

Abstract
Protein kinases are mutated or otherwise rendered constitutively active in numerous cancers where they are attractive therapeutic targets with well over a dozen kinase inhibitors now being used in therapy. While fluorescent sensors have capacity to measure changes in kinase activity, surprisingly they have not been utilized for biomarker studies. A first-generation peptide sensor for ERK based on the Sox fluorophore is described. This sensor called ERK-sensor-D1 possesses high activity toward ERK and more than 10-fold discrimination over other MAPKs. The sensor can rapidly quantify ERK activity in cell lysates and monitor ERK pathway engagement by BRAF and MEK inhibitors in cultured melanoma cell lines. The dynamic range of the sensor assay allows ERK activities that have potential for profound clinical consequences to be rapidly distinguished.
Keywords: ERK, biomarker, melanoma
Protein kinases are mutated or otherwise rendered constitutively active in numerous cancers where they are attractive therapeutic targets with well over a dozen kinase inhibitors now being used in therapy.1 Melanoma is a common and deadly form of skin cancer, accounting for over 76 000 cancer diagnoses and 9000 deaths a year in the United States (seer.cancer.gov). In 2002, it was discovered that more than 50% of melanomas contain a constitutively active mutant of the kinase BRAF called BRAFV600E, with an additional 20% subsequently found to express mutations in NRAS.(2−5) These oncogenes cause inappropriate activation of ERK signaling, which is a dominant driver of human melanoma. Within a decade small molecule kinase inhibitors of BRAF and MEK were developed and clinically validated, showing significant short-term responses in patients with BRAF mutant melanomas.6−11
Despite such initial and often dramatic responses to targeted kinase therapies, it is impossible to predict which patients, even with appropriately genotyped tumors, will respond, and resistance develops quickly.6−8,12 Thus, while BRAF inhibitors have an average response rate of 50%, this ranges from disease progression to complete response.6−8,12 Likewise, although acquired resistance is usually inevitable, the timing can range from a few months to over a year.6−8,12 The basis for these differences is not understood and several unaddressed problems that contribute to these shortcomings include (1) failure to use adaptive dosing guided by pharmacodynamic end points, such as kinase activity inhibition; (2) the inability to presage acquired resistance before macroscopic tumor changes, and (3) a lack of mechanistic understanding regarding early treatment failures. Furthermore, dosing of cancer therapies is largely standardized,13 with no regard for pharmacogenetic variation or functional end points that correlate with response.
As noted above, melanoma is often driven by ERK pathway activation, with high ERK activity, a hallmark of melanomas harboring activating BRAF and NRAS mutations.4,5,14 Vemurafenib and dabrafenib are BRAF inhibitors, and trametinib is a MEK inhibitor used for the treatment of BRAF mutant melanoma. These drugs are clinically effective in the short term and result in ERK pathway inhibition.6,9,12,15,16 Importantly, a high degree of ERK inhibition correlates with clinical response (tumor shrinkage),6,12,17 while ERK pathway reactivation is a hallmark of many melanomas that have acquired resistance to BRAF and MEK inhibitors and consequently progress.18−25 The combination of evidence implicating the functional importance of ERK in melanoma and the prevalence of BRAF and MEK inhibitors in the clinic provide a compelling rationale to develop a biomarker for ERK, potentially as the basis for a new point-of-care assay guiding the treatment of melanoma.
ERK is a member of the mitogen-activated protein kinase (MAPK) family, which consists of three main classes of enzyme; the extracellular signal regulated kinases (ERKs), the Jun N-terminal kinases (JNKs), and the p38 MAPKs.26 We considered the possibility of creating a biomarker assay for ERK by exploiting its mechanism of substrate recognition through recruitment sites. Several years ago, Fernandes et al. described a peptide substrate, which exhibits significant specificity for ERK.27 This peptide is modular in design and contains a D-site joined to a phosphorylation site through a linker. We characterized the kinetic mechanism of phosphorylation of a similar peptide called Sub-D28 by ERK2, which has a different linker, and found that Sub-D is phosphorylated through a sequential mechanism with comparable efficiency to Ets-1,29 one of the most efficient substrates known for ERK2.28
Previously, Sox-based peptide sensors have been reported for a number of protein kinases, including AKT,30,31 MAPKAP-K2,30,31 PKA,30,31 and p38 MAPKα,32 and have been used to detect changes in the activity of the kinases in cell lysates.30,32 While such sensors have capacity to measure changes in kinase activity, surprisingly they have not been utilized to quantify kinase activities in tissue samples for biomarker studies. This may be attributed, in part, to significant phosphorylation by endogenous kinases, which reduces their useful dynamic range. However, with the advent of potent and relatively selective kinase inhibitors the potential now exists for exquisite quantification of cellular kinase activities.
In the present study a first-generation peptide sensor for ERK1/2 based on the Sox fluorophore is described (Scheme 1). This sensor called ERK-Sensor-D1 possesses high activity toward ERK1/2 and more than 10-fold discrimination over other MAPKs. We show that the sensor can rapidly quantify ERK activity in cell lysates and monitor ERK pathway engagement by BRAF and MEK inhibitors in cultured melanoma cell lines. Importantly, when used with specific MAPK inhibitors, the enhanced dynamic range of the sensor allows ERK activities that have potential for profound clinical consequences to be rapidly distinguished.
Scheme 1. Schematic Representation of ERK-Sensor-D1: (QRKTLQRRNLKGLNLNL-XXX-TGPLSPC(Sox)PF).
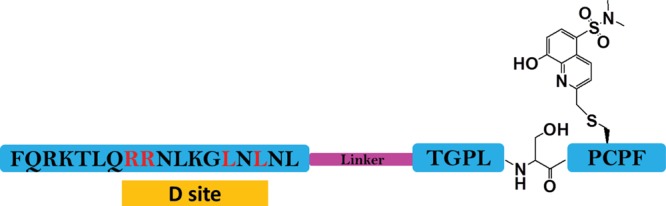
The sox group is introduced on the cysteine at the +2 position relative to the serine phosphorylation site. The D site and the consensus phosphorylation site are connected by a linker (purple) that consists of three repetitive 6-aminohexanoic acid groups.
ERK-Sensor-D1 Is a Selective Fluorescent Substrate of ERK1/2
We reasoned that the following characteristics of a biomarker assay using a fluorescent kinase sensor were desirable: (1) the assay must exhibit a robust fluorescence change upon phosphorylation by the target kinase; (2) the sensor must be an efficient substrate for the target kinase with a kcat/Km of ∼106 M–1 s–1; (3) the assay must exhibit high selectivity for the target kinase; and (4) a stable baseline must be achievable corresponding to zero activity of the target protein kinase.
A combination of protein NMR studies,33 molecular modeling34 and kinetic investigations on ERK228 suggest that the D-site of Sub-D supports efficient phosphorylation through its tethering to the D-recruitment site of ERK (Figure 1A). This tethering promotes the binding of the phosphorylation sequence (TGPLSPCPF) to the active site of ERK2 by virtue of a linker, which ultimately facilitates phosphorylation of the serine through a mechanism of proximity-induced catalysis.35 We reasoned that Sub-D might form the basis for a fluorescent sensor so we investigated its ability to discriminate between isoforms of JNK1/2/3, p38 MAPK α/β/γ/δ, and ERK1/2, which we had purified and activated using previously reported procedures. We found that it exhibits >10-fold discrimination (a higher kcat/Km) for ERK1/2 over the other MAPKs when assayed at 500 μM MgATP (Table 1 and Figure 1B).
Figure 1.
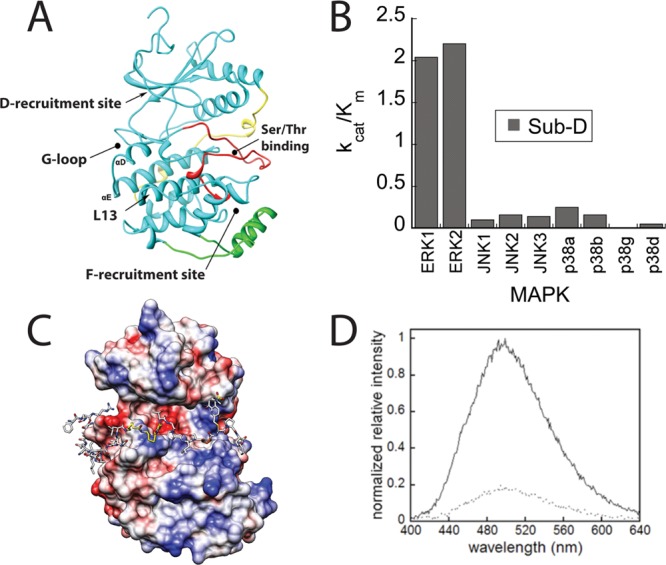
ERK-sensor-D1 is a fluorescent substrate for ERK. A. Ribbon diagram of activated ERK2 (PDB: 2ERK) showing binding site for substrate Ser/Thr-Pro motifs (which become phosphorylated). Also indicated is the D recruitment site. B. kcat/Km for the phosphorylation of Sub-D (QRKTLQRRNLKGLNLNL-XXX-TGPLSPGPF) (X = 6-aminohexanoic acid) by recombinant MAPKs. C. Molecular Model of ERK-sensor-D1 (QRKTLQRRNLKGLNLNL-XXX-TGPLSPC(Sox)PF) bound to ERK2. The flexible X3 linker (colored yellow) joins the D-site34 to the phosphorylation motif (Ser-Pro indicated). Coulombic surface representation was performed in Chimera using default parameters. D. Fluorescence emission spectra of ERK-sensor-D1 before and after overnight phosphorylation with ERK2. Fluorescence emission spectra of fully phosphorylated (solid lines) and unphosphorylated peptides (dotted lines) were taken.
Table 1. Kinetic Parameters for MAPK Phosphorylation of Peptide Substratesa.
| Sub-Db |
ERK-sensor-D1c |
|||
|---|---|---|---|---|
| MAPKa | Km (μM) | kcat (s–1) | Km (μM) | kcat (s–1) |
| ERK1 | 2.3 ± 0.8 | 4.7 ± 0.2 | 3 ± 0.5 | 5.3 ± 0.14 |
| ERK2 | 5.9 ± 1.6 | 13 ± 1 | 6 ± 0.5 | 12.8 ± 0.3 |
| JNK1 | 6.0 ± 1.2 | 0.6 ± 0.03 | 2 ± 0.3 | 0.3 ± 0.01 |
| JNK2 | 7.3 ± 1.2 | 1.2 ± 0.1 | 3.6 ± 0.6 | 0.4 ± 0.01 |
| JNK3 | 6.5 ± 1.0 | 0.9 ± 0.1 | 3.2 ± 0.5 | 0.4 ± 0.02 |
| p38α | 6.3 ± 1.5 | 1.6 ± 0.1 | 9.0 ± 2 | 1.1 ± 0.05 |
| p38β | 9.8 ± 2.8 | 1.6 ± 0.1 | 10.4 ± 1.6 | 2.03 ± 0.09 |
| p38γ | >250 | >250 | ||
| p38δ | 108 ± 10 | 6 ± 0.3 | >250 | |
Assays were performed at 500 μM MgATP.
32P-[γ]-ATP-based assay.
Fluorescence assay
Encouraged by these results we synthesized a fluorescent derivative using the approach of the Imperiali laboratory36 and incorporated the Sox fluorophore to Sub-D through covalent attachment to a Cys residue introduced at the P + 2 position to generate ERK-Sensor-D1(Scheme 1).36 To verify that the incorporation of a Sox-modified Cys does not influence the predicted binding mode of the peptide, ERK-Sensor-D1 was modeled onto the surface of ERK2 using a molecular docking approach as described previously (Figure 1C).28 This model supports the notion that the Sox moiety adopts a conformation that does not interfere with the recognition of the peptide substrates by ERK2.
ERK-Sensor-D1 was assessed for its ability to undergo a change in fluorescence (excitation 360 nm; emission 492 nm) following exhaustive incubation with and without ERK2. Integration of the emission spectra indicated that the fluorescence intensity increases by 5-fold upon phosphorylation (Figure 1D). The peptide was then assessed for linearity in its emission spectrum over a concentration of 0–200 μM. It exhibited excellent linearity, consistent with the notion that over the concentration range it does not self-associate in solution to influence the fluorescence emission spectrum. Having shown that ERK-Sensor-D1 exhibits robust changes in fluorescence over a wide concentration range, we examined its specificity toward the same MAPKs used for Sub-D and found ERK-Sensor-D1 to exhibit a similar ability to discriminate between the MAPKs (see Table 1 and Figure 2A). These experiments establish that ERK-Sensor-D1 is phosphorylated in vitro with high efficiency (at a similar rate to Ets-1 a bona fide protein substrate of ERK29), exhibits a 5-fold change in fluorescence emission, and is more than 10-fold selective for ERK over other MAPKs.
Figure 2.

Monitoring ERK2 activity in cell lysates using ERK-sensor-D1. (A) kcat/Km for the phosphorylation of ERK-sensor-D1 by recombinant MAPKs. (B) Western blotting analysis of MAPKs in HEK 293 cell lysates after treatment with U0126 (1 h) and or PMA (15 min) where indicated. (C) Activity of HEK293T cell lysates toward ERK-sensor-D1. (D) Effect of MAPK inhibitors on the activity of HEK293T cell lysates toward ERK-sensor-D1. Cells were incubated with or without PMA or MEK inhibitor U0126. Cell lysates were incubated with BIRB796/JNK-In-8 or SCH772984 as indicated. Bars labeled with * or # correspond to the assays shown in panel C.
ERK-Sensor-D1 Activity in Cell Lysates
We then decided to test whether the sensor could be used to monitor ERK activity in cell lysates. We chose to first test the sensor using lysates from HEK293T cells, which exhibit robust ERK activation when treated with phorbol-12 myristate 13-acetate (PMA). Thus, cells were serum starved for 24 h then incubated with or without the MEK inhibitor37 U0126 (10 μM) for 1 h to suppress ERK activity in the cells, in order to provide a baseline activity toward the sensor under conditions where cellular ERK activity is low. Cells were then treated with or without PMA (10 nM) for 10–15 min before lysis. The extent of ERK, JNK, and p38 activation was evaluated by Western blot analysis using antibodies specific to the active forms of each kinase. The blots indicated that all three MAPKS were activated by the PMA treatment (Figure 2B), while incubation with U0126 completely blocked the activation of ERK under all conditions. As Western blotting revealed that both JNK and p38 are active in HEK293T cells, we tested whether they could be inhibited in the assay. Gratifyingly, the inclusion of 1 μM BIRB79638 and 1 μM JNK-In-839 (which are potent and selective inhibitors of p38α/p38β and JNK, respectively) was found to completely suppress the activity of recombinant forms of active p38 and JNK in the fluorescent assay with no effect on the observed activity of ERK2 (data not shown).
We then evaluated the ability of the sensor to report on the activity of ERK using aliquots of cell lysates diluted into assay buffer. When lysate from PMA-stimulated cells was compared to lysate from serum starved U0126-treated cells, a clear reproducible difference in the activity toward the sensor was seen (Figure 2C). Interestingly, inclusion of BIRB796 and JNK-In-8 had only a small effect on the signal, suggesting that the p38 and JNK activity in these lysates contributes only marginally to the phosphorylation of the sensor (Figure 2D). We also found that immunoprecipitation of ERK5 from lysates of sorbitol-treated cells, in which ERK5 was active, had no effect on the observed signal, suggesting that ERK5 does not significantly contribute to the signal in the assay (data not shown).
We noted that an appreciable “baseline” signal was obtained using lysates from cells incubated with U0126 in which ERK activity was suppressed. This signal corresponded to approximately 20% of the signal obtained using lysates from PMA-stimulated cells where ERK was activated. When 1 μM of the ERK inhibitor SCH77298440 was introduced into the assays of lysates from U0126-treated cells, a similar signal was obtained compared to assays where SCH772984 was absent (Figure 2D). As this inhibitor is highly specific for ERK and completely blocks ERK activity in the assay (data not shown), it served as a convenient means of obtaining a baseline reference for quantifying ERK activity in lysates. These experiments provide support for the notion that the sensor can be used to quantify the activity in ERK in cell lysates.
Quantifying Clinically Relevant ERK Suppression
The experiments described above demonstrate the potential of applying the sensor toward the measurement of ERK activity in cell lysates. However, for application of a sensor in assessing ERK activity as a pharmacodynamic biomarker, it was important to assess whether the activity of ERK could be compared between samples and whether ERK could be quantified accurately in cells treated with therapeutic levels of clinically relevant inhibitors of the ERK pathway where significant suppression of cell viability is seen.
First we evaluated the ability of the sensor to be calibrated against active recombinant ERK2, as calibration of the assay allows one to estimate the concentration of active ERK in a given cell lysate, enabling comparison between different tissue samples. When we added recombinant ERK to cell lysates we obtained a good linear plot of activity versus the concentration of recombinant ERK2 (0–1000 pM) (Figure 3A).
Figure 3.
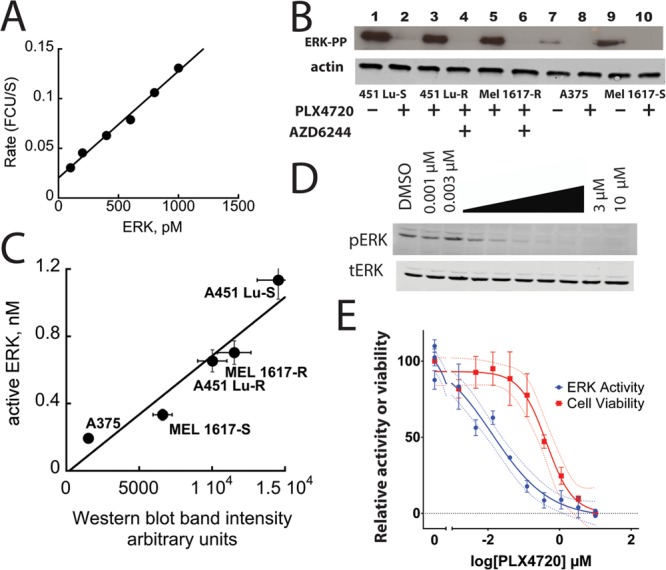
Correlating ERK activity to melanoma cell viability. (A) An ERK2 calibration curve was obtained after adding different concentrations of purified active ERK2 to serum free HEK 293 cell lysates containing BIRB 796 and JNK-In-8. (B) Western blot analysis of various melanoma cell lysates treated with PLX4720 and/or AZD6244/selumetinib (1 μM, as indicated) using an antibody that recognizes active ERK. (C) Plot of ERK activity versus band intensity from panel B. ERK activity is determined by subtracting backgrounds obtained from paired control samples where ERK activity is absent. (D) Dose-dependent inhibition of ERK in BRAF mutant MEL1617 melanoma cells treated with PLX4720 by Western blot. (E) Correlation of the degree of ERK inhibition (measured by the sensor) to the inhibition of cell proliferation (Cell Titer Glo).
We then used the sensor to evaluate the activity of ERK in five melanoma cell lines expressing BRAF(V600E), which had been treated with different combinations of inhibitors of BRAF(V600E) and/or MEK. These treatments generated five sets of cell lysates expressing variable levels of active ERK each paired with a cell lysate in which active ERK was suppressed. PLX4720 is a precursor of PLX4032 (Vemurafenib), a BRAF(V600E) inhibitor approved by the FDA for the treatment of late-stage melanoma. AZD6244/Selumetinib is a MEK inhibitor currently under investigation. Expression of BRAF(V600E) results in constitutive activation of ERK in melanoma cells. Therefore, the measurement of ERK activity by the sensor represents a useful indicator of its ability to report on the suppression of ERK activity by ERK pathway clinical inhibitors. We used an antibody that specifically recognizes the doubly phosphorylated form of ERK to estimate its activity in each cell lysate by determining the intensity of the appropriate immuno-active band obtained by Western blotting.
Figure 3B shows Western blots of cell lysates from the following cell lines (451Lu-S, 451Lu-R, MEL1617-R, A375, and MEL1617-S) treated with 1 μM PLX4720 and/or AZD6244/Selumetinib where indicated. PLX4720 completely suppressed constitutive ERK activity in 451Lu-S, A375, and MEL1617-S cell lines. It has little effect on ERK activity in the 451Lu-R and MEL 1617-R cells, which are metastatic melanoma cell lines derived from the corresponding cells that exhibit resistance to PLX4720. However, as shown in Figure 3B these cells are sensitive to AZD6244/Selumetinib, as judged by the suppression of ERK activity. To evaluate the activity of ERK in each cell line, the rate of change of sensor fluorescence was obtained (in the presence of the JNK and p38 inhibitors) and subtracted from the value obtained for the fluorescence change of the sensor in the corresponding paired lysate where the ERK activity was judged, by Western blotting, to be negligible. As we had calibrated the sensor using highly purified recombinant ERK2 (Figure 3A) we were able to provide an estimate of the concentration of active ERK in each cell lysate based on this calibration. The intensity of the bands from the Western blot also provided an estimate of the relative concentration of active ERK2 in each lysate. In general the two approaches agreed quite well and exhibited a linear correlation of 0.75 ± 0.14 when plotted as the molarity of active ERK (determined by the sensor) versus band intensity (Western blotting) (Figure 3C). Minor deviations in the correlation may be attributed to the nature of the Western blotting experiment, which is a multistep process and is prone to variability.
It has been estimated that drugs targeting the ERK pathway must suppress ERK activity by approximately 80% to exhibit clinical efficacy.12 Thus, to be effective at discriminating between clinically effective and ineffective target engagement requires an ability to distinguish between relatively small differences in low cellular activities of ERK. To assess the potential for our sensor to evaluate the clinical efficacy of a drug we performed a dose-dependent inhibition of ERK in BRAF mutant MEL1617-S melanoma cells treated with PLX4720 by Western blot (Figure 3D) and correlated the degree of ERK inhibition (measured by the sensor) to the inhibition of cell proliferation (Cell Titer Glo) (Figure 3E). By comparing the signal obtained in the absence of drug to the signal obtained from samples treated with a high concentration of drug (where ERK is completely inhibited) the activity of ERK (as judged by the sensor readout) can be determined as a function of drug concentration. This experiment suggested that >90% of ERK inhibition can be accurately measured using the assay and showed that such inhibition is required to achieve the EC50 in suppression of proliferation (Figure 3E). Similar results were obtained when assessing A375 cells (Figure S1, Supporting Information). These data highlight the wide dynamic range of the sensor-based assay, which appears to exceed that of the standard Western blot, which is unable to distinguish between the multiple levels of ERK inhibition observed in the kinase assay (Figures 3D and S1, Supporting Information). This is critically important as distinguishing between 80% versus over 90% inhibition may have profound clinical consequences, potentially reflecting either resistance or inadequate dosing.
Conclusions
A motivation for the current work was to evaluate the potential for using a sensor to measure ERK activity as a biomarker. Personalized dosing is seen as a critical area for improvement, as dosing of cancer therapies is largely standardized,13 with no regard for pharmacogenetic variation or functional end points that correlate with a response. Evaluation during therapy is important because, while acquired resistance is an inevitable consequence of targeted therapies, the timing remains unpredictable. Furthermore, recurrent disease can be explosive and is usually identified only during radiographic imaging of tumor masses, making it difficult to implement an appropriate salvage therapy within time.41
The experiments reported here provide support for the notion that a peptide-based sensor may be used to quantify ERK activity in patient tissue samples. Currently, available methods such as immunohistochemistry performed on archival tissue can be notoriously unreliable and nonquantitative, with extreme sensitivity to fixation methods and processing variation across sites.42 Studies are currently ongoing to evaluate the ability of the sensor to quantify ERK activity in primary melanoma tumors.
Glossary
Abbreviations
- Sox
8-hydroxy-5-(N,N-dimethylsulfonamido)-2-methylquinoline
- Ets
murine (His6-tagged)Ets1(1–138)
- ERK-sensor-D1
FQRKTLQRRLKGLNLNL-XXX-TGPLSPC(SOX)PF
Supporting Information Available
Reagents and detailed experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
Support by NIH (GM059802, GM106137, and CA167505), the Welch Foundation (F-1390 and F-1691), CPRIT (RP101501, RP140648, and RP140649), a Cyrus Scholar Award, and the DXB Biosciences Research Fund is acknowledged.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhang C.; Habets G.; Bollag G. Interrogating the kinome. Nat. Biotechnol. 2011, 29, 981–3. [DOI] [PubMed] [Google Scholar]
- Davies H.; Bignell G. R.; Cox C.; Stephens P.; Edkins S.; Clegg S.; Teague J.; Woffendin H.; Garnett M. J.; Bottomley W.; Davis N.; Dicks E.; Ewing R.; Floyd Y.; Gray K.; Hall S.; Hawes R.; Hughes J.; Kosmidou V.; Menzies A.; Mould C.; Parker A.; Stevens C.; Watt S.; Hooper S.; Wilson R.; Jayatilake H.; Gusterson B. A.; Cooper C.; Shipley J.; Hargrave D.; Pritchard-Jones K.; Maitland N.; Chenevix-Trench G.; Riggins G. J.; Bigner D. D.; Palmieri G.; Cossu A.; Flanagan A.; Nicholson A.; Ho J. W.; Leung S. Y.; Yuen S. T.; Weber B. L.; Seigler H. F.; Darrow T. L.; Paterson H.; Marais R.; Marshall C. J.; Wooster R.; Stratton M. R.; Futreal P. A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–54. [DOI] [PubMed] [Google Scholar]
- Dhomen N.; Marais R. BRAF signaling and targeted therapies in melanoma. Hematol. Oncol. Clin. North Am. 2009, 23, 529–45. [DOI] [PubMed] [Google Scholar]
- Colombino M.; Capone M.; Lissia A.; Cossu A.; Rubino C.; De Giorgi V.; Massi D.; Fonsatti E.; Staibano S.; Nappi O.; Pagani E.; Casula M.; Manca A.; Sini M.; Franco R.; Botti G.; Caraco C.; Mozzillo N.; Ascierto P. A.; Palmieri G. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–9. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Choi J. W.; Kim Y. S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br. J. Dermatol. 2011, 164, 776–84. [DOI] [PubMed] [Google Scholar]
- Flaherty K. T.; Puzanov I.; Kim K. B.; Ribas A.; McArthur G. A.; Sosman J. A.; O’Dwyer P. J.; Lee R. J.; Grippo J. F.; Nolop K.; Chapman P. B. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P. B.; Hauschild A.; Robert C.; Haanen J. B.; Ascierto P.; Larkin J.; Dummer R.; Garbe C.; Testori A.; Maio M.; Hogg D.; Lorigan P.; Lebbe C.; Jouary T.; Schadendorf D.; Ribas A.; O’Day S. J.; Sosman J. A.; Kirkwood J. M.; Eggermont A. M.; Dreno B.; Nolop K.; Li J.; Nelson B.; Hou J.; Lee R. J.; Flaherty K. T.; McArthur G. A. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosman J. A.; Kim K. B.; Schuchter L.; Gonzalez R.; Pavlick A. C.; Weber J. S.; McArthur G. A.; Hutson T. E.; Moschos S. J.; Flaherty K. T.; Hersey P.; Kefford R.; Lawrence D.; Puzanov I.; Lewis K. D.; Amaravadi R. K.; Chmielowski B.; Lawrence H. J.; Shyr Y.; Ye F.; Li J.; Nolop K. B.; Lee R. J.; Joe A. K.; Ribas A. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty K. T.; Infante J. R.; Daud A.; Gonzalez R.; Kefford R. F.; Sosman J.; Hamid O.; Schuchter L.; Cebon J.; Ibrahim N.; Kudchadkar R.; Burris H. A.; Falchook G.; Algazi A.; Lewis K.; Long G. V.; Puzanov I.; Lebowitz P.; Singh A.; Little S.; Sun P.; Allred A.; Ouellet D.; Kim K. B.; Patel K.; Weber J. Combined BRAF and MEK inhibition in melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A.; Grob J. J.; Demidov L. V.; Jouary T.; Gutzmer R.; Millward M.; Rutkowski P.; Blank C. U.; Miller W. H. Jr.; Kaempgen E.; Martin-Algarra S.; Karaszewska B.; Mauch C.; Chiarion-Sileni V.; Martin A. M.; Swann S.; Haney P.; Mirakhur B.; Guckert M. E.; Goodman V.; Chapman P. B. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–65. [DOI] [PubMed] [Google Scholar]
- Long G. V.; Trefzer U.; Davies M. A.; Kefford R. F.; Ascierto P. A.; Chapman P. B.; Puzanov I.; Hauschild A.; Robert C.; Algazi A.; Mortier L.; Tawbi H.; Wilhelm T.; Zimmer L.; Switzky J.; Swann S.; Martin A. M.; Guckert M.; Goodman V.; Streit M.; Kirkwood J. M.; Schadendorf D. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 1087–95. [DOI] [PubMed] [Google Scholar]
- Bollag G.; Hirth P.; Tsai J.; Zhang J.; Ibrahim P. N.; Cho H.; Spevak W.; Zhang C.; Zhang Y.; Habets G.; Burton E. A.; Wong B.; Tsang G.; West B. L.; Powell B.; Shellooe R.; Marimuthu A.; Nguyen H.; Zhang K. Y.; Artis D. R.; Schlessinger J.; Su F.; Higgins B.; Iyer R.; D’Andrea K.; Koehler A.; Stumm M.; Lin P. S.; Lee R. J.; Grippo J.; Puzanov I.; Kim K. B.; Ribas A.; McArthur G. A.; Sosman J. A.; Chapman P. B.; Flaherty K. T.; Xu X.; Nathanson K. L.; Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty K. T.; Yasothan U.; Kirkpatrick P. Vemurafenib. Nat. Rev. Drug Discovery 2011, 10, 811–2. [DOI] [PubMed] [Google Scholar]
- Solus J. F.; Kraft S. Ras, Raf, and MAP kinase in melanoma. Adv. Anat. Pathol. 2013, 20, 217–26. [DOI] [PubMed] [Google Scholar]
- Tsai J.; Lee J. T.; Wang W.; Zhang J.; Cho H.; Mamo S.; Bremer R.; Gillette S.; Kong J.; Haass N. K.; Sproesser K.; Li L.; Smalley K. S.; Fong D.; Zhu Y. L.; Marimuthu A.; Nguyen H.; Lam B.; Liu J.; Cheung I.; Rice J.; Suzuki Y.; Luu C.; Settachatgul C.; Shellooe R.; Cantwell J.; Kim S. H.; Schlessinger J.; Zhang K. Y.; West B. L.; Powell B.; Habets G.; Zhang C.; Ibrahim P. N.; Hirth P.; Artis D. R.; Herlyn M.; Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 3041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G.; Tsai J.; Zhang J.; Zhang C.; Ibrahim P.; Nolop K.; Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 2012, 11, 873–86. [DOI] [PubMed] [Google Scholar]
- Trunzer K.; Pavlick A. C.; Schuchter L.; Gonzalez R.; McArthur G. A.; Hutson T. E.; Moschos S. J.; Flaherty K. T.; Kim K. B.; Weber J. S.; Hersey P.; Long G. V.; Lawrence D.; Ott P. A.; Amaravadi R. K.; Lewis K. D.; Puzanov I.; Lo R. S.; Koehler A.; Kockx M.; Spleiss O.; Schell-Steven A.; Gilbert H. N.; Cockey L.; Bollag G.; Lee R. J.; Joe A. K.; Sosman J. A.; Ribas A. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–74. [DOI] [PubMed] [Google Scholar]
- Nazarian R.; Shi H.; Wang Q.; Kong X.; Koya R. C.; Lee H.; Chen Z.; Lee M. K.; Attar N.; Sazegar H.; Chodon T.; Nelson S. F.; McArthur G.; Sosman J. A.; Ribas A.; Lo R. S. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen C. M.; Boehm J. S.; Kim S. Y.; Thomas S. R.; Wardwell L.; Johnson L. A.; Emery C. M.; Stransky N.; Cogdill A. P.; Barretina J.; Caponigro G.; Hieronymus H.; Murray R. R.; Salehi-Ashtiani K.; Hill D. E.; Vidal M.; Zhao J. J.; Yang X.; Alkan O.; Kim S.; Harris J. L.; Wilson C. J.; Myer V. E.; Finan P. M.; Root D. E.; Roberts T. M.; Golub T.; Flaherty K. T.; Dummer R.; Weber B. L.; Sellers W. R.; Schlegel R.; Wargo J. A.; Hahn W. C.; Garraway L. A. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J.; Vultur A.; Lee J. T.; Somasundaram R.; Fukunaga-Kalabis M.; Cipolla A. K.; Wubbenhorst B.; Xu X.; Gimotty P. A.; Kee D.; Santiago-Walker A. E.; Letrero R.; D’Andrea K.; Pushparajan A.; Hayden J. E.; Brown K. D.; Laquerre S.; McArthur G. A.; Sosman J. A.; Nathanson K. L.; Herlyn M. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraiso K. H.; Fedorenko I. V.; Cantini L. P.; Munko A. C.; Hall M.; Sondak V. K.; Messina J. L.; Flaherty K. T.; Smalley K. S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R.; Morikawa T.; Shee K.; Barzily-Rokni M.; Qian Z. R.; Du J.; Davis A.; Mongare M. M.; Gould J.; Frederick D. T.; Cooper Z. A.; Chapman P. B.; Solit D. B.; Ribas A.; Lo R. S.; Flaherty K. T.; Ogino S.; Wargo J. A.; Golub T. R. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Moriceau G.; Kong X.; Lee M. K.; Lee H.; Koya R. C.; Ng C.; Chodon T.; Scolyer R. A.; Dahlman K. B.; Sosman J. A.; Kefford R. F.; Long G. V.; Nelson S. F.; Ribas A.; Lo R. S. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran R. B.; Ebi H.; Turke A. B.; Coffee E. M.; Nishino M.; Cogdill A. P.; Brown R. D.; Della Pelle P.; Dias-Santagata D.; Hung K. E.; Flaherty K. T.; Piris A.; Wargo J. A.; Settleman J.; Mino-Kenudson M.; Engelman J. A. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discovery 2012, 2, 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. R.; Fridlyand J.; Yan Y.; Penuel E.; Burton L.; Chan E.; Peng J.; Lin E.; Wang Y.; Sosman J.; Ribas A.; Li J.; Moffat J.; Sutherlin D. P.; Koeppen H.; Merchant M.; Neve R.; Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Gibson T. B.; Robinson F.; Silvestro L.; Pearson G.; Xu B.-E.; Wright A.; Vanderbilt C.; Cobb M. H. MAP Kinases. Chem. Rev. 2001, 101, 2449–2476. [DOI] [PubMed] [Google Scholar]
- Fernandes N.; Bailey D. E.; VanVranken D. L.; Allbritton N. L. Use of docking peptides to design modular substrates with high efficiency for mitogen-activated protein kinase extracellular signal-regulated kinase. ACS Chem. Biol. 2007, 2, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Warthaka M.; Yan C.; Kaoud T. S.; Ren P.; Dalby K. N. Examining docking interactions on ERK2 with modular peptide substrates. Biochemistry 2011, 50, 9500–9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas W. F.; Dalby K. N. Transient protein-protein interactions and a random-ordered kinetic mechanism for the phosphorylation of a transcription factor by extracellular-regulated protein kinase 2. J. Biol. Chem. 2002, 277, 12532–40. [DOI] [PubMed] [Google Scholar]
- Stains C. I.; Tedford N. C.; Walkup T. C.; Lukovic E.; Goguen B. N.; Griffith L. G.; Lauffenburger D. A.; Imperiali B. Interrogating signaling nodes involved in cellular transformations using kinase activity probes. Chem. Biol. 2012, 19, 210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults M. D.; Carrico-Moniz D.; Imperiali B. Optimal Sox-based fluorescent chemosensor design for serine/threonine protein kinases. Anal. Biochem. 2006, 352, 198–207. [DOI] [PubMed] [Google Scholar]
- Stains C. I.; LukovicÌ E.; Imperiali B. A p38α-Selective chemosensor for use in unfractionated cell lysates. ACS Chem. Biol. 2011, 6, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piserchio A.; Warthaka M.; Devkota A. K.; Kaoud T. S.; Lee S.; Abramczyk O.; Ren P.; Dalby K. N.; Ghose R. Solution NMR insights into docking interactions involving inactive ERK2. Biochemistry 2011, 50, 3660–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Warthaka M.; Yan C.; Kaoud T. S.; Piserchio A.; Ghose R.; Ren P.; Dalby K. N. A model of a MAPK*substrate complex in an active conformation: a computational and experimental approach. PloS One 2011, 6, e18594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey M. A.; Callaway K.; Barnes R.; Wilson B.; Dalby K. N. Proximity-induced catalysis by the protein kinase ERK2. J. Am. Chem. Soc. 2005, 127, 10494–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LukovicÌ E.; GonzaÌ lez-Vera J. A.; Imperiali B. Recognition-domain focused chemosensors: Versatile and efficient reporters of protein kinase activity. J. Am. Chem. Soc. 2008, 130, 12821–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncia J. V.; Santella J. B. 3rd; Higley C. A.; Pitts W. J.; Wityak J.; Frietze W. E.; Rankin F. W.; Sun J. H.; Earl R. A.; Tabaka A. C.; Teleha C. A.; Blom K. F.; Favata M. F.; Manos E. J.; Daulerio A. J.; Stradley D. A.; Horiuchi K.; Copeland R. A.; Scherle P. A.; Trzaskos J. M.; Magolda R. L.; Trainor G. L.; Wexler R. R.; Hobbs F. W.; Olson R. E. MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett. 1998, 8, 2839–44. [DOI] [PubMed] [Google Scholar]
- Kuma Y.; Sabio G.; Bain J.; Shpiro N.; Marquez R.; Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–9. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; Laughlin J. D.; Park H.; LoGrasso P. V.; Patricelli M.; Nomanbhoy T. K.; Sorger P. K.; Alessi D. R.; Gray N. S. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris E. J.; Jha S.; Restaino C. R.; Dayananth P.; Zhu H.; Cooper A.; Carr D.; Deng Y.; Jin W.; Black S.; Long B.; Liu J.; Dinunzio E.; Windsor W.; Zhang R.; Zhao S.; Angagaw M. H.; Pinheiro E. M.; Desai J.; Xiao L.; Shipps G.; Hruza A.; Wang J.; Kelly J.; Paliwal S.; Gao X.; Babu B. S.; Zhu L.; Daublain P.; Zhang L.; Lutterbach B. A.; Pelletier M. R.; Philippar U.; Siliphaivanh P.; Witter D.; Kirschmeier P.; Bishop W. R.; Hicklin D.; Gilliland D. G.; Jayaraman L.; Zawel L.; Fawell S.; Samatar A. A. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery 2013, 3, 742–50. [DOI] [PubMed] [Google Scholar]
- Ascierto P. A.; Simeone E.; Grimaldi A. M.; Curvietto M.; Esposito A.; Palmieri G.; Mozzillo N. Do BRAF inhibitors select for populations with different disease progression kinetics?. J. Transl. Med. 2013, 11, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A. F.; Dragovich T.; Ihle N. T.; Williams R.; Fenoglio-Preiser C.; Powis G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin. Cancer Res. 2005, 11, 4338–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.