionally immortalized WT STHdhQ7striatal neuronal progenitor cells expressing endogenous normal Htt, with seven glutamines, and homozygous mutant STHdhQ111 striatal neuronal progenitor cell lines expressing endogenous mutant Htt, with 111 glutamines, were a kind donation from Marcy MacDonald, Massachusetts General Hospital, Boston, and grown as described previously (51). Cells were plated in 24- or 96-well tissue culture plates. The following day, the cells were transfected with an expression plasmid containing the cDNA of human KDM5D (NCBI Nucleotide database, accession no. BM455812), which was directionally cloned into the expression vector pCMV-Sport6. Transfection was carried out using FuGENE transfection reagent (Promega) according to the manufacturer’s instructions. Empty expression vector, pCMV, was used as a control. Metabolic assays were performed 24 h after shifting the cells to 39 °C by adding the XTT [2,3-Bis-(2-Methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, disodium salt] reagent (Sigma–Aldrich), followed by absorbance readings at 490 nm. Successful transfection of KDM5D was assessed by measuring overexpression of Kdm5d protein using Western blotting with a specific anti-Kdm5d antibody (1:1,000 dilution; Acris). Significant differences in metabolic activity were determined by one-way ANOVA, followed by Dunnett’s posttest using GraphPad software.
Western Blotting.
Western blot assays were performed as described previously (9) using a specific anti-Kdm5d antibody (1:1,000 dilution; Acris) and anti-H3K4me3 (1:5,000 dilution; Millipore), and were normalized to anti-GAPDH (1:1,000 dilution; Abcam) or total histone H3 (1:8,000 dilution; Millipore). Chemiluminescent signals were visualized using Western blotting luminol reagent (Pierce), and bands were captured and quantified using Fluorochem E image analyzer (Cell Biosciences).
Supplementary Material
Acknowledgments
We thank Giovanni Coppola for performing the microarray studies. These studies were funded by NIH Grant U01NS063953.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE56963).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415195112/-/DCSupplemental.
References
- 1.Cohen-Carmon D, Meshorer E. Polyglutamine (polyQ) disorders: The chromatin connection. Nucleus. 2012;3(5):433–441. doi: 10.4161/nucl.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deutsch SI, Rosse RB, Mastropaolo J, Long KD, Gaskins BL. Epigenetic therapeutic strategies for the treatment of neuropsychiatric disorders: Ready for prime time? Clin Neuropharmacol. 2008;31(2):104–119. doi: 10.1097/WNF.0b013e318067e255. [DOI] [PubMed] [Google Scholar]
- 3.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7(10):854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 4.Cantley MD, Haynes DR. Epigenetic regulation of inflammation: Progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology. 2013;21(4):301–307. doi: 10.1007/s10787-012-0166-0. [DOI] [PubMed] [Google Scholar]
- 5.Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77(2):126–135. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- 6.Malvaez M, et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. 2013;110(7):2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herman D, et al. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2(10):551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 8.Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283(51):35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia H, Kast RJ, Steffan JS, Thomas EA. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: Implications for the ubiquitin-proteasomal and autophagy systems. Hum Mol Genet. 2012;21(24):5280–5293. doi: 10.1093/hmg/dds379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia H, et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. 2012;46(2):351–361. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas EA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA. 2008;105(40):15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23(28):9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hockly E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2003;100(4):2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardian G, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280(1):556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 15.Peart MJ, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(10):3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Ribchester RR, et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. 2004;20(11):3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- 18.Lodi R, et al. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann Neurol. 2000;48(1):72–76. [PubMed] [Google Scholar]
- 19.Arenas J, et al. Complex I defect in muscle from patients with Huntington’s disease. Ann Neurol. 1998;43(3):397–400. doi: 10.1002/ana.410430321. [DOI] [PubMed] [Google Scholar]
- 20.Chaturvedi RK, et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet. 2009;18(16):3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barreto G, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 22.Carey N, Marques CJ, Reik W. DNA demethylases: A new epigenetic frontier in drug discovery. Drug Discov Today. 2011;16(15-16):683–690. doi: 10.1016/j.drudis.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Hu XV, et al. Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc Natl Acad Sci USA. 2010;107(34):15087–15092. doi: 10.1073/pnas.1009025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown SE, Suderman MJ, Hallett M, Szyf M. DNA demethylation induced by the methyl-CpG-binding domain protein MBD3. Gene. 2008;420(2):99–106. doi: 10.1016/j.gene.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Nakao M. Epigenetics: Interaction of DNA methylation and chromatin. Gene. 2001;278(1-2):25–31. doi: 10.1016/s0378-1119(01)00721-1. [DOI] [PubMed] [Google Scholar]
- 26.Ng CW, et al. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci USA. 2013;110(6):2354–2359. doi: 10.1073/pnas.1221292110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui L, et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Lee JM, et al. Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet. 2007;3(8):e135. doi: 10.1371/journal.pgen.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greer EL, et al. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479(7373):365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hauser MT, Aufsatz W, Jonak C, Luschnig C. Transgenerational epigenetic inheritance in plants. Biochim Biophys Acta. 2011;1809(8):459–468. doi: 10.1016/j.bbagrm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruden DM, Lu X. Hsp90 affecting chromatin remodeling might explain transgenerational epigenetic inheritance in Drosophila. Curr Genomics. 2008;9(7):500–508. doi: 10.2174/138920208786241207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pembrey ME. Male-line transgenerational responses in humans. Hum Fertil (Camb) 2010;13(4):268–271. doi: 10.3109/14647273.2010.524721. [DOI] [PubMed] [Google Scholar]
- 34.Pembrey ME, et al. ALSPAC Study Team Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet. 2006;14(2):159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- 35.Carone BR, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143(7):1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng SF, et al. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. 2010;467(7318):963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- 37.Hajkova P, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452(7189):877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oswald J, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10(8):475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 39.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet. 2005;14(Spec No 1):R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- 40.Shao GB, et al. Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell Dev Biol Anim. 2014;50(7):603–613. doi: 10.1007/s11626-014-9741-6. [DOI] [PubMed] [Google Scholar]
- 41.Greer EL, et al. A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Reports. 2014;7(1):113–126. doi: 10.1016/j.celrep.2014.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Junien C, Nathanielsz P. Report on the IASO Stock Conference 2006: Early and lifelong environmental epigenomic programming of metabolic syndrome, obesity and type II diabetes. Obes Rev. 2007;8(6):487–502. doi: 10.1111/j.1467-789X.2007.00371.x. [DOI] [PubMed] [Google Scholar]
- 43.Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab. 2010;21(4):214–222. doi: 10.1016/j.tem.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beconi M, et al. Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PLoS ONE. 2012;7(9):e44498. doi: 10.1371/journal.pone.0044498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen JY, et al. Effects of the Pimelic Diphenylamide Histone Deacetylase Inhibitor HDACi 4b on the R6/2 and N171-82Q Mouse Models of Huntington’s Disease. PLoS Curr. 2013;5:5. doi: 10.1371/currents.hd.ec3547da1c2a520ba959ee7bf8bdd202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Committee on Care and Use of Laboratory Animals . Guide for the Care and Use of Laboratory Animals. Natl Inst Health; Bethesda: 1996. DHHS Publ No (NIH) 85-23. [Google Scholar]
- 47.Chang HS, Anway MD, Rekow SS, Skinner MK. Transgenerational epigenetic imprinting of the male germline by endocrine disruptor exposure during gonadal sex determination. Endocrinology. 2005;147(12):5524–5541. doi: 10.1210/en.2006-0987. [DOI] [PubMed] [Google Scholar]
- 48.Weber M, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37(8):853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 49.Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. 2006;1(1):7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- 50.Bontempi B, Whelan KT, Risbrough VB, Lloyd GK, Menzaghi F. Cognitive enhancing properties and tolerability of cholinergic agents in mice: A comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology. 2003;28(7):1235–1246. doi: 10.1038/sj.npp.1300150. [DOI] [PubMed] [Google Scholar]
- 51.Trettel F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9(19):2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.