

Abstract
The Ser/Thr protein kinase, RSK, is associated with oncogenesis, and therefore, there are ongoing efforts to develop RSK inhibitors that are suitable for use in vivo. SL0101 is a natural product that demonstrates selectivity for RSK inhibition. However, SL0101 has a short biological half-life in vivo. To address this issue we designed a set of eight cyclitol analogues, which should be resistant to acid catalyzed anomeric bond hydrolysis. The analogues were synthesized and evaluated for their ability to selectively inhibit RSK in vitro and in cell-based assays. All the analogues were prepared using a stereodivergent palladium-catalyzed glycosylation/cyclitolization for installing the aglycon. The l-cyclitol analogues were found to inhibit RSK2 in in vitro kinase activity with a similar efficacy to that of SL0101, however, the analogues were not specific for RSK in cell-based assays. In contrast, the d-isomers showed no RSK inhibitory activity in in vitro kinase assay.
Keywords: Ser/Thr protein kinase, cyclitol, RSK inhibition, SL0101, de novo synthesis
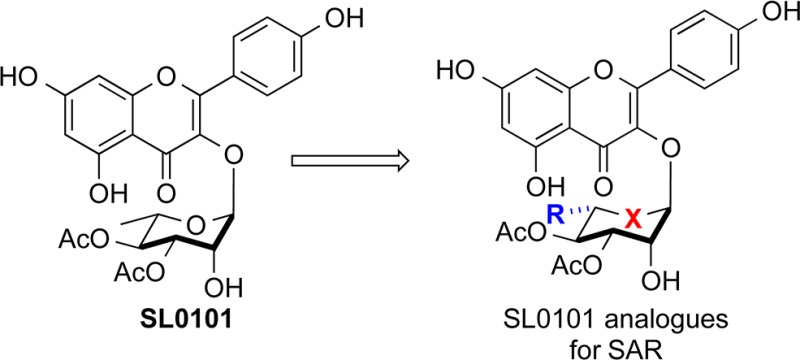
The Ser/Thr kinases, RSK, have emerged as a potential drug target for numerous cancers.1 A number of RSK inhibitors have been identified2−9 and of these the kaempferol l-rhamnoside SL0101 (1a) is the only allosteric inhibitor of RSK (Figure 1),10 which most likely accounts for its specificity.7 RSK is unusual in that it contains two nonidentical kinase domains.12 On the basis of the crystal structure of SL0101 in complex with the RSK2 N-terminal kinase domain (NTKD) we generated the derivative, C3″/C4″-diacetate with a C6″-n-propyl substituent 2, which has a 50-fold higher affinity for RSK than SL0101.13−202 In an effort to further explore the structure–activity relationship of SL0101 as it relates to RSK inhibition, we targeted for synthesis cyclitol (aka, 5a-carbasugar) analogues of SL0101 (e.g., 3 and 4, Figure 2).14,15 We hypothesized that the cyclitol analogues (3a–c) (i.e., sans-anomeric stabilization) would serve as exact conformational mimics of the natural sugar; whereas the enantiomeric analogues (ent)-3a–c serve as control molecules. Finally, to further test the importance of the C6″ alkyl group, we envisioned preparing and evaluating the desmethyl cyclitol analogue 4.
Figure 1.

Structures of SL0101 (1a) and top analogue 2.
Figure 2.
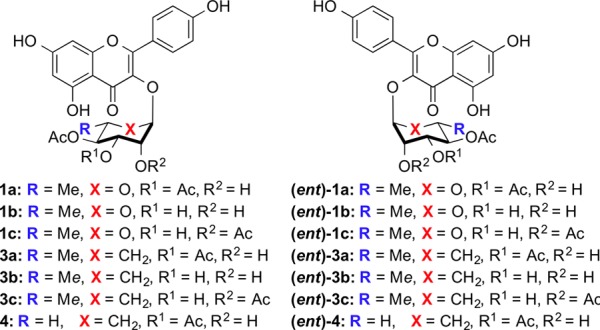
Structure of d-/l-SL0101 analogues 1–4.
We have been developing practical and generalizable approaches to both pyranose16−24 and 5a-carbasugar14,15,25,26 and have reported synthetic approaches to SL0101 and its derivatives. The general approach to these analogues is outlined in Scheme 1. The technology that enables this approach was the use of a Pd-catalyzed glycosylation25−29 or cyclitolization14,15 and subsequent post-glycosylation transformations. Using the Pd-catalyzed glycosylation, the desired pyranose analogues 1a–c and (ent)-1a–c were produced in five to seven steps from pyranones α-l-5 and α-d-5, respectively. Using the related Pd-catalyzed cyclitolization and in the same number of steps, the desired cyclitol analogues 3a–c and (ent)-3a–c were produced from the corresponding enones α-l-6 and α-d-6.
Scheme 1. Enantiodivergent Synthesis of SL0101 Analogues 1 and 3.
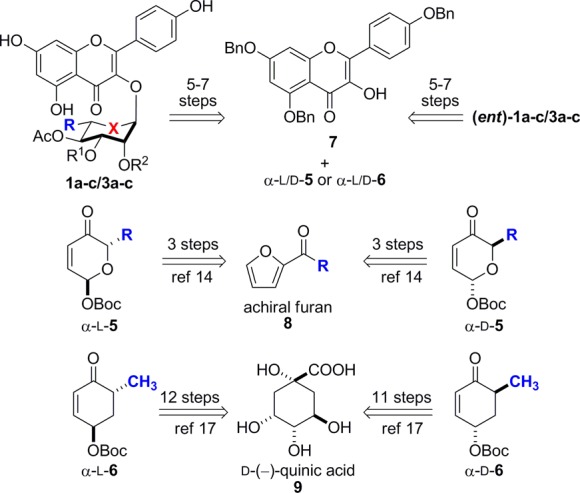
The key to the success of this approach is the reliance of an enantio-divergent (i.e., d/l) and highly stereocontrolled synthesis of both glycosyl- and cyclitol-donors from readily available intermediates (8 and 9, Scheme 1). For instance, the pyranose glycosyl donors were readily prepared in three steps from achiral acylfuran intermediate 8. In contrast, the carbasugar cyclitol donor 6 was significantly more difficult to prepare. Like the pyranones 5, the cyclitol 6 can also be prepared from a single intermediate, d-quinic acid 9. Thus, in 11 steps, d-quinic acid was converted into α-d-enone α-d-6, whereas in a related 12-step sequence quinic acid can be also converted into its enantiomeric enone, α-l-6.
With access to the cyclitol analogues 3, we next pursued the de novo asymmetric synthesis of the desmethyl cyclitol analogues 4 and (ent)-4 (Schemes 3 and 4). Interestingly, the removal of the C6″-methyl group greatly simplifies the analogue synthesis. The simplicity of this approach is enabled by the use of the Trost asymmetric allylation of 7 with meso-1,4-bis-benzoate 10 to form either enantiomer of cyclitol 11.30−32 Thus, by appropriate choice of the chiral ligand, cyclitol 11 (via (S,S)-DACH) or its enantiomer (ent)-11 (via (R,R)-DACH) was prepared in only one stereodivergent step. The enantiomeric excess of 12 and (ent)-12 were determined to be >96% ee by Mosher ester analysis. This was accomplished by converting 12 and (ent)-12 into their corresponding Mosher ester and integrating resolved diasteromeric vinyl protons in the 1H NMR (see Supporting Information).
Scheme 3. Synthesis of SL0101 Cyclitol Analogue 4.

Scheme 4. Synthesis of SL0101 Cyclitol Analogue (ent)-4.

Scheme 2. Enantiodivergent Cyclitolization of Aglycon 7.

With the C1″/C4″ stereochemistry installed in 11, the C4″ benzoate was transformed into an acetate (Scheme 3), via a hydrolysis and acylation sequence (11 to 13). Using an Upjohn dihydroxylation (1% OsO4/NMO),33 the C2″/C3″-hydroxyl groups were stereoselectively installed in 14. The required C3″ acetate was regioselectively installed by means of the Taylor catalysis (14 to 15).34−37Finally hydrogenolysis was used for a global debenzylation of 15 to give the desired cyclitol analogue 4. Using an identical sequence, the enantiomer 11 was converted into the enantiomeric analogue (ent)-4 (Scheme 4).
Using purified recombinant RSK2 enzyme in an in vitro kinase assay, the analogues (ent)-1a, 3a–c, (ent)-3a–c, 4 and (ent)-4 were evaluated for their ability to inhibit RSK2 kinase activity.38 Nonlinear regression analysis was used to fit the data (Table 1). Regardless of substitution, we found an absolute requirement for the l-isoform, as none of the d-isoforms displayed any RSK2 kinase inhibitory activity at concentrations ≤30 μM.
Table 1. In Vitro Potency of SL0101 (1a) and Analoguesa.
| name | RSK2 IC50 (μM) | RSK2 IC50p(1a) |
|---|---|---|
| SL0101 (1a) | 0.37 ± 0.13 | |
| (ent)-1a | N.D. | |
| 3a | 0.27 ± 0.15 | 0.0404 |
| (ent)-3a | N.D. | |
| 3b | 1.23 ± 0.62 | <0.0001 |
| (ent)-3b | N.D. | |
| 3c | 1.60 ± 0.66 | <0.0001 |
| (ent)-3c | N.D. | |
| 4 | 8.93 ± 1.02 | <0.0001 |
| (ent)-4a | N.D. |
RSK2 IC50: concentration needed for 50% RSK2 inhibition (n > 2; quadruplicate: mean; S.D., p(1a) Student’s t test compared to SL0101(1a)). N.D.: no inhibition detected.
Interestingly, we found that replacing the ring oxygen in the rhamnose ring with a methylene did not interfere with in vitro RSK2 inhibitory activity (Table 1). In fact, the cyclitol analogue 3a was a slightly better inhibitor of RSK than SL0101, albeit the difference is unlikely to be biologically meaningful. In contrast, the cyclitols with varied acetate substitution (3b and 3c) had higher IC50s. This trend was consistent to what was observed for the related rhamnose sugar analogues (1b and 1c).39 The C6″ methyl group proved to be important for activity, as the desmethyl analogue 4 was a poor inhibitor. Even in the less active desmethyl series, the importance of the sugar absolute stereochemistry could be seen, as 4 was significantly more active than its enantiomer (ent)-4. This result is consistent with our crystal structure of the RSK2 NTKD/SL0101 complex. Specifically, we observed SL0101 in a specific 3D orientation with the C6″ methyl group residing in a key hydrophobic pocket10 and that alkyl substitution of the C6″ alkyl group (Me to n-Pr) increased the affinity for RSK2.13
The ability of the analogues to inhibit the proliferation of the breast cancer cell line, MCF-7, was compared to that obtained with the immortalized nontransformed human breast cell line, MCF-10A. We have found that a preferential ability to inhibit MCF-7 compared to MCF-10A proliferation correlates with RSK inhibition.13,38−41 The cyclitol analogue 3b inhibited both cell lines to the same extent, which suggests that it does not specifically inhibit RSK (Figure 3). In contrast, the enantiomer (ent)-3b showed no inhibition of either cell line. At 25 μM the analogue 3a inhibited MCF-7 proliferation by ∼60% but also significantly inhibited MCF-10A proliferation (Figure 4A). For comparison, at 100 μM SL0101 (1a) inhibited MCF-7 proliferation by ∼60% but had no effect on MCF-10A proliferation. Both analogues 3a and 3c were able to completely inhibit proliferation of MCF-7 cells at ∼50 μM, but they also significantly inhibited MCF-10A proliferation at that concentration (Figure 4). For both 3a and 3c the dose response differed by ∼3-fold between MCF-7 and MCF-10A cells. This modest differential effect suggests that the inhibitors are not specific for RSK, as we have found that MCF-10A proliferation is not dependent on RSK.
Figure 3.

C3″,C4″ acetates are essential for preferential inhibition of MCF-7 proliferation. The inhibitors were added at time 0, and ATP content was measured after 48 h of treatment. Values are the fold proliferation as a % of the control (n ≥ 2 in triplicate; mean, S.D.; *p < 0.01 in a Student’s t test compared to the appropriate cell line in the presence of vehicle).
Figure 4.
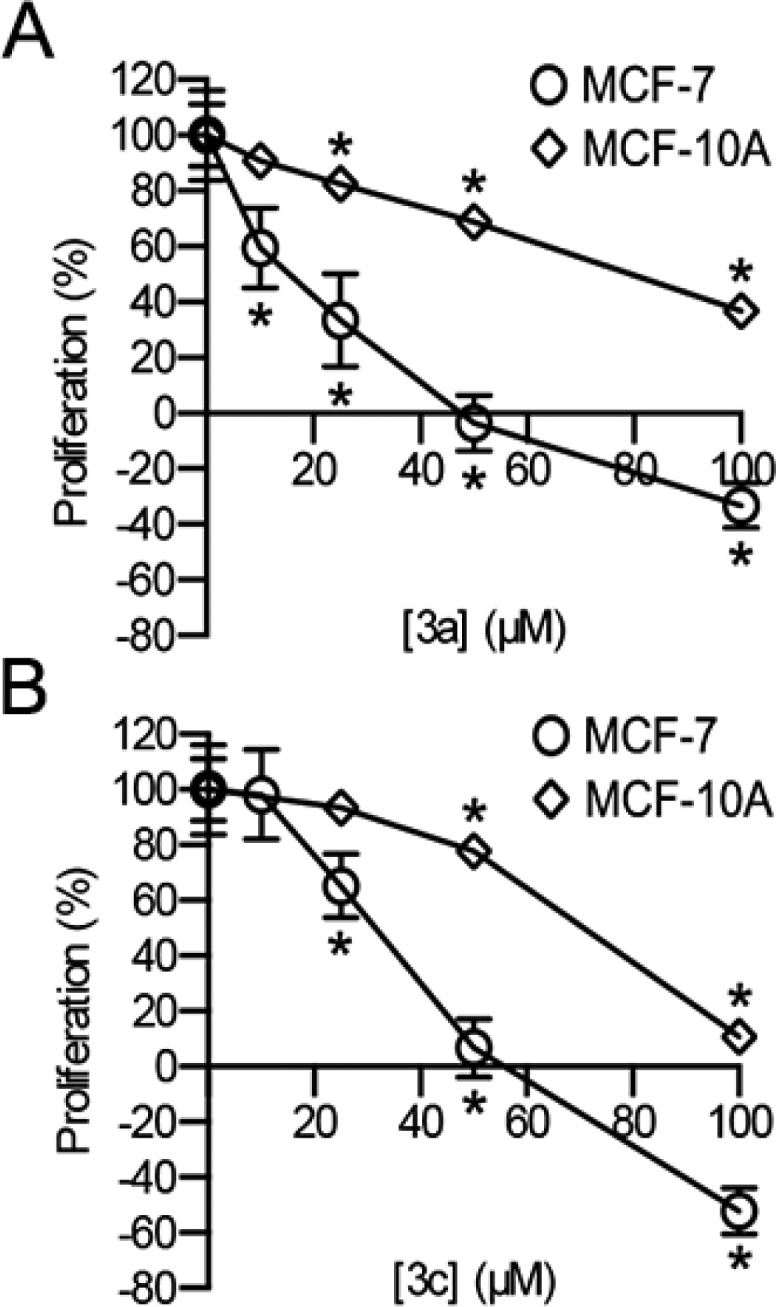
Efficacy and specificity of analogues 3a and 3c for inhibition of RSK. As described in Figure 3 (n ≥ 2 in triplicate; mean, S.D.; *p < 0.01 in a Student’s t test compared to control).
To further investigate the specificity of 3a and 3c for inhibition of RSK we determined their ability to inhibit known RSK substrates in comparison to SL0101. We tested the compounds 3a and 3c at 50 μM, which is the cytostatic concentration. Lysates were generated from MCF-7 cells that had been treated with the mitogen, phorbol myristate acetate (PMA) after a pretreatment with inhibitor or vehicle. Inhibition of RSK is known to result in an increase in the phosphorylation of eukaryotic elongation factor 2 (p-eEF2) via release of the RSK-induced repression of eEF2 kinase.42 As expected SL0101 dramatically enhanced p-eEF2 levels, but 3a and 3c induced only a minor increase (Figure 5A). To further evaluate whether the analogues could alter RSK biomarkers we used an antibody against a phosphorylation motif, which is recognized by a subset of the AGC family of kinases, which includes RSK. SL0101 decreased the intensity of a band at ∼65 and ∼27 kDa, but 3a and 3c did not alter the phosphorylation pattern compared to the PMA control (Figure 5B). We have also determined that RSK regulates the levels of the oncogene, cyclin D1.43 In agreement with our previous observations SL0101 decreased cyclin D1 levels, whereas 3a and 3c had no effect (Figure 5A). We conclude that 3a and 3c are not specific for RSK inhibition in cell-based assays.
Figure 5.

Evaluation of 3a and 3c as RSK-specific inhibitors in MCF-7 cells. MCF-7 cells were treated with PMA after pretreatment with the indicated inhibitors. Lysates of the cells were immunoblotted. The arrows indicate bands whose intensity decreases upon treatment of cells with SL0101 (1a).
To obtain insight into kinases that 3a and 3c could target we used antibodies that detect the phosphorylation motif of protein kinase A (PKA), protein kinase C (PKC), and tyrosine kinases. Cyclitols 3a and 3c did not alter the phosphorylation pattern obtained with antibodies to the PKC and tyrosine kinase phosphorylation motifs (Figure S1, Supporting Information). However, 3a and 3c resulted in the partial increase in the intensity of a band at ∼90 kDa. In contrast, SL0101 dramatically increased the intensity of this band compared to PMA. The PKA motif antibody is able to detect phosphorylations generated by RSK, and therefore, observing changes with SL0101 is expected. On the basis of our immunoblot analysis, 3a and 3c do not inhibit kinases that prefer an Arg at the -5 position but do inhibit kinases that prefer an Arg at the -3 and -2 positions from the Ser or Thr phosphorylation site. This information narrows down the possible candidate kinases from within the AGC kinase family that 3a and 3c target.
In conclusion, using a Pd-catalyzed glycosylation or cyclitolization in combination with post-glycosylation transformation, an enantiomerically diverse set of SL0101 analogues were prepared and evaluated as RSK inhibitors. Replacement of the l-rhamno-sugar with a l-rhamno-5a-carbasugar did not substantially alter the ability of the analogues to inhibit RSK kinase activity in vitro; however, the compounds demonstrated off-target effects in cell-based assays. Further efforts aimed at identifying cyclitol analogues that specifically target RSK inhibition are ongoing and will be reported in due course.
Supporting Information Available
Experimental details for synthetic procedure and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∇ Co-first authors; the order is alphabetical.
This work was supported by NIH (GM09025901 to G.A.O.), NSF (CHE-0749451 to G.A.O.), and Susan G. Komen Breast Cancer Foundation (#IIR12223770 to D.A.L.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eisinger-Mathason T. S.; Andrade J.; Lannigan D. A. RSK in tumorigenesis: connections to steroid signaling. Steroids 2010, 75, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronchik I.; Appleton B. A.; Basham S. E.; Crawford K.; Del Rosario M.; Doyle L. V.; Estacio W. F.; Lan J.; Lindvall M. K.; Luu C. A.; Ornelas E.; Venetsanakos E.; Shafer C. M.; Jefferson A. B. Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Mol. Cancer Res. 2014, 12, 803–812. [DOI] [PubMed] [Google Scholar]
- Cohen M. S.; Hadjivassiliou H.; Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat. Chem. Biol. 2007, 3, 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. S.; Zhang C.; Shokat K. M.; Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. L.; Gussio R.; Smith J. A.; Lannigan D. A.; Hecht S. M.; Scudiero D. A.; Shoemaker R. H.; Zaharevitz D. W. Homology model of RSK2 N-terminal kinase domain, structure-based identification of novel RSK2 inhibitors, and preliminary common pharmacophore. Bioorg. Med. Chem. 2006, 14, 6097–6105. [DOI] [PubMed] [Google Scholar]
- Lowe H. I.; Facey C. O.; Toyang N. J.; Bryant J. L. Specific RSK kinase inhibition by dibenzyl trisulfide and implication for therapeutic treatment of cancer. Anticancer Res. 2014, 34, 1637–1641. [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; McLauchlan H.; Klevernic I.; Arthur J. S.; Alessi D. R.; Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costales A.; Mathur M.; Ramurthy S.; Lan J.; Subramanian S.; Jain R.; Atallah G.; Setti L.; Lindvall M.; Appleton B. A.; Ornelas E.; Feucht P.; Warne B.; Doyle L.; Basham S. E.; Aronchik I.; Jefferson A. B.; Shafer C. M. 2-Amino-7-substituted benzoxazole analogs as potent RSK2 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1592–1596. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Xue M.; Zhao X.; Yuan J.; Liu X.; Huang J.; Zhao Z.; Li H.; Xu Y. Substituted indolin-2-ones as p90 ribosomal S6 protein kinase (RSK2) inhibitors: Molecular docking simulation and structure-activity relationship analysis. Bioorg. Med. Chem. 2013, 21, 1724–1734. [DOI] [PubMed] [Google Scholar]
- Utepbergenov D.; Derewenda U.; Olekhnovich N.; Szukalska G.; Banerjee B.; Hilinski M. K.; Lannigan D. A.; Stukenberg P. T.; Derewenda Z. S. Insights into the inhibition of the p90 ribosomal S6 kinase (RSK) by the flavonol glycoside SL0101 from the 1.5 Å crystal structure of the N-terminal domain of RSK2 with bound inhibitor. Biochemistry 2012, 51, 6499–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehn U.; Hauge C.; Frank S. R.; Jensen C. J.; Duda K.; Nielsen J. V.; Cohen M. S.; Johansen J. V.; Winther B. R.; Lund L. R.; Winther O.; Taunton J.; Hansen S. H.; Frodin M. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 35, 511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum R.; Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [DOI] [PubMed] [Google Scholar]
- Mrozowski R. M.; Vemula R.; Wu B.; Zhang Q.; Schroeder B. R.; Hilinski M. K.; Clark D. E.; Hecht S. M.; O’Doherty G. A.; Lannigan D. A. Improving the affinity of SL0101 for RSK using structure-based design. ACS Med. Chem. Lett. 2012, 4, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y. L.; Wu B.; Zhang Q.; Kang S.-W.; Rojanasakul Y.; O'Doherty G. A. C5′-Alkyl Substitution Effects on Digitoxigenin α-l-Glycoside Cancer Cytotoxicity. ACS Med. Chem. Lett. 2011, 2, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrozowski R. M.; Sandusky Z. M.; Vemula R.; Wu B.; Zhang Q.; Lannigan D. A.; O'Doherty G. A. De Novo Synthesis and Biological Evaluation of C6'-Substituted C4'-Amide Analogues of SL0101. Org. Lett. 2014, 16, 5996–5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O’Doherty G. A. De novo asymmetric synthesis of SL0101 and its analogues via a palladium-catalyzed glycosylation. Org. Lett. 2006, 8, 5149–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O’Doherty G. A. Synthesis of SL0101 carbasugar analogues: carbasugars via Pd-catalyzed cyclitolization and post cyclitolization transformations. Org. Lett. 2010, 12, 2986–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj S. O.; Sharif E. U.; Akhmedov N. G.; O’Doherty G. A. De novo asymmetric synthesis of the mezzettiaside family of natural products via the iterative use of a dual B-/Pd-catalyzed glycosylation. Chem. Sci. 2014, 5, 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljahdali A. Z.; Shi P.; Zhong Y.; O’Doherty G. A.. De Novo Asymmetric Synthesis of the Pyranoses. In Advances in Carbohydrate Chemistry and Biochemistry; Horton D., Ed.; Elsevier: New York, 2013; Vol. 69, pp 55–123. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Keranen M. D.; O’Doherty G. A. Syntheses of d- and l-mannose, gulose, and talose via diastereoselective and enantioselective dihydroxylation reactions. J. Org. Chem. 1999, 64, 2982–2983. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Keranen M. D.; Nguyen H.; Young V. G.; O’Doherty G. A. Syntheses of four d- and l-hexoses via diastereoselective and enantioselective dihydroxylation reactions. Carbohydr. Res. 2000, 328, 17–36. [DOI] [PubMed] [Google Scholar]
- For its use in oligosaccharide synthesis, seeBabu R. S.; Zhou M.; O’Doherty G. A. De novo synthesis of oligosaccharides using a palladium-catalyzed glycosylation reaction. J. Am. Chem. Soc. 2004, 126, 3428–3429. [DOI] [PubMed] [Google Scholar]
- Ahmed M. M.; O’Doherty G. A. De novo synthesis of galacto-sugar d-lactones via a catalytic osmium/palladium/osmium reaction sequence. Tetrahedron Lett. 2005, 46, 3015–3019. [Google Scholar]
- Ahmed M. M.; Berry B. P.; Hunter T. J.; Tomcik D. J.; O’Doherty G. A. De novo enantioselective syntheses of galacto-sugars and deoxy sugars via the iterative dihydroxylation of dienoate. Org. Lett. 2005, 7, 745–748. [DOI] [PubMed] [Google Scholar]
- Guo H.; O’Doherty G. A. De novo asymmetric synthesis of the anthrax tetrasaccharide by a palladium-catalyzed glycosylation reaction. Angew. Chem., Int. Ed. 2007, 46, 5206–5208. [DOI] [PubMed] [Google Scholar]
- Zhou M.; O’Doherty G. A. De novo synthesis of the trisaccharide subunit of landomycins A and E. Org. Lett. 2008, 10, 2283–2286. [DOI] [PubMed] [Google Scholar]
- Shan M.; O’Doherty G. A. Synthesis of cyclitol sugars via Pd-catalyzed cyclopropanol ring opening. Synthesis 2008, 19, 3171–3179. [DOI] [PubMed] [Google Scholar]
- Shan M.; Sharif E. U.; O’Doherty G. A. Total synthesis of jadomycin A and carbasugar analogue of jadomycin B. Angew. Chem., Int. Ed. 2010, 49, 9492–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu R. S.; O’Doherty G. A. A palladium-catalyzed glycosylation reaction: the de novo synthesis of natural and unnatural glycosides. J. Am. Chem. Soc. 2003, 125, 12406–12407. [DOI] [PubMed] [Google Scholar]
- Babu R. S.; O’Doherty G. A. Palladium catalyzed glycosylation reaction: de-novo synthesis of trehalose analogues. J. Carbohydr. Chem. 2005, 24, 169–177. [Google Scholar]
- Guo H.; O’Doherty G. A. De novo asymmetric synthesis of anthrax tetrasaccharide and analogue. J. Org. Chem. 2008, 73, 5211–5220. [DOI] [PubMed] [Google Scholar]
- Trost B. M. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 2003, 103, 2921–2944. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Machacek M. R.; Aponick A. Predicting the stereochemistry of diphenylphosphino benzoic acid (DPPBA)-based palladium-catalyzed asymmetric allylic alkylation reactions: a working model. Acc. Chem. Res. 2006, 39, 747–760. [DOI] [PubMed] [Google Scholar]
- VanRheenen V.; Kelly R. C.; Cha D. Y. An improved catalytic OsO4 oxidation of olefins to -1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17, 1973–1976. [Google Scholar]
- Chan L.; Taylor M. S. Regioselective alkylation of carbohydrate derivatives catalyzed by a diarylborinic acid derivative. Org. Lett. 2011, 13, 3090–3093. [DOI] [PubMed] [Google Scholar]
- Gouliaras C.; Lee D.; Chan L.; Taylor M. S. Regioselective activation of glycosyl acceptors by a diarylborinic acid-derived catalyst. J. Am. Chem. Soc. 2011, 133, 13926–13929. [DOI] [PubMed] [Google Scholar]
- Lee D.; Taylor M. S. Borinic acid-catalyzed regioselective acylation of carbohydrate derivatives. J. Am. Chem. Soc. 2011, 133, 3724–3727. [DOI] [PubMed] [Google Scholar]
- Lee D.; Williamson C. L.; Chan L.; Taylor M. S. Regioselective, borinic acid-catalyzed monoacylation, sulfonylation and alkylation of diols and carbohydrates: expansion of substrate scope and mechanistic studies. J. Am. Chem. Soc. 2012, 134, 8260–8267. [DOI] [PubMed] [Google Scholar]
- Smith J. A.; Poteet-Smith C. E.; Xu Y.; Errington T. M.; Hecht S. M.; Lannigan D. A. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005, 65, 1027–1034. [PubMed] [Google Scholar]
- Smith J. A.; Maloney D. J.; Clark D. E.; Xu Y.; Hecht S. M.; Lannigan D. A. Influence of rhamnose substituents on the potency of SL0101, an inhibitor of the Ser/Thr kinase, RSK. Bioorg. Med. Chem. 2006, 14, 6034–6042. [DOI] [PubMed] [Google Scholar]
- Hilinski M. K.; Mrozowski R. M.; Clark D. E.; Lannigan D. A. Analogs of the RSK inhibitor SL0101: optimization of in vitro biological stability. Bioorg. Med. Chem. Lett. 2012, 22, 3244–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A.; Maloney D. J.; Hecht S. M.; Lannigan D. A. Structural basis for the activity of the RSK-specific inhibitor, SL0101. Bioorg. Med. Chem. 2007, 15, 5018–5034. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li W.; Williams M.; Terada N.; Alessi D. R.; Proud C. G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger-Mathason T. S.; Andrade J.; Groehler A. L.; Clark D. E.; Muratore-Schroeder T. L.; Pasic L.; Smith J. A.; Shabanowitz J.; Hunt D. F.; Macara I. G.; Lannigan D. A. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol. Cell 2008, 31, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.