

Abstract
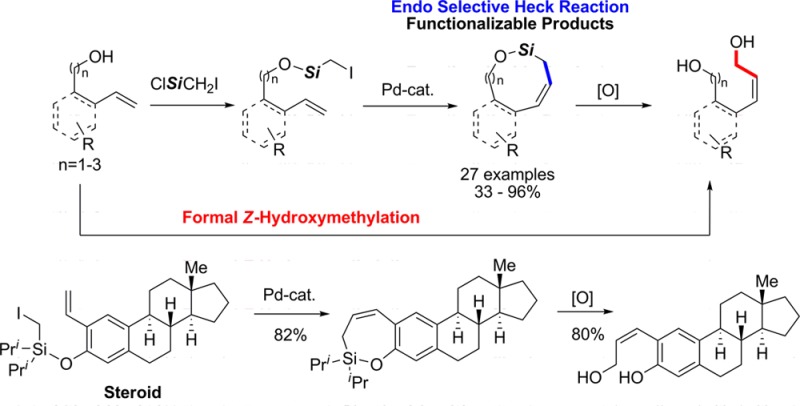
A palladium (Pd)-catalyzed endo-selective Heck reaction of iodomethylsilyl ethers of phenols and aliphatic alkenols has been developed. Mechanistic studies reveal that this silyl methyl Heck reaction operates via a hybrid Pd-radical process and that the silicon atom is crucial for the observed endo selectivity. The obtained allylic silyloxycycles were further oxidized into (Z)-alkenyldiols.
The Mizoroki–Heck reaction is a fundamental synthetic transformation that has been extensively used for synthesis of valuable alkenes.1 Although Heck reaction of sp2 halides is well established, the analogous reaction of sp3 halides, mainly due to a premature β-hydride elimination problem, is much less developed. In his seminal work, Fu solved this problem by employing NHC ligands, which promote migratory insertion over a competitive β-hydride elimination path (Scheme 1, a).2 Moreover, Alexanian3 reported an elegant alkyl Heck reaction where a premature β-hydride elimination problem was suppressed by redirecting reaction into a hybrid Pd-radical pathway.4 Both methods feature conventional exo-trig-cyclization pathway (Scheme 1, a). While, methods for selective endo-trig-Heck processes employing sp2 halides are scarcely reported,5 to the best of our knowledge, there are no reports of an endo-selective alkyl Heck reaction. Herein, we report an endo-selective silyl methyl Heck reaction of iodomethylsilyl ethers of phenols and aliphatic alkenols (Scheme 1, b). The use of the silyl tether allows for selective endo cyclization without substrate bias and enables post-modification of the obtained silyloxycycles. Moreover, by design, the employed substrates do not have the issue of premature β-hydride elimination.
Scheme 1. Intramolecular Alkyl Heck Reaction.

Halomethylsilanes have been used as a tool for a formal hydro–hydroxymethylation of allylic alcohols via a reductive radical cyclization, which is generally exo-selective.6 Conversely, in 1990, Koreeda reported that bromomethylsilyl tethered homoallylic alcohols underwent endo radical cyclization over the expected exo cyclization.7 Accordingly, we hypothesized that if this unique endo-selectivity outcome would translate into a Heck-type reaction, it would allow for a selective generation of valuable (Z)-allylic silanes (Scheme 1, b).8
To this end, we first tested our hypothesis on a conformationally biased silyl-tethered vinylphenol 1a (Table 1). Gratifyingly, it was found that under the optimized conditions,91a smoothly cyclized into 7-membered siloxycycle 2a in 79% yield with delightfully high endo-selectivity (endo:exo = 33:1, Table 1, entry 1). Next, the generality of this transformation was examined. It was found that the regiochemical outcome was not affected by electronic properties of substituents around the arene ring (entries 2–8).10 Notably, this methodology enabled the synthesis of medium size rings via 8-endo and 9-endo cyclization generating 2i and 2j, respectively, in reasonable yields. Moreover, this Heck reaction tolerates substitution at the halomethyl moiety, as secondary bromomethylsilane 1k produced aryl-substituted allylsilane 2k in good yield. However, it was found that the regioselectivity of the cyclization is sensitive to the substitution pattern of the pendent olefin. Hence, cyclization of substrates possessing substituents at the α-position produced endo products, 2l and 2m, selectively. In contrast, substitution at the β-position of olefin reversed the regioselectivity trend producing 2n and 2o, the products of the exo-trig cyclization, exclusively (entries 14, 15).
Table 1. Scope of Benzene-Tethered Substrates.

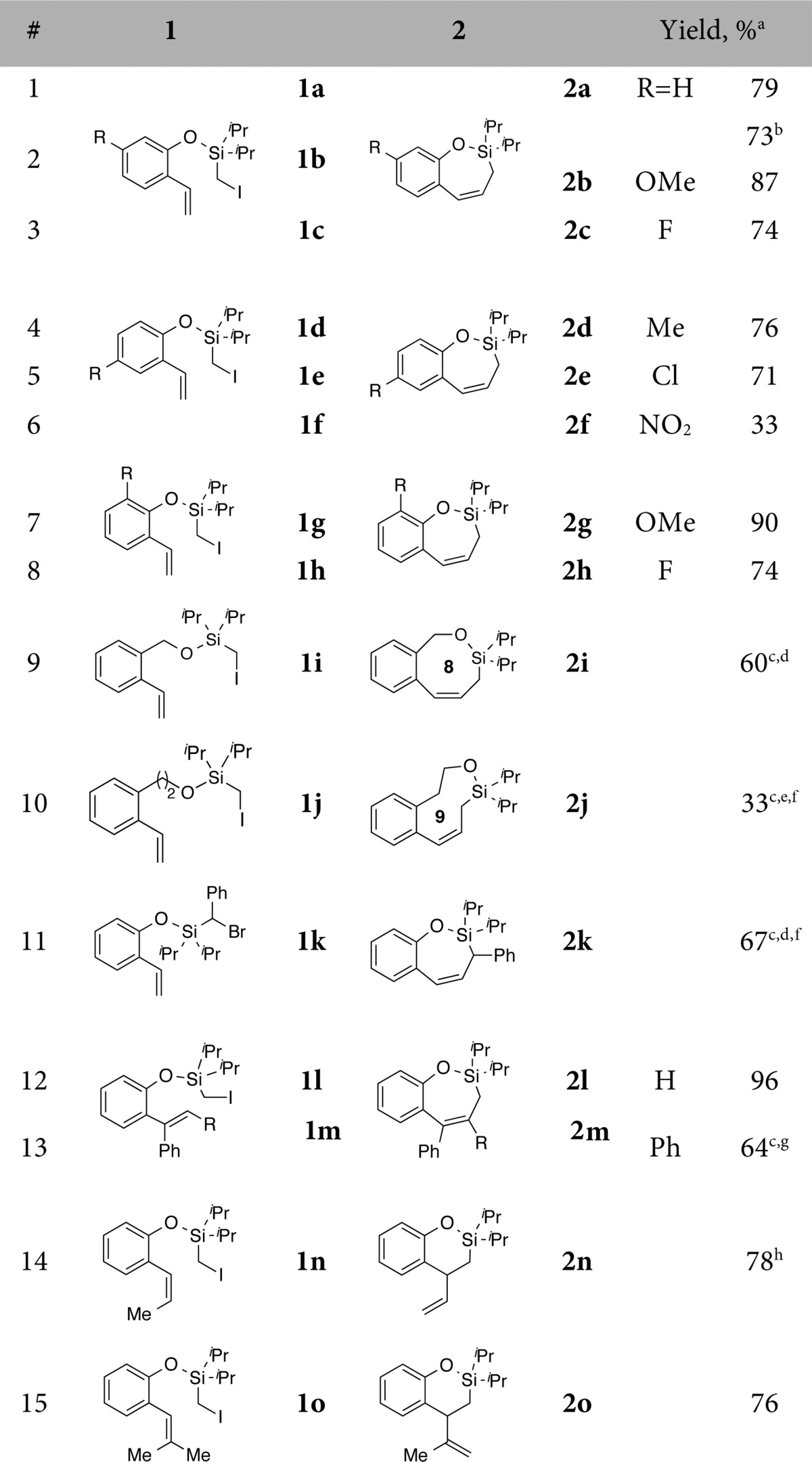
Isolated yields.
Reaction performed at 3.8 mmol scale.
Reaction performed at 120 °C.
NMR ratio of product to hydro-dehalogenation side product is 12:1.
NMR ratio of product to hydro-dehalogenation side product is 1.6:1.
Reaction time = 36 h.
Ag(OTf) was used as an additive.
Mixture of isomers.
After developing the silyl methyl Heck reaction of arene-tethered substrates, we were eager to test this methodology on more challenging systems such as aliphatic alkenols (Table 2). The regiochemical outcome of their cyclization seemed more ambiguous since (a) they are sterically unbiased and hence are less predisposed to the endo cyclization; and (b) due to the availability of alternative site for β-hydride elimination (vide infra), the reaction could also lead to the formation of homoallylic silane products. Indeed, the experiments revealed that the Heck reaction of homoallylic alcohol 3a under the optimized conditions resulted in formation of siloxycycle 4a with high endo-selectivity (endo:exo = 50:1), albeit in moderate yield (entry 1). Introduction of bulkier groups, such as nPent- (3b) and Ph- (3c) at the α-position of the alcohol, led to an increase of overall efficiency (entries 2, 3). Interestingly, substrates possessing two geminal substituents at the α-position of the tethered alcohols efficiently produced cyclic allylic silanes 4d and 4e. Remarkably, medium sized rings 4h and 4i were obtained via 8-endo-trig and 9-endo-trig cyclization in 85% and 44% yield, respectively. Applying this methodology to naturally occurring Isopulegol resulted in the formation of two endo products, from which 4j was isolated as the major isomer.
Table 2. Scope of Aliphatic Alkenols.


Isolated yields.
DABCO was used instead of iPr2NEt.
Ag(OTf) was not used as an additive.
Major product shown, ratio of major product to homoallylic side product is 7:1.
Major product shown, ratio of major product to homoallylic side product is 17:1.
Reaction performed at 130 °C.
Major product shown, ratio of major product to hydro-dehalogenation side product is 1:1.
Major product shown, ratio of major product to allylic side product is 3.5:1. Isomers were separated.
Naturally, after establishing the scope of the silyl methyl Heck reaction, we were eager to determine whether this transformation proceeds via a classical Heck-type or a hybrid Pd-radical mechanism.3,4 Our studies using radical traps such as BHT and TEMPO3,11 were inconclusive.9 Therefore, the nature of this cyclization was examined by a radical clock test.12 It was found that 5a underwent smooth cyclization with a subsequent ring opening of the cyclopropyl ring producing a 1:1 mixture of dienes, 7a (8a) and 9a in 58% yield with no cyclopropyl-containing product 6a detected (eq 1). In order to verify whether cyclopropane ring opening occurs via a radical- or a Pd-mediated β-C elimination process,13 the cyclization of phenyl-containing substrate 5b was tested. If the Pd-mediated cyclization is operative, then formation of a mixture of 7b and 8b would be expected.13 However, reaction of 5b under the optimized conditions produced 7b as the sole isomer, thus, strongly supporting the radical cyclization pathway.12,14
 |
Based on these preliminary mechanistic studies, the silyl methyl Heck reaction is proposed to proceed via a hybrid-Pd-radical process, introduced by Alexanian.3,15 A catalytic cycle is depicted in Scheme 2. Oxidative addition of 3 followed by homolysis produces radical A and the Pd(I) species. In cyclization of substrates possessing substituent(s) at the β-position of the alkene (e.g., 1n,o), a 6-exo-trig cyclization occurred, which subsequently leads to the products 2n,o. However, with substrates possessing terminal double bond, A undergoes selective endo-trig cyclization to produce cyclic alkyl radical B. This regioselectivity trend could be attributed to the combination of factors, including the elongated Si–C bond,6 slower relative rate of competitive exo cyclization,6c and favorable stability of the endo transition state proposed for radical cyclizations of halomethylsilanes.6c,7 Next, recombination of B with Pd(I) produces alkylpalladium species C, which upon β-hydride elimination produces reaction products 4. Apparently, C has two alternative β-hydrogens, Hh and Ha, elimination of which leads to homoallylic and allylic silanes, respectively. Although, the origins of the observed selectivity at this point are not clearly understood, it is apparent that sterics plays an important role during the β-hydride elimination event,8 where increasing substitution pattern favors allylic silane formation.
Scheme 2. Hybrid Pd-Radical Catalytic Cycle.

Finally, we turned our attention to the synthetic utility of products obtained via this novel silyl methyl Heck reaction (Scheme 3).16 Thus, ring opening of silyloxycycle 2a with MeLi, produced 11 in 70% yield. Its subsequent intramolecular Hosomi–Sakurai reaction with 1,1-dimethoxycyclohexane generated spiro benzofuran 12 in excellent yield (Scheme 3, a).17 Alternatively, 2a can be smoothly oxidized into (Z)-allylic alcohol 10 in good yield, which highlights employment of halomethylsilanes as a tool for a formal (Z)-hydroxymethylation of alkenols.18 Next, we applied this methodology to a cascade process, where a silyl methyl Heck reaction of dienol 1p produced tricyclic compound 2p in excellent yield (Scheme 3, b). Notably, due to the influence of the silicon atom in the first cyclization event, this cascade reaction of 1p occurs via an endo/exo cyclization protocol, whereas most carbon analogs produce exo/exo cyclized products.19 Compound 2p was further oxidized into indenediol 13 in good yield (Scheme 3, b). Lastly, silyl methyl Heck reaction of vinyl steroid substrate 1q efficiently underwent 7-endo-trig cyclization (Scheme 3, c), thus illustrating usefulness of this methodology for a late stage modification of complex molecules. Oxidation of silyl steroid 2q produced (Z)-allylic alcohol 14 in good yield.
Scheme 3. Synthetic Utility of the Silyl Methyl Heck Reaction.
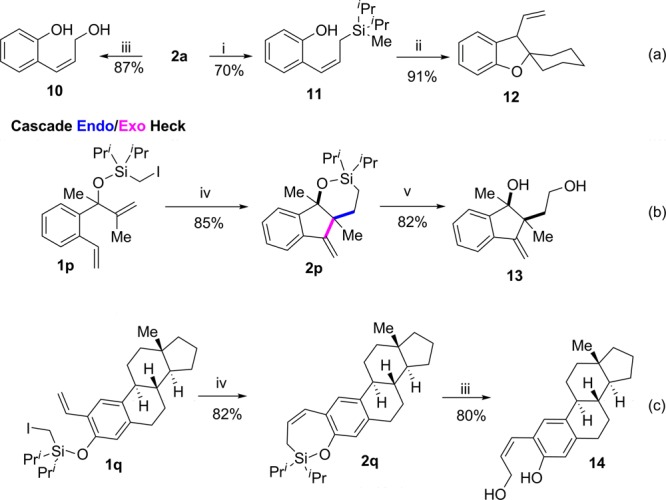
(i) 3 equiv MeLi, Et2O, 0 °C, 1 h. (ii) 1.1 equiv 1,1-Dimethoxycyclohexane, 1.1 equiv BF3OEt2, DCM, −78 °C to rt, 1 h. (iii) 10 equiv 50% H2O2, 12 equiv KHCO3, 2 equiv KF, DMF, 70 °C, 12 h. (iv) 10 mol % Pd(OAc)2, 20 mol % L, 2.2 equiv iPr2NEt, PhMe, 75 °C, 12 h. (v) 10 equiv tBuOOH, 12 equiv KH, 5 equiv TBAF, NMP, rt, 12 h.
In summary, we have shown the use of iodomethylsilane as a tether for the endo-selective alkyl Heck reaction of phenols and aliphatic alkenols. Mechanistic studies suggest this unique reaction occurs via a hybrid Pd-radical process, and the silicon atom is crucial for the observed endo selectivity. Generality of this new methodology was further showcased by employment of natural and complex alkenol substrates and by a unique silyl methyl endo/exo cascade Heck cyclization. The obtained siloxycycles were further functionalized via the intramolecular Hosomi–Sakurai reaction to produce spiro benzofuran skeleton. In addition, the obtained silyl ethers were oxidized to form (Z)-allylic alcohols. We envision this protocol may become a useful tool for a formal (Z)-hydroxymethylation of a broad range of alkenols.
Acknowledgments
We thank National Institutes of Health (GM-64444) and National Science Foundation (CHE-1362541) for financial support of this work.
Supporting Information Available
Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For review of the Heck reaction, see:; The Mizoroki-Heck Reaction; Oestreich M., Ed.; John Wiley & Sons: West Sussex, U.K., 2009. [Google Scholar]
- Firmansjah L.; Fu G. C. J. Am. Chem. Soc. 2007, 129, 11340. [DOI] [PubMed] [Google Scholar]
- Bloome K. S.; McMahen R. L.; Alexanian E. J. J. Am. Chem. Soc. 2011, 133, 20146. [DOI] [PubMed] [Google Scholar]
- For carbonylative radical Heck-type reactions of alkyl iodides, see:; a Bloome K. S.; Alexanian E. J. J. Am. Chem. Soc. 2010, 132, 12823. [DOI] [PubMed] [Google Scholar]; For radical cobalt-catalyzed coupling of alkyl iodides with alkenes, see:; b Weiss M. E.; Kreis L. M.; Lauber A.; Carreira E. M. Angew. Chem., Int. Ed. 2011, 50, 11125. [DOI] [PubMed] [Google Scholar]; For intermolecular radical Heck reactions of alkyl halides, see:; c Zou Y.; Zhou J. Chem. Commun. 2014, 50, 3725. [DOI] [PubMed] [Google Scholar]; d McMahon C. M.; Alexanian E. J. Angew. Chem., Int. Ed. 2014, 53, 5974. [DOI] [PubMed] [Google Scholar]
- a Hegedus L. S.; Sestrick M. R.; Michaelson E. T.; Harrington P. J. J. Org. Chem. 1989, 54, 4141. [Google Scholar]; b Owczarczyk Z.; Lamaty F.; Vawter E. J.; Negishi E. J. Am. Chem. Soc. 1992, 114, 10091. [Google Scholar]; c Rigby J. H.; Hughes R. C.; Heeg M. J. J. Am. Chem. Soc. 1995, 117, 7834. [Google Scholar]; d Iimura S.; Overman L. E.; Paulini R.; Zakarian A. J. Am. Chem. Soc. 2006, 128, 13095. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Klein J. E. M. N.; Muller-Bunz H.; Ortin Y.; Evans P. Tetrahedron Lett. 2008, 49, 7187. [Google Scholar]; f Kim S. H.; Lee S.; Lee H. S.; Kim J. N. Tetrahedron Lett. 2010, 51, 6305. [Google Scholar]; g Kotoku N.; Sumii Y.; Kobayashi M. Org. Lett. 2011, 13, 3514. [DOI] [PubMed] [Google Scholar]; h Gao P.; Cook S. P. Org. Lett. 2012, 14, 3340. [DOI] [PubMed] [Google Scholar]
- For reviews of silyl tethered reactions, see:; a Bols M.; Skrydstrup T. Chem. Rev. 1995, 95, 1253. [Google Scholar]; b Fensterbank L.; Malacria M.; Sieburth S. M. Synthesis 1997, 813. [Google Scholar]; For initial reports see:; c Wilt J. W. J. Am. Chem. Soc. 1981, 103, 5251. [Google Scholar]; d Nishiyama H.; Kitajima T.; Matsumoto M.; Itoh K. J. Org. Chem. 1984, 49, 2298. [Google Scholar]; e Stork G.; Sofia M. J. J. Am. Chem. Soc. 1986, 108, 6826. [Google Scholar]; f Blaszykowski C.; Dhimane A.-L.; Fensterbank L.; Malacria M. Org. Lett. 2003, 5, 1341. [DOI] [PubMed] [Google Scholar]; g Friestad G. K.; Massari S. E. J. Org. Chem. 2004, 69, 863. [DOI] [PubMed] [Google Scholar]; For recent reports, see:; h Huang C.; Gevorgyan V. J. Am. Chem. Soc. 2009, 131, 10844. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Huang C.; Gevorgyan V. Org. Lett. 2010, 12, 2442. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Ghavtadze N.; Melkonyan F. S.; Gulevich A.; Huang C.; Gevorgyan V. Nat. Chem. 2014, 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreeda M.; Hamann L. G. J. Am. Chem. Soc. 1990, 112, 8175. [Google Scholar]
- For pioneering work on the silyl Heck reaction leading to allylic silanes, see:; a McAtee J. R.; Martin S. E.; Ahneman D. T.; Johnson K. A.; Watson D. A. Angew. Chem., Int. Ed. 2012, 51, 3663. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martin S. E. S.; Watson D. A. J. Am. Chem. Soc. 2013, 135, 13330. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McAtee J. R.; Yap G. P.; Watson D. A. J. Am. Chem. Soc. 2014, 136, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See SI for details.
- See SI for endo:exo NMR ratios.
- a Phapale V. B.; Buñuel E.; García-Iglesias M.; Cárdenas D. J. Angew. Chem., Int. Ed. 2007, 46, 8790. [DOI] [PubMed] [Google Scholar]; b Schley N. D.; Fu G. C. J. Am. Chem. Soc. 2014, 136, 16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Newcomb M.; Toy P. H. Acc. Chem. Res. 2000, 33, 449. [DOI] [PubMed] [Google Scholar]; b Baldwin J. E. Chem. Rev. 2003, 103, 1197. [DOI] [PubMed] [Google Scholar]
- Satoh T.; Miura M. Top. Organomet. Chem. 2005, 14, 1. [Google Scholar]
- A radical pathway is also consistent with the results of additional experiments suggested by a referee, which include trapping a side hydro-dehalogenation product of 3i with deuterium radical arising from toluene-d8, and cyclization of analog of 1a possessing a geometrically defined deuterium-labeled alkene moiety. See SI for details.
- For a review on hybrid palladium–radical reactions, see:Jahn U. Top. Curr. Chem. 2012, 320, 363–362. [DOI] [PubMed] [Google Scholar]
- a Jiménez-González L.; García-Muñoz S.; Álvarez-Corral M.; Muñoz-Dorado M.; Rodríguez-García I. Chem.—Eur. J. 2006, 12, 8762. [DOI] [PubMed] [Google Scholar]; b Jiménez-González L.; García-Muñoz S.; Álvarez-Corral M.; Muñoz-Dorado M.; Rodríguez-García I. Chem.—Eur. J. 2007, 13, 557. [DOI] [PubMed] [Google Scholar]
- For general transformations using allylic silanes, see:; a Hosomi A.; Endo H.; Sakurai H. Chem. Lett. 1976, 941. [Google Scholar]; b Masse C. E.; Panek J. S. Chem. Rev. 1995, 95, 1293. [Google Scholar]; c Brook M. A.Silicon in Organic, Organometallic, and Polyer Chemistry. Wiley, Chichester, 2000. [Google Scholar]; d Denmark S. E.; Fu J. Chem. Rev. 2003, 103, 2763. [DOI] [PubMed] [Google Scholar]
- Fujiwara T.; Yanai K.; Shimane K.; Takamori M.; Takeda T. Eur. J. Org. Chem. 2001, 2001, 155. [Google Scholar]
- For a recent review of the cascade Heck processes, see:; a Muzart J. Tetrahedron 2013, 69, 6735. [Google Scholar]; For selected examples, see:; b Kucera D. J.; O’Connor S. J.; Overman L. E. J. Org. Chem. 1993, 58, 5304. [Google Scholar]; c Fox M. E.; Li C.; Marino J. P.; Overman L. E. J. Am. Chem. Soc. 1999, 121, 5467. [Google Scholar]; d Monks B. M.; Cook S. P. J. Am. Chem. Soc. 2012, 134, 15297. [DOI] [PubMed] [Google Scholar]; e Hu J.; Hirao H.; Li Y.; Zhou J. Angew. Chem., Int. Ed. 2013, 52, 8676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.