

Abstract
Lenvatinib is an oral multikinase inhibitor that selectively inhibits vascular endothelial growth factor (VEGF) receptors 1 to 3 and other proangiogenic and oncogenic pathway-related receptor tyrosine kinases. To elucidate the origin of the potency of lenvatinib in VEGF receptor 2 (VEGFR2) inhibition, we conducted a kinetic interaction analysis of lenvatinib with VEGFR2 and X-ray analysis of the crystal structure of VEGFR2–lenvatinib complexes. Kinetic analysis revealed that lenvatinib had a rapid association rate constant and a relatively slow dissociation rate constant in complex with VEGFR2. Co-crystal structure analysis demonstrated that lenvatinib binds at its ATP mimetic quinoline moiety to the ATP binding site and to the neighboring region via a cyclopropane ring, adopting an Asp-Phe-Gly (DFG)-“in” conformation. These results suggest that lenvatinib is very distinct in its binding mode of interaction compared to the several approved VEGFR2 kinase inhibitors.
Keywords: Receptor tyrosine kinase, kinase inhibitor, VEGFR2, X-ray crystallography, kinetic interaction analysis
Vascular endothelial growth factor (VEGF) is a crucial regulator of both physiologic and pathologic angiogenesis and is essential for tumor growth and metastasis.1,2 Several small molecule tyrosine kinase inhibitors (TKIs) of VEGF receptor 2 (VEGFR2) have been approved for the treatment of several types of cancer, including renal cell carcinoma,3 hepatocellular carcinoma,4 and colorectal carcinoma.5
All of the currently approved VEGFR2 TKIs bind to the ATP-binding site. They are categorized into either type I or type II inhibitors on the basis of the conformation of VEGFR2 in complex with them, as revealed by cocrystal structure analysis. Type I inhibitors target the active form of VEGFR2, which is characterized by an open conformation of the activation loop.6 This conformation is referred to as “DFG-in” from the orientation of the conserved triad Asp-Phe-Gly (DFG) at the beginning of the activation loop. Inhibitors that target the inactive, DFG-out conformation, in which DFG motif is flipped out relative to its orientation in the active state, are classified as type II inhibitors.6 Generally, type I inhibitors have more rapid association and dissociation kinetics, whereas type II inhibitors have slow binding kinetics, leading to a prolonged residence time (i.e., the period of time for which a compound occupies a target).7−9
Lenvatinib is an oral multikinase inhibitor that selectively inhibits VEGFR1 to 3 and other proangiogenic and prooncogenic receptor tyrosine kinases, including fibroblast growth factor receptors 1 to 4, platelet-derived growth factor receptor α, KIT, and RET.10−12 Lenvatinib is currently being evaluated in several clinical trials, including phase 3 clinical trials in patients with thyroid cancer or hepatocellular carcinoma. Recently, lenvatinib achieved the primary end point in a phase 3 clinical trial in patients with differentiated thyroid cancer.13 To gain insight into the mode of interaction of lenvatinib with VEGFR2, we analyzed the kinetics of interaction between lenvatinib and VEGFR2 and also the crystal structure of VEGFR2 complexed with lenvatinib.
The affinity between inhibitors and target proteins is influenced by the balance between association rate and dissociation rate.9 To clarify which parameter was the key driver in the affinity of lenvatinib for VEGFR2, we determined kinetic parameters for the interaction by using the Proteros reporter displacement assay. This assay is based on the use of a reporter probe that is designed to bind to the site of interest in the target enzyme.14 Proximity between reporter and target results in the emission of an optical signal. Signal loss caused by probe displacement by inhibitors is monitored to determine the kinetic parameters. We also determined kinetic parameters for sorafenib and sunitinib with VEGFR2, which, from their cocrystal structures, are representative type II and type I VEGFR2 inhibitors, respectively.15 The kinetic parameters for the interactions of sorafenib and sunitinib with VEGFR2 clearly demonstrated the reported features of the interaction modes of these inhibitors16 (Table 1). Sorafenib had slow binding kinetics with a residence time of 64 min. In contrast, sunitinib had fast binding kinetics; its residence time was below the detection limit of the VEGFR2 reporter displacement assay (2.9 min), and we could not determine association rate constant (kon) and dissociation rate constant (koff) values. Lenvatinib had intermediate characteristics compared with those of these two representative inhibitors. The kon and koff values of lenvatinib were, respectively, 61 times and 3.8 times those of sorafenib. The residence time of lenvatinib was 17 min—longer than that of sunitinib. Lenvatinib likely associates rapidly with VEGFR2 and resides a relatively longer time than sunitinib. These differences are also mirrored in the equilibrium dissociation constant (Kd) values of lenvatinib, sorafenib, and sunitinib in complex with VEGFR2, which were 2.1, 33, and 30 nmol/L, respectively. Because of its fast association rate and prolonged residence time, lenvatinib was more than 10 times as potent as sorafenib and sunitinib against VEGFR2.
Table 1. Parameters of Kinetic Interaction with VEGFR2a.
| compd | Kd (nmol/L) | kon (s–1 × M–1) | koff (s–1) | residence time (min) |
|---|---|---|---|---|
| lenvatinib | 2.1 ± 0.1 | 4.8 × 105 ± 1.4 × 104 | 9.9 × 10–4 ± 9.0 × 10–5 | 17 ± 2 |
| sorafenib | 33 ± 2.6 | 7.9 × 103 ± 1.5 × 102 | 2.6 × 10–4 ± 2.5 × 10–5 | 64 ± 6 |
| sunitinib | 30 ± 1.7 | >1.9 × 105 | >57 × 10–4 | <2.9 |
Kd = equilibrium dissociation constant, calculated by using the Cheng–Prusoff equation from IC50-like values obtained from the percent displacement values at time point, at which system has reached equilibrium; kon = association rate constant; displacement traces at each compound concentration were fitted according to a monoexponential equation to yield kobs. Secondary plot of kobs over compound concentration was linearly fitted to generate kon; koff = dissociation rate constant (= Kd × kon); residence time = 1/koff. From standard errors of IC50 and kon calculated in regression analysis for curve fitting, errors for Kd, koff, and residence time were calculated by Gaussian error propagation.
Next, we analyzed the crystal structure of a complex of VEGFR2 with either lenvatinib or sorafenib. First, we purified recombinantly expressed human VEGFR2 kinase domain (Leu814–Asn1162) and incubated it with lenvatinib or sorafenib. Single crystals of VEGFR2 were obtained by cocrystallization. The structures were solved at a resolution of 1.57 Å (lenvatinib) or 1.90 Å (sorafenib), revealing the details of the modes of binding of each inhibitor to VEGFR2 (Figure 1a–d). The overall structure of VEGFR2 in complex with sorafenib adopted a bilobar architecture characteristic of the eukaryotic protein kinase family. Situated in the cleft formed between the N-terminal and the C-terminal lobe, sorafenib bound to both the ATP-binding site (common sites for protein kinases) and the neighboring nonconserved allosteric region.17 The DFG motif adopted a DFG-out conformation (inactive form) in the VEGFR2–sorafenib complex (Figure 1d). These results for sorafenib agree well with those reported by McTigue et al.18
Figure 1.

Overall structure of VEGFR2 with each ligand. (a) VEGFR2 complex with lenvatinib; (b) VEGFR2 complex with sorafenib. Each ligand is shown as a CPK model colored according to the chemical atom type; carbon atoms are green for lenvatinib and blue for sorafenib. One monomer of VEGFR2 is represented as a ribbon. The beta-sheet is shown in blue, the helixes in red, and other components in gray. Binding pocket of VEGFR2 complexes. (c) VEGFR2 complex with lenvatinib; (d) VEGFR2 complex with sorafenib. The ligand and neighboring protein side chains are shown as stick models colored according to the chemical atom type, as described above. Carbon atoms of the DFG domain are salmon colored. Residue names at ATP-binding sites are indicated by red letters, and neighboring regions by black letters. Gatekeeper residue, Val916, is indicated by blue-gray letters and Asn923, which makes a water-mediated interaction with lenvatinib, is indicated by maroon letters in italics. Interaction scheme of VEGFR2 complexes. (e) VEGFR2 complex with lenvatinib; (f) VEGFR2 complex with sorafenib. Each ligand is shown as a ball and stick model; residues with some interaction with each ligand are circled by a different color according to the type of interaction: those involved in hydrogen bonding, charge, or polar interactions are in pink circles. Residues involved in van der Waals interactions are in green circles. Water molecules bridging lenvatinib and Asn923 are shown in light blue circles. Hydrogen bond interactions with amino acid side chains are represented by blue dashed arrows directed toward the electron donor. Hydrogen bond interactions with amino acid main chains are represented by green dashed arrows directed toward the electron donor, and black dashed arrows indicate hydrogen bond interaction with water molecules. Pi interactions are represented by orange lines indicating the interaction.
X-ray analysis of the crystal structure of VEGFR2–lenvatinib complexes demonstrated that the mode of binding of lenvatinib to VEGFR2 differed partly from that of sorafenib to VEGFR2. Lenvatinib was also situated in the cleft between the two lobes. However, the conformation of VEGFR2 complexed with lenvatinib was less closed, possibly because of the different conformation of the DFG motif. There was a notable difference in the conformation of the DFG motif and the N-terminal part of the activation loop between the VEGFR2–lenvatinib complex and the VEGFR2–sorafenib complex (Figure 1a,b). The DFG motif adopted the DFG-in conformation in the VEGFR2–lenvatinib complex (Figure 1c), unlike the DFG-out one in the VEGFR2–sorafenib complex (Figure 1d). The latter conformation might result from the presence of a large residue of 4-chloro-3-(trifluoromethyl) phenyl of sorafenib occupying the neighboring region. In contrast, the small substituent cyclopropane ring of lenvatinib might allow this inhibitor to bind to VEGFR2 in a DFG-in conformation. The induced-fit mechanism is the most common mechanism of slow binding by enzyme inhibitors.19 For higher-affinity binding with sorafenib, VEGFR2 changes conformation from DFG-in to DFG-out. This conformational change may be the cause of the low kon of sorafenib. In contrast, this conformational change is not present in the case of lenvatinib binding. This likely explains the large difference in kon between these compounds.
The amino acid residues located in the vicinity of lenvatinib or sorafenib at a maximum distance of 3.9 Å are summarized in Table S3, Supporting Information. These amino acid residues belonged to the ATP-binding site, a gatekeeper residue, and the neighboring region. Among the total of 25 amino acid residues, 16 were common to lenvatinib- and sorafenib-binding sites.
Lenvatinib and sorafenib bound at their cores, from the urea group to the quinoline ring (lenvatinib) and pyridine (sorafenib), to the ATP-binding site (Figure 1c,d). These cores occupied nearly identical positions. On the basis of a distance of less than 3.5 Å between donor and acceptor atoms, we identified a total of four specific hydrogen bonds of lenvatinib to the main-chain atoms of Cys919 and Asp1046 and the side-chain atoms of Glu885; there was a total of five bonds in these locations in the case of sorafenib (Figure 1e,f). The hydrogen bond from the nitrogen atom of Asp1046 to the oxygen atom of the urea group of the ligand was present in both structures. Sorafenib formed two hydrogen bonds to the hinge region, whereas there was only one in the case of lenvatinib. However, lenvatinib formed additional hydrophilic interactions with an amide group in the hinge region (Figure 1e,f). The glycine-rich loop was completely ordered in the structure with sorafenib, but it was mostly flexible in the structure with lenvatinib. In the structure with sorafenib, the turn of this loop was supported by the residues Leu1049 to Ala1050 following the DFG motif. These residues were completely flexible in the structure with lenvatinib.
In the VEGFR2–lenvatinib complex, the 6-carbamoyl substituent of the quinoline ring of lenvatinib generated two possible hydrophilic interactions with the main and side chains of Asn923, bridged by water molecules; we did not identify these in the VEGFR2–sorafenib complex (Figure 1e,f). Hydrogen bonding with the hinge regions is likely an indispensable interaction. Examination of the Connolly surface of the binding sites (Figure 2) clearly showed that lenvatinib completely occupied the adenine ring binding site and had strong hydrophobic interactions within the entrance region. The entrance region is frequently used to gain selectivity because of its diversity of sequence and conformation; it also might positively influence the residence time.20,21
Figure 2.

Binding pocket of VEGFR2 in complex with each ligand. Each ligand is presented as a CPK model, and the Connolly surface areas of the binding sites are presented as a mesh. The adenine ring binding site is indicated by the blue circle and the entrance region by the red circle. (a) View from the N-terminal domain of VEGFR2 complexed with lenvatinib. (b) View from the N-terminal domain of VEGFR2 complexed with sorafenib. (c) View from the entrance region of VEGFR2 complexed with lenvatinib. (d) View from the entrance region of VEGFR2 complexed with sorafenib. Panels (a) and (b) show that lenvatinib occupies a wider area at the adenine ring binding site than does sorafenib. It is also clear from panels (c) and (d) that lenvatinib binds more effectively at the entrance region than does sorafenib.
Lenvatinib also bound to the region neighboring the ATP binding site via the moiety adjacent to the urea group, i.e., the cyclopropane ring; sorafenib bound to the neighboring region via a 4-chloro-3-(trifluoromethyl) phenyl ring. These interactions may help to prolong the residence time in complex with VEGFR2. As a representative Type I inhibitor, sunitinib had no interaction with the neighboring region and a short residence time (Table 1).18 The cyclopropane ring of lenvatinib had a CH–pi interaction with the phenyl ring of Phe1047 (Figure 3). Although the CH–pi interaction is one of weak molecular forces, it sometimes plays an important role in ligand–protein binding.22,23 The CH–pi interaction may help to prolong the residence time of lenvatinib in complex with VEGFR2.
Figure 3.
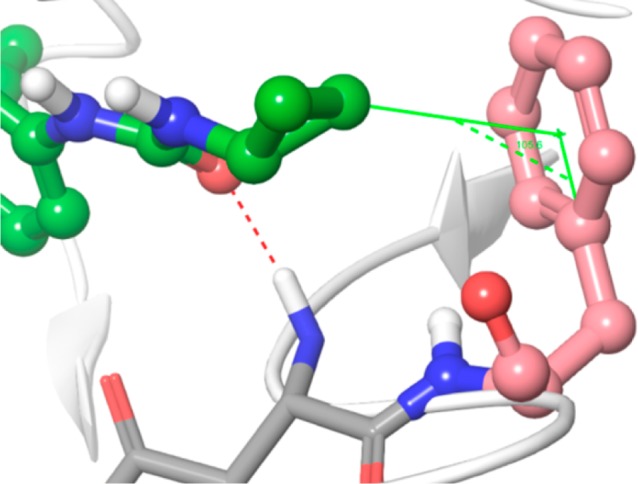
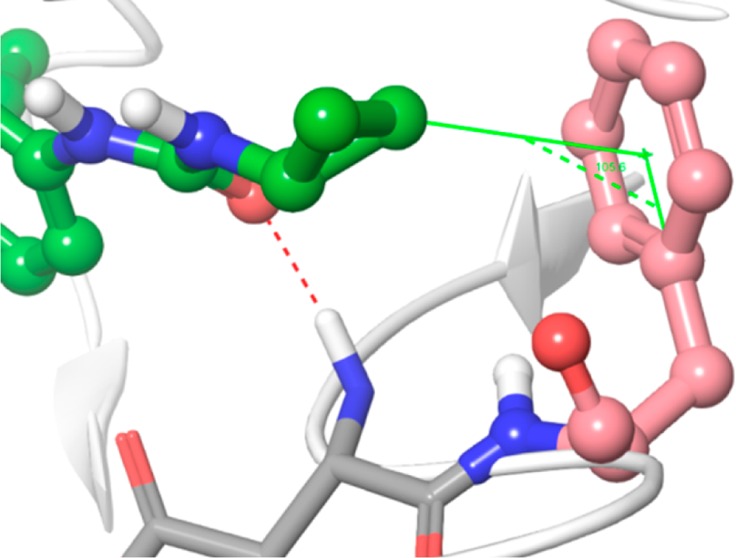
Close-up view of cyclopropane ring and Phe1047 (DFG motif). Lenvatinib is presented in a ball and stick model; carbon atoms are shown in green and the other atoms in default atom colors. VEGFR2 is illustrated as a white ribbon model. The distance between the cyclopropane ring and the centroid of the benzene ring of Phe1047 (salmon colored) is 3.43 Å and the angle is 105.6°.
The results of these cocrystal structure analyses demonstrated that although both lenvatinib and sorafenib bound to not only the ATP-binding site on VEGFR2 but also the neighboring nonconservative allosteric region, the VEGFR2 structure complexed with sorafenib had a DFG-out conformation, whereas, interestingly, VEGFR2 complexed with lenvatinib had a DFG-in conformation. The long residence time of sorafenib may result from the need for conformational change from DFG-out to DFG-in in the dissociation of sorafenib from VEGFR2. This type of retrograde induced-fit model has recently been reviewed by Copeland.19 As lenvatinib binds to VEGFR2 in a DFG-in conformation, no conformational change-related energy barrier to dissociation contributes to the residence time. Interactions of the carbamoyl of lenvatinib with Asn923 and of the cyclopropane ring with the region neighboring the ATP binding site may instead help to prolong the residence time of lenvatinib. Most inhibitors fitting to DFG-in kinases can bind only to the ATP-binding site, resulting in low kinase selectivity, whereas those that fit DFG-out kinases bind to both the ATP-binding site and allosteric regions, resulting in high kinase selectivity. Lenvatinib could bind to both the ATP-binding site and the neighboring allosteric region of VEGFR2 in DFG-in conformation, and it has high kinase selectivity comparable to that of sorafenib.24 In addition to type I and type II inhibitors, two other categories of kinase inhibitor have been proposed on the basis of their modes of interaction.25 Type III inhibitors bind to a site proximal to the ATP-binding site without interacting with the hinge region. Type IV inhibitors bind to a site distal to the ATP-binding site and induce conformational changes that render the kinase inactive. Results in this study have shown that mode of interaction of lenvatinib was different from any of these types, and we consider that lenvatinib should be classified as a different type of kinase inhibitor that binds both the ATP-binding site and the neighboring allosteric region in kinases, with DFG-in conformation.26,27 We propose to classify such compounds as type V inhibitors (Table 2).
Table 2. Comparison of General Properties of Kinase Inhibitorsa.
| type I | type II | type III | type IV | type V | |
|---|---|---|---|---|---|
| DFG conformation | in | out | out | ND | in |
| binding region | ATP-binding site | ATP-binding
site and neighboring region |
neighboring region |
allosteric site not adjacent to ATP-binding site |
ATP-binding site and neighboring region |
| ATP competitive | yes | yes | no | no | yes |
| selectivity | usually low | high | high | high | high |
| association kinetics | rapid | slow | slow | ND | rapid |
| dissociation kinetics | rapid | slow | slow | ND | relatively slow |
ND, not determined.
In conclusion, a kinetic interaction analysis and cocrystal structure analysis revealed the distinct mode of interaction of lenvatinib with VEGFR2. Lenvatinib had a rapid association rate constant in complex with VEGFR2 and targeted VEGFR2 in a DFG-in conformation. In addition, lenvatinib interacted with the region neighboring the kinase ATP-binding site of VEGFR2. This interaction may help to prolong the residence time compared with that of type I inhibitors. To our knowledge, lenvatinib is the first type V kinase inhibitor to have been evaluated in phase 3 clinical trials. Further clinical investigation may be verified in order to explore the association of clinical activity with the distinct binding mode of lenvatinib.
Acknowledgments
We thank ELSS, Inc., for providing editorial support funded by Eisai, Co., Ltd.
Glossary
Abbreviations
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- TKI
tyrosine kinase inhibitor
- Kd
equilibrium dissociation constant
- kon
association rate constant
- koff
dissociation rate constant
Supporting Information Available
Experimental procedures and Tables S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: VEGFR2-lenvatinib complex, 3WZD, and VEGFR2-sorafenib complex, 3WZE.
Author Contributions
∥ These authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): K.O., M.I.K., Y.F., T.M., A.T., A.I., and J.M. are employees of Eisai Co., Ltd.
Supplementary Material
References
- Folkman J. Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease. Nat. Med. 1995, 1, 27–31. [DOI] [PubMed] [Google Scholar]
- Carmeliet P.; Jain R. K. Angiogenesis in Cancer and Other Diseases. Nature 2000, 407, 249–257. [DOI] [PubMed] [Google Scholar]
- Rini B. I.; Escudier B.; Tomczak P.; Kaprin A.; Szczylik C.; Hutson T. E.; Michaelson M. D.; Gorbunova V. A.; Gore M. E.; Rusakov I. G.; Negrier S.; Ou Y.-C.; Castellano D.; Lim H. Y.; Uemura H.; Tarazi J.; Cella D.; Chen C.; Rosbrook B.; Kim S.; Motzer R. J. Comparative Effectiveness of Axitinib Versus Sorafenib in Advanced Renal Cell Carcinoma (AXIS): A Randomised Phase 3 Trial. Lancet 2011, 378, 1931–1939. [DOI] [PubMed] [Google Scholar]
- Llovet J. M.; Ricci S.; Mazzaferro V.; Hilgard P.; Gane E.; Blanc J. F.; de Oliveira A. C.; Santoro A.; Raoul J. L.; Forner A.; Schwartz M.; Porta C.; Zeuzem S.; Bolondi L.; Greten T. F.; Galle P. R.; Seitz J. F.; Borbath I.; Haussinger D.; Giannaris T.; Shan M.; Moscovici M.; Voliotis D.; Bruix J. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [DOI] [PubMed] [Google Scholar]
- Grothey A.; Van Cutsem E.; Sobrero A.; Siena S.; Falcone A.; Ychou M.; Humblet Y.; Bouche O.; Mineur L.; Barone C.; Adenis A.; Tabernero J.; Yoshino T.; Lenz H. J.; Goldberg R. M.; Sargent D. J.; Cihon F.; Cupit L.; Wagner A.; Laurent D. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. Rational Design of Inhibitors That Bind to Inactive Kinase Conformations. Nat. Chem. Biol. 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Tummino P. J.; Copeland R. A. Residence Time of Receptor-Ligand Complexes and Its Effect on Biological Function. Biochemistry 2008, 47, 5481–5492. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. Can Binding Kinetics Translate to a Clinically Differentiated Drug? From Theory to Practice. Lett. Drug Des. Discovery 2006, 3, 569–574. [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- Matsui J.; Yamamoto Y.; Funahashi Y.; Tsuruoka A.; Watanabe T.; Wakabayashi T.; Uenaka T.; Asada M. E7080, a Novel Inhibitor That Targets Multiple Kinases, Has Potent Antitumor Activities against Stem Cell Factor Producing Human Small Cell Lung Cancer H146, Based on Angiogenesis Inhibition. Int. J. Cancer 2008, 122, 664–671. [DOI] [PubMed] [Google Scholar]
- Matsui J.; Funahashi Y.; Uenaka T.; Watanabe T.; Tsuruoka A.; Asada M. Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 Via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [DOI] [PubMed] [Google Scholar]
- Okamoto K.; Kodama K.; Takase K.; Sugi N. H.; Yamamoto Y.; Iwata M.; Tsuruoka A. Antitumor Activities of the Targeted Multi-Tyrosine Kinase Inhibitor Lenvatinib (E7080) against RET Gene Fusion-Driven Tumor Models. Cancer Lett. 2013, 340, 97–103. [DOI] [PubMed] [Google Scholar]
- Schlumberger M.; Tahara M.; Wirth L. J.; Robinson B.; Brose M. S.; Elisei R.; Dutcus C. E.; de las Heras B.; Zhu J.; Habra M. A.; Newbold K.; Shah M. H.; Hoff A. O.; Gianoukakis A. G.; Kiyota N.; Taylor M. H.; Kim S.-B.; Krzyzanowska M. K.; Sherman S. I. A Phase 3, Multicenter, Double-Blind, Placebo-Controlled Trial of Lenvatinib (E7080) in Patients with 131I-Refractory Differentiated Thyroid Cancer (SELECT). ASCO Meet. Abstr. 2014, 32, LBA6008. [Google Scholar]
- Neumann L.; Ritscher A.; Müller G.; Hafenbradl D. Fragment-Based Lead Generation: Identification of Seed Fragments by a Highly Efficient Fragment Screening Technology. J. Comput.-Aided Mol. Des. 2009, 23, 501–511. [DOI] [PubMed] [Google Scholar]
- Iwata H.; Imamura S.; Hori A.; Hixon M. S.; Kimura H.; Miki H. Biochemical Characterization of TAK-593, a Novel VEGFR/PDGFR Inhibitor with a Two-Step Slow Binding Mechanism. Biochemistry 2011, 50, 738–751. [DOI] [PubMed] [Google Scholar]
- Iwata H.; Imamura S.; Hori A.; Hixon M. S.; Kimura H.; Miki H. Biochemical Characterization of a Novel Type-II VEGFR2 Kinase Inhibitor: Comparison of Binding to Non-Phosphorylated and Phosphorylated VEGFR2. Bioorg. Med. Chem. 2011, 19, 5342–5351. [DOI] [PubMed] [Google Scholar]
- Traxler P.; Furet P. Strategies toward the Design of Novel and Selective Protein Tyrosine Kinase Inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [DOI] [PubMed] [Google Scholar]
- McTigue M.; Murray B. W.; Chen J. H.; Deng Y. L.; Solowiej J.; Kania R. S. Molecular Conformations, Interactions, and Properties Associated with Drug Efficiency and Clinical Performance among VEGFR TK Inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 18281–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2013; pp 294–299. [PubMed] [Google Scholar]
- Schneider E. V.; Bottcher J.; Huber R.; Maskos K.; Neumann L. Structure-Kinetic Relationship Study of CDK8/CycC Specific Compounds. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8081–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J. J. Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J. Med. Chem. 2007, 50, 409–424. [DOI] [PubMed] [Google Scholar]
- Nishio M. CH/Pai Hydrogen Bonds in Crystals. CrystEngComm 2004, 6, 130. [Google Scholar]
- Hirabayashi A.; Mukaiyama H.; Kobayashi H.; Shiohara H.; Nakayama S.; Ozawa M.; Miyazawa K.; Misawa K.; Ohnota H.; Isaji M. Structure-Activity Relationship Studies of 5-Benzylaminoimidazo[1,2-C]Pyrimidine-8-Carboxamide Derivatives as Potent, Highly Selective ZAP-70 Kinase Inhibitors. Bioorg. Med. Chem. 2009, 17, 284–294. [DOI] [PubMed] [Google Scholar]
- Tohyama O.; Matsui J.; Kodama K.; Hata-Sugi N.; Kimura T.; Okamoto K.; Minoshima Y.; Iwata M.; Funahashi Y. Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. J. Thyroid Res. 2014, 2014, 638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard J. R.; Klüter S.; Grütter C.; Getlik M.; Rabiller M.; Rode H. B.; Rauh D. A New Screening Assay for Allosteric Inhibitors of cSrc. Nat. Chem. Biol. 2009, 5, 394–396. [DOI] [PubMed] [Google Scholar]
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53, 2681–2694. [DOI] [PubMed] [Google Scholar]
- Blanc J.; Geney R.; Menet C. Type II Kinase Inhibitors: An Opportunity in Cancer for Rational Design. Anticancer Agents Med. Chem. 2013, 13, 731–747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.