

Abstract
This contribution features a small molecule that binds TrkC (tropomyosin receptor kinase C) receptor that tends to be overexpressed in metastatic breast cancer cells but not in other breast cancer cells. A sensitizer for 1O2 production conjugated to this structure gives 1-PDT for photodynamic therapy. Isomeric 2-PDT does not bind TrkC and was used as a control throughout; similarly, TrkC– cancer cells were used to calibrate enhanced killing of TrkC+ cells. Ex vivo, 1- and 2-PDT where only cytotoxic when illuminated, and 1-PDT, gave higher cell death for TrkC+ breast cancer cells. A 1 h administration-to-illumination delay gave optimal TrkC+/TrkC–-photocytotoxicity, and distribution studies showed the same delay was appropriate in vivo. In Balb/c mice, a maximum tolerated dose of 20 mg/kg was determined for 1-PDT. 1- and 2-PDT (single, 2 or 10 mg/kg doses and one illumination, throughout) had similar effects on implanted TrkC– tumors, and like those of 2-PDT on TrkC+ tumors. In contrast, 1-PDT caused dramatic TrkC+ tumor volume reduction (96% from initial) relative to the TrkC– tumors or 2-PDT in TrkC+ models. Moreover, 71% of the mice treated with 10 mg/kg 1-PDT (n = 7) showed full tumor remission and survived until 90 days with no metastasis to key organs.
Keywords: photodynamic therapy (PDT), tropomyosin receptor kinase C (TrkC), metastatic breast cancer, histochemistry, theranostic, imaging
Introduction
Agents that selectively associate with cell surface receptors overexpressed on tumor cells can be used to deliver therapeutics. This strategy is referred to here as active to distinguish it from other forms of targeting (e.g., where the agent is intended to directly cause a therapeutic effect by binding cell surface receptor or is designed to target intracellular pathways upregulated in cancer cells).1,2
The most widely investigated active targeting agents are humanized monoclonal antibodies (hmAbs), but mAbs in general have limitations.3 First, they have poor permeation into solid tumors, only a few mAbs enter cells,4 most do not,5 and even cell-permeable mAbs may not reach the intracellular target.6 Moreover, mAbs can have undesirable immunogenic effects, circulation times, and they are further restricted by cost and shelf life issues.7,8
Active targeting via small molecules that selectively bind to receptors on tumor cells can have advantages relative to hmAbs with respect to cell internalization and affordability; however, relatively few small molecule targeting entities are known. Folic acid9−12 and Arg-Gly-Asp peptidomimetics13−17 are probably the most widely appreciated examples, but there are not many more besides these. There are no clinically approved small molecule active targeting agents for delivering therapeutics to breast cancer.18
TrkC, a cell surface receptor, and its natural ligand neurotrophin-3 were reported to play an essential role in breast cancer growth and metastasis;19,20 suppression of TrkC expression in highly metastatic mammary carcinoma cells inhibited their growth in vitro and their ability to metastasize from the mammary gland to the lung in vivo.21 This paper features a novel molecular fragment A (Figure 1) for active targeting of breast cancer types that overexpress TrkC.22−24 TrkC-targeting molecules 1, containing fragments A (blue in Figure 1), elicit only weak functional effects.23 One fragment A is not enough to bind TrkC adequately, but two joined as shown give good affinity.23 The scaffold part in molecules 1 (shown in black) supports the two TrkC-binding entities and the BODIPY cargoes (colored purple and red here). The BODIPY dyes are similar, except that those without iodines are highly fluorescent, while ones with are only weakly fluorescent and act as sensitizers for singlet oxygen production. Thus, compound 1-F (F = fluorescent) is designed for cell imaging and histology, while 1-PDT is intended for use in PDT.25,26
Figure 1.
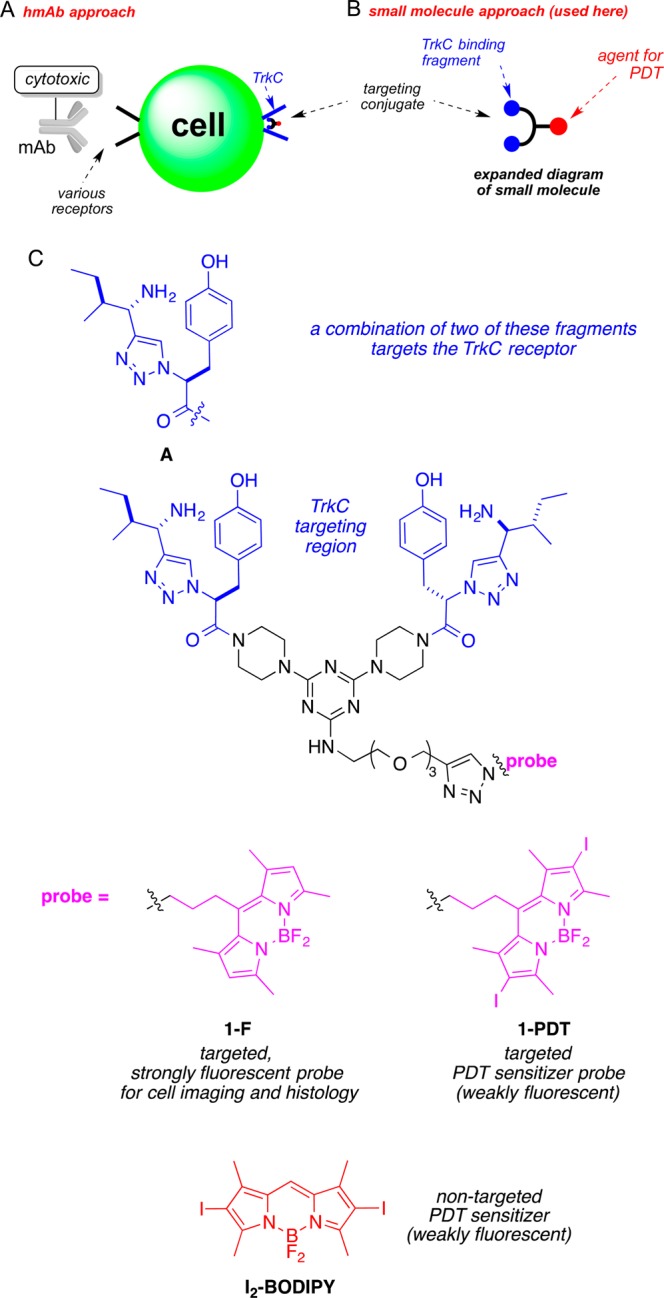
Fundamentals of active targeting. (A) mAb conjugates have limited cell permeabilities, but (B) many small molecule conjugates can. (C) Structures of the targeted compounds featured in this work, 1-F and 1-PDT, and the parent iodinated BODIPY, I2-BODIPY.
Singlet oxygen is very reactive, half-life 1 ns to 1 μs, hence the effect of PDT agents illuminated in an aerobic environment is to generate 1O2 that kills cells in a highly localized area. Some boron dipyrromethene (BODIPY) based systems can have excellent attributes for PDT with high extinction coefficients, favorable light-to-dark toxicity ratios, high antitumor efficacies in vivo, and good body clearance,27−31 but they are not inherently inclined to localized in tumors; accumulation of the sensitizers in tumors is important for PDT. Experiments described in this paper were undertaken to explore the effects of coupling molecular fragments A that can bind TrkC receptors expressed on breast cancer cells and deliver a BODIPY-based PDT agent to them. As far as we are aware, there are no other agents, in the clinic or in the literature, on experimental modalities that actively target TrkC+ with a small molecular fragment that binds this receptor conjugated to a therapeutic.
Results
Compound 1-F Selectively Stains TrkC-Expressing Tissue and Is Internalized by TrkC+ Cells
We hypothesized that agent 1-F could be used to stain tissue that express TrkC+ (e.g., from biopsies) and TrkC+ tumors and metastases during surgeries. In histochemistry on a commercial array of human breast cancer sections using 1-F, all 36 malignant tissues showed evidence for expression of TrkC (23% with unambiguous staining in the cytoplasm and cell membrane, 65% same but not as clear, and in 12% not all the tumor cells stained); conversely, none of the normal breast tissue showed significant staining in the cytoplasm and cell membrane just as fluorescent anti-TrkC mAb did (Figure 2A). We infer 1-F has potential for histochemistry and as a surgical marker for TrkC-expressing cancers.21
Figure 2.

Compound 1-F stains in TrkC+ tumor tissue and is internalized TrkC+ cells. (A) Histochemical stains for a library of 96 breast tissue slices were performed using 1-F (top) and anti-TrkC antibody as control (bottom), and the three illustrative ones shown here illustrate staining of the malignant tumor, whereas normal tissue is not stained. No staining was observed in the tissues without the small molecule probe or mAb. (B) Cell imaging on 4T1 cells shows 1-F was internalized into lysosomes just as the natural TrkC ligand NT3 is.
Intracellular imaging studies featuring 1-F on murine 4T1 cells (Figure 2B) showed compound 1-F is internalized and partially colocalizes with a lysosome tracker dye. Thus, 1-F localizes into the lysosome, just as the natural TrkC ligand “neurotrophin-3” (NT3) does when it is internalized via the TrkC receptor,32 implying 1-F also enters the cell via TrkC. Similar experiments were performed using (the less fluorescent agent) 1-PDT, and the outcome was much the same (Supporting Information, Figure S3). Moreover, import of 1-PDT at 2 h was more than 2-PDT into 4T1 cells and that uptake of 1-PDT was suppressed by pretreatment with the natural TrkC ligand, NT3.
Compound 1-PDT Selectively Kills TrkC-Expressing Cells In Tissue Culture
Binding NT3 to TrkC on the surface of living cells causes growth and survival.33,34 Some common breast cell lines used for cancer research express TrkC (e.g., human Hs578t and murine 4T1)21,35 but not in others (e.g., SKBR3, 67NR, and MCF-10A).21,36 TrkC is expressed in the majority of metastatic breast tumors.37
Figure 3A shows photocytotoxicities of 1-PDT are greater for the TrkC+ breast cancer cell lines HS578t and 4T1 than the immortalized, TrkC–, breast cell lines 67NR and MCF-10A. Comparison of photocytotoxicities for the targeted PDT agent 1-PDT with the nontargeted form 2-PDT, and I2-BODIPY on TrkC+ expressing cells (Figure 3B,C) shows the targeted agent is more photocytotoxic (IC50 = 0.325 μM in 4T1; IC50 = 0.285 μM in HS578t) than the control ones, 2-PDT and I2-BODIPY (undetermined IC50), which does not bind TrkC. These results suggest that 1-PDT induces selective photocytotoxicity in TrkC-expressing cells via TrkC receptor targeting. Some photocytotoxicity was observed for 2-PDT on the human HS578t but not on the murine 4T1 cells, indicating the scrambled control 2-PDT might be binding to some other receptor on the human line.
Figure 3.

1-PDT is photocytotoxic in TrkC+ cell lines. (A) Photocytotoxicities for 1-PDT are more for the following breast cells, murine metastatic 4T1 and human metastatic, HS578t; compared with the following breast cell lines, murine nonmetastatic, 67NR; human immortalized MCF-10A. (B) Photocytotoxicities on the 4T1 and HS578t cells were enhanced for 1-PDT compared to the scramble control 2-PDT featuring an isomer of the targeting fragment that does not adhere to TrkC+ cells and control I2-BODIPY. (C) Structure of 2-PDT. (D) Photocytotoxicities for 1-PDT on 4T1 and HS578t cells are dose dependent (red bars) and suppressed by fixed concentrations of competing: (i) natural ligand NT3 (blue) and (ii) targeting ligand without a PDT group “IY-IY-TEG”. Data shown are mean ± SEM of three independent experiments. *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001 vs control using One-Way ANOVA ((A) TrkC– cell line, (B) I2-BODIPY, (D) red bars).
Figure 3D shows the photocytotoxicities of 1-PDT on TrkC+ cells can be suppressed by the natural TrkC ligand (NT3) or the targeting agent without any BODIPY dye attached, “IY-IY-TEG” (structure in Supporting Information, Figure S1). When interpreting this data, it is important to note that the concentration of NT3 is constant throughout, so 1-PDT only becomes noticeably competitive with the small molecule ligand at higher concentrations (e.g., > ca. 0.1 μM for human cells, Figure 3D, right side).
All the photocytotoxicity experiments described above involved adding the test compounds for 2 h incubation, washed off before illuminating the cells. Effects of prolonging the incubation on the cell viability in PDT revealed that more selective cell-growth inhibition was achieved for TrkC+ 4T1 compared to TrkC– 67NR at 2 h incubation (Supporting Information, Figure S2), and the difference becomes less noticeable when 4 and 6 h incubation was used; thus a shorter incubation time is optimal for selective photokilling by the TrkC-seeking conjugate. For a full comparison, the same time course experiments for the untargeted 2-PDT and the parent iodinated BODIPY, I2-BODIPY, were also performed (Supporting Information, Figure S2). As expected, both compounds caused increased photocytotoxicity with increasing incubation time. However, the cell viability observed between 4T1 and 67NR cells were similar across the different time points for incubation, implying no selective binding to cell surface receptors (Supporting Information, Figure S2).
Maximal Tolerated Dose (MTD) of 1-PDT is 20 mg/kg in a Murine Model
1-PDT at 20, 30, and 100 mg/kg was administered to mice intravenously via the tail vein, and toxicity was evaluated based on typical symptoms (apathy, horrent fur, diarrhea, behavior changes, and loss of body weight). All mice receiving doses of 30 mg/kg or more experienced motility and balancing difficulties and died 1–3 h post drug administration. However, 20 mg/kg 1-PDT was well tolerated and gave no signs of toxicity and death up to 17 days of post-treatment (Figure 4), and no sign of gross organ toxicity was found in autopsies performed at the end of study. No death or signs of toxicity were observed in mice receiving equivalent doses of 2-PDT and BODIPY. These results suggest 20 mg/kg is the maximum tolerated dose (MTD) for 1-PDT. If, after further studies, these compounds were to be used therapeutically, then the dose should be significantly less than this; however, modifications to the dye structures are required first (see below).
Figure 4.
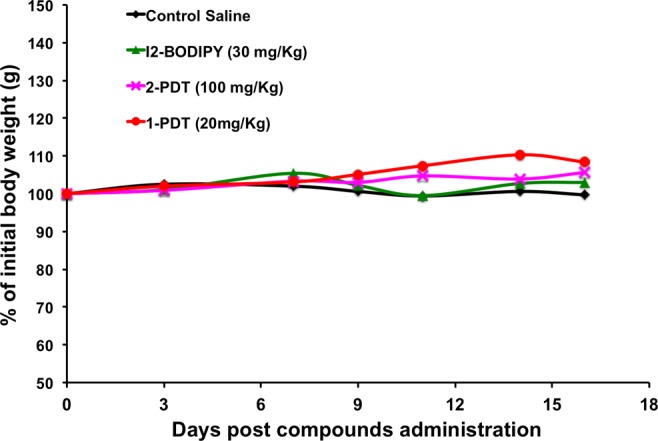
1-PDT was not toxic to mice at 20 mg/kg. Healthy 7–8 weeks old Balb/c female mice were administered intravenously via tail vein respectively with 1-PDT and 2-PDT at 20, 30, and 100 mg/kg (I2-BODIPY content equivalent to 6.25, 10, and 30 mg/kg, respectively, i.e., corrected for MW), and the parent I2-BODIPY (30 mg/kg). The mice were then kept in the dark and observed for 16 days. Data represent the average body weight (grams) of two mice/treatment group.
Agent 1-PDT Accumulated Maximally in Tumor Tissues 1 h after Administration and Cleared from the Body 72 h Post-Treatment
The biodistribution of 1-PDT and the isomeric non-TrkC targeting control 2-PDT were monitored in 4T1 tumor bearing mice (n = 3) up to 72 h. Significant and prolonged accumulation of 1-PDT was observed in tumor (Figure 5). At 1 h post administration, the fluorescence intensity of tumors in mice treated with 1-PDT was 2.1× higher (897000 ± 135800) than the corresponding intensities for tumors treated with the nontargeted control 2-PDT (416000 ± 43000) (p < 0.05, Student’s t test). The 1-PDT dye intensity in tumor tissue remained significantly higher compared with 2-PDT for up to 6 h, but there were no significant differences at 24 h onward. Trends in the 1-PDT tumor distribution were significantly different to that of 2-PDT, indicating selective accumulation of 1-PDT in the tumor. Maximum accumulation of 1-PDT at 1 h post administration led us to adopt a drug-to-light interval of 1 h in determining 1-PDT’s in vivo antitumor efficacies in the subsequent studies.
Figure 5.

1-PDT demonstrated significant and prolonged accumulation in tumor tissue for up to 6 h and cleared from the body 72 h postadministration. 4T1-tumor bearing female Balb/c mice were treated at 10 mg/kg via the tail vein. Mice (n = 3) were sacrificed at 0, 0.25, 1, 3, 6, 24, 48, 72 h. (A) Organs and tissues (tumor, draining lymph nodes, spleen, kidney, liver, lung, skin, and eye) were harvested, and (B) fluorescence intensities in each organ were imaged using an in vivo imager (data represent mean ± SEM of three mice at each time point). * p < 0.05; ** p < 0.01; for 1-PDT vs 2-PDT.
A large amount of 1-PDT accumulated in the liver (ca. 20-fold more than in the tumor), then in the kidney and lung within the first 3 h post administration (Figure 5), but these accumulations dissipated swiftly in the subsequent monitoring period. A similar pattern of accumulation was found for 2-PDT. Swift clearance of both compounds in these organs is typical of small molecular weight compounds27 and indicates that the accumulation in these organs was random and not due to TrkC receptor binding.
Nonselective accumulation of 1-PDT and 2-PDT was also observed in lymphoid organs such as spleen and lymph node at a much lower level (Figure 5); others have observed no significant TrkC expression for these organs.38 Interestingly, 2-PDT but not 1-PDT was found to accumulate significantly in the lymph node for a prolonged period of time. The eye has relatively impermeable blood capillaries and low TrkC receptor expression,39 whereas murine eyes express relatively few TrkC receptors, concentrated mainly at nerve bundle portions.40
1-PDT at 10 mg/kg Gave Effective Eradication of 4T1 Tumor with 96% Average Tumor Regression
Compound targeting efficacies and effects on TrkC+ tumors were assessed in the following experiments. Aggressive TrkC+ murine breast carcinoma (4T1) cancer cells were subcutaneously injected to the murine mammary fat pad and then treated with 1-PDT, 2-PDT, and I2-BODIPY when the tumor sizes reached 80 mm3. In a control, mice inoculated with TrkC– murine breast carcinoma 67NR cancer cells were also treated using these compounds.
Agent 1-PDT significantly reduced tumor growth after illumination when treated at 2 and 10 mg/kg (equivalent to 0.6 and 3.0 mg/kg of I2-BODIPY, respectively). Inflammation and erythema surrounded the irradiated tumor region was observed in the 1-PDT treated groups at 4–6 days post PDT but was less pronounced or not observed in the control groups (Figure 6A). Subsequently, tumor sizes were drastically reduced in the 1-PDT treated groups 4–6 d postillumination (61% and 96% maximum tumor reduction in mice treated with 2 and 10 mg/kg of compound compared to pretreatment tumor size, Figure 6B). In contrast, 2-PDT and I2-BODIPY treatment induced only moderate tumor size reduction within the first 6 d after illumination (20% reduction for mice treated with 10 mg/kg of 2-PDT, and 11% reduction for mice treated with 10 mg/kg of I2-BODIPY, Figure 6B). At day 13, both 2-PDT and I2-BODIPY treated mice showed rapid tumor regrowth at the necrotic tumor tissue periphery while tumor regrowth in 1-PDT treated mice was minimal and delayed (Figure 6A,B). More importantly, 1 out of 7 mice (14%), treated with 2 mg/kg, and 5 out of 7 mice (71%), treated with 10 mg/kg of 1-PDT, healed from eschar by day 13–15 after illumination and showed no palpable tumor for up to 90 d post-treatment. Such total tumor remission was not found in both the 2-PDT and I2-BODIPY treated groups. To confirm the targeting ability of 1-PDT on TrkC+ cells in vivo, the compound efficacy in the non-TrkC expressing 67NR tumor cell line in mice was examined and compared to that in the 4T1 model; as expected, neither 1-PDT nor 2-PDT at 10 mg/kg fully eradicated the 67NR tumors in mice (Figure 6C); the tumor volumes were reduced at day 4–6 days postillumination but regrew at day 9.
Figure 6.
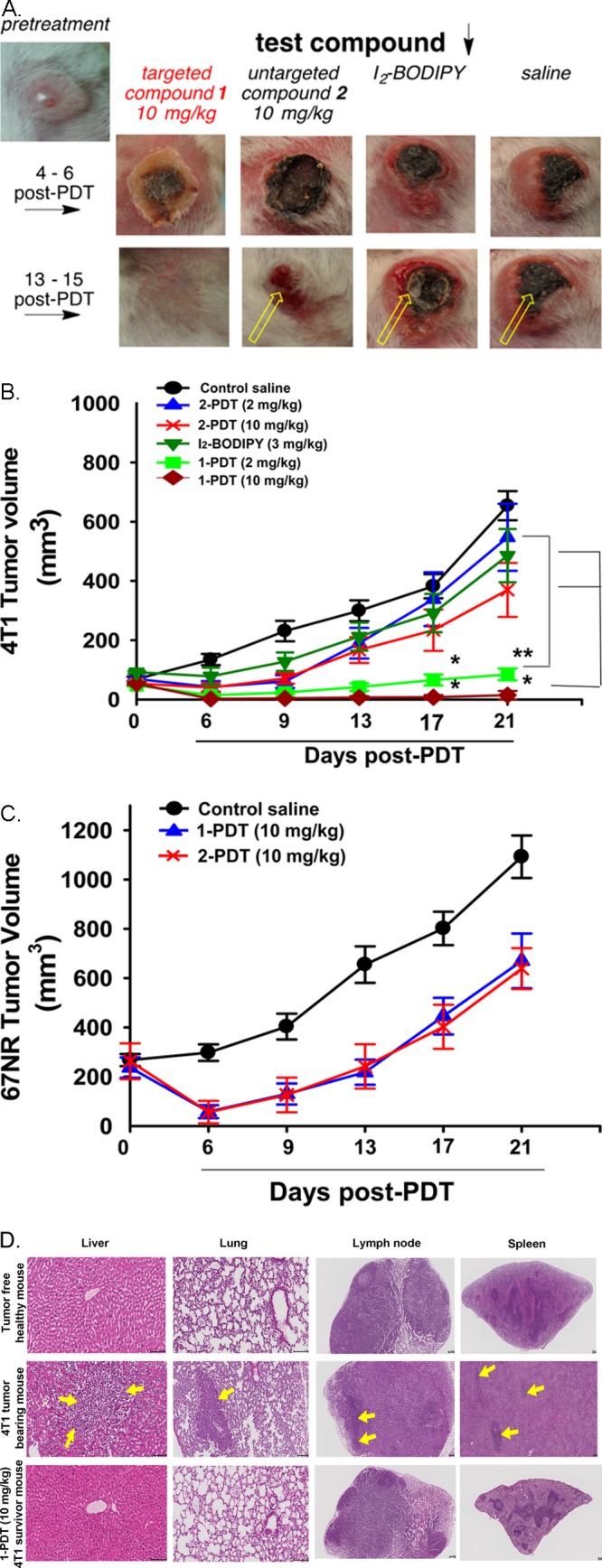
1-PDT effectively suppressed the growth of TrkC+ (4T1) tumor but not in TrkC– (67NR). (A) Regrowth of TrkC+ 4T1 tumor (yellow arrow) in female Balb/C mice receiving 2-PDT (10 mg/kg), I2-BODIPY (3.0 mg/kg), and saline controls but not in mice receiving 1-PDT (10 mg/kg). (B) Significant dose dependent mean tumor volume reduction and delayed tumor regrowth in TrkC+ 4T1 tumor bearing mice receiving 2 and 10 mg/kg 1-PDT as compared to rapid tumor growth in mice receiving the control substances. (C) 1-PDT gave impermanent and nonselective antitumor effect (resembled that with 2-PDT) in mice bearing TrkC– 67NR tumor. Photoactivation was conducted at 100 J/cm2 with a fluence rate of 0.16 W/cm2 1 h after intravenous injection of the compounds. All graphs showed mean tumor volume ± SEM (n = 7). * p < 0.05, ** p < 0.005, for I2-BODIPY vs 1-PDT and 2-PDT group using One-Way ANOVA. (D) There were no tumor metastases in 1-PDT treated survivor mice post 90 d. Mice treated with 10 mg/kg 1-PDT that survived up to 90 d with no palpable primary tumor found were metastases free in all the major organs assessed (liver, lung, draining lymph node, and spleen, representative histological images). Control (tumor free healthy and 4T1 tumor burden mice) results were included for comparison (yellow arrow = 4T1 tumor metastases). Scale bar: 100 μm. The current results had been verified by certified veterinary pathologist.
Mice Surviving Treatment with 1-PDT Showed Complete Remission up to 90 Days Postillumination, with No Metastasis Development
All 1-PDT treated mice that showed complete tumor regression remained disease free with no palpable tumor at the primary site up to 90 d. Tumors from 4T1 cancer cells are known to be aggressive and typically metastasize to lymph nodes, liver, and lung, even in the early stages of the disease.41 Thus, at 90 d postillumination, the surviving mice that were physically active were sacrificed for major organ/tissue histopathology by a certified veterinary pathologist. H&E staining showed no 4T1 tumor metastases in all the examined organs (liver, lung, draining lymph node, spleen, kidney, and heart) of the 10 mg/kg 1-PDT treated survivor mice and tumor free control mouse (Figure 6D). However, in 4T1 tumor bearing control mice, tumor metastases were found in liver, lung, lymph nodes, and spleen, with extramedullary hematopoiesis observed in this animal (Figure 6D). These results show effective eradication of TrkC expressing 4T1 tumor by 1-PDT post PDT treatment in the survivor mice.
Discussion
The assertion that agent 1-PDT targets TrkC+ breast cancer cells is supported by the observation that it has a more profound effect on these tumors than the isomeric compound, 2-PDT, and the PDT agent without any appendage, i.e., I2-BODIPY. This is consistent with the significant in vivo selective accumulation of 1-PDT in TrkC+ tumors 1 h after administration, as calibrated relative to levels of 2-PDT at the same time. Moreover, 1-PDT, 2-PDT, and I2-BODIPY have comparable effects on tumors from TrkC– cell lines.
The data outlined above are consistent with ex vivo studies on cells stably transfected with TrkC,42 but those experiments did not involve breast cancer cells. In this article, we report selective photocytotoxicity of 1-PDT ex vivo correlates with natural levels of TrkC+ expression in breast cell lines. The cell studies established that a 4–6 h interval between treatment and illumination decreased the selectivity for TrkC-expressing cells relative to a 2 h interval, perhaps due to relatively slower, and nonselective, interaction with cells, something that could be anticipated from the literature.43,44 Consequently, we used a 1 h interval between injection and light treatment for the in vivo work.
It is remarkable that 1-PDT at 10 mg/kg in vivo caused, on average, 96% tumor volume reduction in the mice bearing TrkC+ tumor at day 6 post-PDT. Among these mice, 71% showed full remission and were tumor-free for 90 days after therapy, and histology indicated no metastasis development in these animals. The fact that 1-PDT was ineffective for suppressing TrkC– (67NR) tumors in mice supports the overall assertion that this compound targets TrkC+ expressing tissue in vivo.
Long- and short-term toxicities of agents like 1-PDT must be considered in the context of experimental therapeutics. In the dark, the TrkC-targeting fragments featured are capable of transducing signals similar to NT3 upon binding to TrkC,23 so it is conceivable that in the long term 1-PDT might induce tumorigenesis just as NT3 does.20,21,45 However, this was not the case in the extended time course of these experiments because, in the dark, mice receiving saline control or 2-PDT (no TrkC binding) have comparable tumor volumes, with no significant differences, to the 1-PDT group (Supporting Information, Figure S5).
Another relevant long-term toxicity issue relates to photosensitivity. Intravenous administration of photosensitizers can result in accumulation in different tissues and undesirable photosensitivities.46 Adverse photosensitivity is common in PDT,47,48 but targeting PDT agents to tumors that express TrkC should alleviate some of these effects.
Short-term toxicities for agents that bind TrkC could be anticipated because NT3 promotes neuronal cell survival, differentiation, and synapse transmission, and antagonism at the TrkC receptor might have undesirable effects.
Short-term toxicities of agent 1-PDT are a concern because, while doses of 20 mg/kg were tolerated, 30 mg/kg was not. A claim that high doses of neurotrophins (including NT3) promote relatively rapid excitotoxic necrosis of neurons49 might be pertinent. In any event, 1-PDT requires further structural modifications because the light wavelength to excite this is optimally around 520 nm whereas PDT agents should absorb above 700 nm if they are to be addressed at more than 1 cm tissue penetration.50−53 Consequently, 1-PDT is a prototype for other compounds, currently under development in our laboratories, that involve the same targeting fragments differently disposed around other PDT active dyes. It is anticipated that the short-term toxicities of the second-generation systems will be structure dependent.
Overall, the data presented here demonstrate the potential of targeting TrkC+ tumors with PDT agents. Excellent therapeutic indices can be achieved because PDT is spatially restricted to the illumination area, and active targeting accumulates the agents in the tumors. This study features dosing with a targeted-PDT agent alone, but there is also the intriguing possibility of combination therapies featuring Trk inhibitors currently in trials as chemotherapeutic agents (e.g., Lestauritinib54 and PLX7486 (http://clinicaltrials.gov/show/NCT01804530)).
Materials and Methods
Histochemistry
Two slides of human breast cancer tissue microarrays (BRC962) were purchased from US Biomax, Inc. The arrays include 36 cases of breast cancers and 12 cases of normal, reactive, and benign tumor tissues of the breast in duplicates. The slides were transferred to a xylene bath for 10 min and then rehydrated in two changes of fresh absolute ethanol for 7 min each. Excess liquid was shaken off, and the slides were incubated in fresh 90%, 70% ethanol then water for 7 min each. The slides were washed in two changed of PBS for 5 min each, then incubated with PBS containing 4% BSA for 30 min. The tissues were rinsed with PBS and incubated again in two changed of PBS for 5 min each. 1-F solution in 4% PBS/BSA and anti-TrkC antibody were added to separated slide and incubated overnight at 4 °C. The slides were rinsed twice in PBS, then in water (10 min each). Then the slide contained 1-F was mounted in permanent mounting media with DAPI (Vector) and incubated at 4 °C for 4 h. The slide contained anti-TrkC antibody was incubated with rodamine-tag primary antibody for 30 min and then rinsed twice in PBS and in water (10 min each) before putting in the mounting media. Two slides were imaged with a Zeiss Stallion dual detector imaging system consisting of an Axiovert 200 M inverted fluorescence microscope, CoolSnap HQ digital cameras, and Intelligent Imaging Innovations (3I) software. Digital images of 1-F and DAPI were captured with a C-APO 63X/1.2 W CORR D = 0.28M27 objective with the following filter sets: exciter BP470/20, dichroic FT 493, emission BP 505–530 for 1-F, emission BP 565–615 for rodamine and exciter G 365, dichroic FT 395, emission BP 445/50 for DAPI. Sequential optical sections (Z-stacks) from the basal-to-apical surfaces of the cell were acquired. Digital image acquisition was initiated approximately 1 μm below the basal surface of the cells, and optical slices were collected at 0.5 μm steps through the apical surface of the cells with a high numerical objective lens (C-APO 63X/1.2 W CORR D = 0.28M27). These wide-field images were subjected to deconvolution using 3I software.
Cell Culture
4T1, MCF-10A, and Hs578t (ATCC) and 67NR (Barbara Ann Karmanos Cancer Institute, Detroit, MI) cells were cultured on 75 cm2 culture flasks in Dulbecco’s Modified Eagle Medium/nutrient mixture F-12 (DMEM/F12, Sigma Chemical, St. Louis, MO) supplemented with 10% FBS. All cells were cultures in a humidified incubator at 37 °C with 5% CO2 and 95% air.
Fluorescence Microscopy
Intracellular localization of the 4T1 cells was measured using a Zeiss 510 META NLO Multiphoton system containing of an Axiovert 200 MOT microscope. Throughout, digital images were captured with a 40×/1.3 oil objective. 1-F was excited at 488 nm, and the emission BP used was 500–530 nm; for LysoTracker red, the excitation was at 543 nm and the emission BP was 565–615 nm.
Sequential optical sections (Z-stacks) from the basal-to-apical surfaces of the cell were acquired, initiated approximately 1 μm below the basal surface of the cells, and optical slices were collected at 0.5 μm steps through their apical surface using a high numerical objective lens (C-APO 63X/1.2 W CORR D = 0.28M27). These wide-field images were subjected to deconvolution using Intelligent Imaging Innovations (3I) software.
Intracellular Localization
4T1 cells were incubated with 1-F, 1 μM, for 12 h at 37 °C. After the cells were washed with PBS, LysoTracker Red (Life Technology, 500 nM) was added and the cells were incubated for 30 min at 37 °C. The cells were washed again with PBS before imaging.
Photoinduced Cytotoxicity Assay
Approximately 5000 cells/well in culture medium containing 10% fetal bovine serum were seeded in a 96-well plate. Cells were allowed to adhere overnight before test compounds were introduced. 1-PDT, 2-PDT, and I2-BODIPY stock solutions (0.02 M in DMSO) were diluted with protein-free medium (PFHM-II), 1 μL stock solution/1 mL PFHM-II, to make master stock solutions. The master stock solutions were further diluted with PFHM-II to the desired final concentrations varying from 0.02 to 10 μM to test on the cells (less than 0.001% DMSO contained in the final solutions). After 2 h of treatments, cells were washed twice with PBS, then culture media without any additives was added (ACAS) before irradiation, irradiated with a light dose of 7.3 J/cm2 from a broad-spectrum halogen light source and fluence rate of 12.2 mW/cm2, then further incubated for 24 h; viabilities were assessed through MTT conversion.37 Briefly, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide MTT (5 mg/mL, in Hank’s balanced salt solution) were added and the cells were incubated for an additional 3 h. The medium was then removed, and 100 μL of DMSO was added to dissolve the formazan crystal formed. The optical density of each well (at 570 nm) was measured with a BioTek Synergy 4 microplate reader. The viability of each cell line in response to the treatment with tested compounds was calculated as % of cell viability = (OD treated/OD control) × 100.
Animal Model
Female 8–10 week old, wild-type BALB/c mice were purchased from Monash University, Malaysia campus (Sunway, Malaysia) for in vivo studies and maintained in the satellite animal facility, Department of Pharmacology, Faculty of Medicine, University of Malaya. All animal experiments were performed according to protocol approved by the Faculty of Medicine Institutional Animal Care and Use Committee, University of Malaya (FOM IACUC) (Ethics Approval no. 2013-05-07/PHA/R/KLV).
The toxicity profiles of 1-PDT and 2-PDT were determined after intravenous administration of these compounds at 20–100 mg/kg to the mice via tail vein. Toxicity was observed based on typical symptoms such as apathy, horrent fur, behavior changes, and loss of body weight for 2 weeks.
Pharmacokinetics and Compound Clearance
4T1 tumor bearing female BALB/c mice with an average tumor volume of 80 mm3 were divided into two groups and intravenously administered with 10 mg/kg of photosensitizer 1-PDT and 2-PDT, respectively. Mice (n = 3 for each time point)55 were then sacrificed at different time points (0, 15 min, 1, 3, 6, 24, 48, and 72 h post compound administration), and major organs such as liver, spleen, lung, kidney, lymph nodes, skin, eye, and tumor tissues were harvested. Organs and tissues were imaged using an In Vivo MS FX PRO (Carestream Molecular Imaging, Woodbridge, CT) with an excitation filter at 530 nm and emission filter at 600 nm. Mice treated with saline were used as control. Fluorescence intensities of each organ and tissue were quantified using Carestream Molecular Imaging software 5.0 (Woodbridge, CT).
Tumor Cells Inoculation and PDT in BALB/c Mouse
The fur of the BALB/c mice was shaved, and murine 4T1 and 67NR cells at a density of 5 × 105 cells in 0.1 mL of medium was orthotopically injected into the mammary fat pad of the mice (18–20 g, 8–10 weeks old), respectively. The mice were then randomly divided into groups for PDT at 8 days post injection, when the tumor size reached 80 mm3.56,57 Compounds (1-PDT, 2-PDT, and I2BODIPY) at 2–10 mg/kg body weight were dissolved in a cocktail of 2.5% ethanol and 2.5% Cremophore EL. The mixture was then further dissolved using saline to a volume of 0.2 mL and administered by intravenous tail vein injection into the mice. The mice were then kept in the dark for 1 h before an anesthesia cocktail of 90 mg/kg of ketamine and 10 mg/kg of xylazine cocktail was administered. Thereafter, PDT was performed using Lumacare LC-122A fiber optic light delivery system (standard fiber optic probe model LUM V, 400–700 nm, Lumacare Medical Group, Newport Beach, CA, USA, with a 500/585 nm bandpass filter from Omega Optical, catalogue no. XF 3105) emitting light at 530 nm. A 4 mm thick glass slide (1.0 mm × 1.2 mm, purchased from Sail Brand, China, catalogue no. 7101) was used as a barrier to avoid direct photothermal effect on tumor. The illuminating spot was positioned at the tumor and the surrounding was covered using black cloth to avoid PDT effect on nontumor parts of body. PDT was conducted at 100 J/cm2 with the fluence rate of 160 mW/cm2. After PDT, the mice were kept in dark and tumor size was measured 3 times per week. Tumor volume changes were determined by caliper measurements with tumor volume, mm3 = (L × W2/2), where L is the longest dimension and W is the shortest dimension.58 During the study, the mean tumor diameter did not exceed 13 mm.
Histology Sample Preparation
Survivor mice were sacrificed at 90 days post therapy, and major organs such as liver, kidney, spleen, draining lymph node, lung, and heart were isolated for histological analysis. The isolated specimens were fixed with 10% formalin solution for a minimum of 48 h at room temperature, following which, tissues were trimmed into representative segments and then dehydrated using an ascending series of alcohol, cleared in xylene, and embedded in paraffin. Microtomy was performed using a Leica RM2255 microtome (Leica Microsystems, Germany). Sections were cut at 5 μm thickness and placed onto appropriately labeled microscope slides. The slides were then stained with haematoxylin and eosin (H&E) and coverslipped, then evaluated by a board certified pathologist at the Institute of Molecular and Cell Biology, Agency for Science, Technology and Research, in Singapore.
Statistical Analysis
In vitro and in vivo experiments were performed to compare the efficacy of the three compounds, and statistical analysis was analyzed using SPSS. Results were analyzed using One-Way ANOVA with Dunnett’s Multiple Comparisons when comparing among the three groups of compounds. Student t test was used to analyze between two groups; differences were considered statistically significant at the p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) levels.
Acknowledgments
We thank Dr. Muthafar Al-Haddawi (Senior Veterinary Pathologist) for histopathological analysis. This project was supported by the National Institutes of Health (GM087981), the Robert A. Welch Foundation (A-1121), and High Impact Research (HIR (UM.C/625/1/HIR/MOHE/MED/17 and UM.C/625/1/HIR/MOHE/MED/33) from the Ministry of Higher Education, Malaysia.
Glossary
Abbreviations Used
- PDT
photodynamic therapy
- TrkC
tropomyosin receptor kinase C
Supporting Information Available
Structure of IY-IY-TEG, comparison of phototoxicities for all compounds at different incubation time, uptake of compounds in TrkC+ cells, biodistribution of 1-PDT in 4T1 tumor bearing mice, and toxicity of 1-PDT in the dark on 4T1 tumor. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Agarwal A.; Saraf S.; Asthana A.; Gupta U.; Gajbhiye V.; Jain N. K. Ligand based dendritic systems for tumor targeting. Int. J. Pharm. 2008, 350, 3–13. [DOI] [PubMed] [Google Scholar]
- Minko T.; Dharap S. S.; Pakunlu R. I.; Wang Y. Molecular targeting of drug delivery systems to cancer. Curr. Drug Targets 2004, 5, 389–406. [DOI] [PubMed] [Google Scholar]
- Krall N.; Scheuermann J.; Neri D. Small Targeted Cytotoxics: Current State and Promises from DNA-Encoded Chemical Libraries. Angew. Chem., Int. Ed. 2013, 52, 1384–1402. [DOI] [PubMed] [Google Scholar]
- Sievers Eric L.; Senter Peter D. Antibody–drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [DOI] [PubMed] [Google Scholar]
- Dennis M. S.; Jin H.; Dugger D.; Yang R.; McFarland L.; Ogasawara A.; Williams S.; Cole M. J.; Ross S.; Schwall R. Imaging Tumors with an Albumin-Binding Fab, a Novel Tumor-Targeting Agent. Cancer Res. 2007, 67, 254–261. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Okeley N. M.; Senter P. D. Antibody–drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [DOI] [PubMed] [Google Scholar]
- Kaur S. Bioanalysis special focus issue on antibody–drug conjugates. Bioanalysis 2013, 5, 981–983. [DOI] [PubMed] [Google Scholar]
- Borsi L.; Balza E.; Bestagno M.; Castellani P.; Carnemolla B.; Biro A.; Leprini A.; Sepulveda J.; Burrone O.; Neri D.; Zardi L. Selective targeting of tumoral vasculature: comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int. J. Cancer 2002, 102, 75–85. [DOI] [PubMed] [Google Scholar]
- Xia W.; Low P. S. Folate-Targeted Therapies for Cancer. J. Med. Chem. 2010, 53, 6811–6824. [DOI] [PubMed] [Google Scholar]
- Low P. S.; Kularatne S. A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Low Philip S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Delivery Rev. 2012, 64, 342–352. [DOI] [PubMed] [Google Scholar]
- Hilgenbrink A. R.; Low P. S. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J. Pharm. Sci. 2005, 94, 2135–2146. [DOI] [PubMed] [Google Scholar]
- Lee S.; Xie J.; Chen X. Peptides and Peptide Hormones for Molecular Imaging and Disease Diagnosis. Chem. Rev. 2010, 110, 3087–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. E.; Rice K. G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garanger E.; Boturyn D.; Dumy P. Tumor targeting with RGD peptide ligands—design of new molecular conjugates for imaging and therapy of cancers. Anti-Cancer Agents Med. Chem. 2007, 7, 552–558. [DOI] [PubMed] [Google Scholar]
- Dunehoo A. L.; Anderson M.; Majumdar S.; Kobayashi N.; Berkland C.; Siahaan T. J. Cell adhesion molecules for targeted drug delivery. J. Pharm. Sci. 2006, 95, 1856–1872. [DOI] [PubMed] [Google Scholar]
- Li F.; Liu J.; Jas G. S.; Zhang J.; Qin G.; Xing J.; Cotes C.; Zhao H.; Wang X.; Diaz L. A.; Shi Z.-Z.; Lee D. Y.; Li K. C. P.; Li Z. Synthesis and Evaluation of a Near-Infrared Fluorescent Non-Peptidic Bivalent Integrin αvβ3 Antagonist for Cancer Imaging. Bioconjugate Chem. 2010, 21, 270–278. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Li Z. Molecular imaging probes for diagnosis and therapy evaluation of breast cancer. Int. J. Biomed. Imaging 2013, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P.; Edkins S.; Davies H.; Greenman C.; Cox C.; Hunter C.; Bignell G.; Teague J.; Smith R.; Stevens C.; O’Meara S.; Parker A.; Tarpey P.; Avis T.; Barthorpe A.; Brackenbury L.; Buck G.; Butler A.; Clements J.; Cole J.; Dicks E.; Edwards K.; Forbes S.; Gorton M.; Gray K.; Halliday K.; Harrison R.; Hills K.; Hinton J.; Jones D.; Kosmidou V.; Laman R.; Lugg R.; Menzies A.; Perry J.; Petty R.; Raine K.; Shepherd R.; Small A.; Solomon H.; Stephens Y.; Tofts C.; Varian J.; Webb A.; West S.; Widaa S.; Yates A.; Brasseur F.; Cooper C. S.; Flanagan A. M.; Green A.; Knowles M.; Leung S. Y.; Looijenga L. H. J.; Malkowicz B.; Pierotti M. A.; Teh B.; Yuen S. T.; Nicholson A. G.; Lakhani S.; Easton D. F.; Weber B. L.; Stratton M. R.; Futreal P. A.; Wooster R. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nature Genet. 2005, 37, 590–592. [DOI] [PubMed] [Google Scholar]
- Ivanov S. V.; Panaccione A.; Brown B.; Guo Y.; Moskaluk C. A.; Wick M. J.; Brown J. L.; Ivanova A. V.; Issaeva N.; El-Naggar A. K.; Yarbrough W. G. TrkC signaling is activated in adenoid cystic carcinoma and requires NT-3 to stimulate invasive behavior. Oncogene 2013, 32, 3698–3710. [DOI] [PubMed] [Google Scholar]
- Jin W.; Kim G.-M.; Kim M.-S.; Lim M.-H.; Yun C.-H.; Jeong J.; Nam J.-S.; Kim S.-J. TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis 2010, 31, 1939–1947. [DOI] [PubMed] [Google Scholar]
- Liu J.; Brahimi F.; Saragovi H. U.; Burgess K. Bivalent Diketopiperazine-Based TrkC Antagonists. J. Med. Chem. 2010, 53, 5044–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Brahimi F.; Angell Y.; Li Y.-C.; Moscowicz J.; Saragovi H. U.; Burgess K. Bivalent Peptidomimetic Ligands of TrkC are Biased Agonists, Selectively Induce Neuritogenesis, or Potentiate Neurotrophin-3 Trophic Signals. ACS Chem. Biol. 2009, 4, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahimi F.; Malakhov A.; Lee H. B.; Pattarawarapan M.; Ivanisevic L.; Burgess K.; Saragovi H. U. A peptidomimetic of NT-3 acts as a TrkC antagonist. Peptides 2009, 30, 1833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison R. R.; Sibata C. H. Oncologic photodynamic therapy photosensitizers: a clinical review. Photodiagn. Photodyn. Ther. 2010, 7, 61–75. [DOI] [PubMed] [Google Scholar]
- Juarranz A.; Jaen P.; Sanz-Rodriguez F.; Cuevas J.; Gonzalez S. Photodynamic therapy of cancer. Basic principles and applications. Clin. Transl. Oncol. 2008, 10, 148–154. [DOI] [PubMed] [Google Scholar]
- Byrne A. T.; O’Connor A. E.; Hall M.; Murtagh J.; O’Neill K.; Curran K. M.; Mongrain K.; Rousseau J. A.; Lecomte R.; McGee S.; Callanan J. J.; O’Shea D. F.; Gallagher W. M. Vascular-targeted photodynamic therapy with BF2-chelated tetraaryl-azadipyrromethene agents: a multi-modality molecular imaging approach to therapeutic assessment. Br. J. Cancer 2009, 101, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. H.; Thivierge C.; Nowak-Sliwinska P.; Han J.; Van den Bergh H.; Wagnieres G.; Burgess K.; Lee H. B. In vitro and in vivo photo-cytotoxicity of boron dipyrromethene derivatives for photodynamic therapy. J. Med. Chem. 2010, 53, 2865–2874. [DOI] [PubMed] [Google Scholar]
- Yogo T.; Urano Y.; Ishitsuka Y.; Maniwa F.; Nagano T. Highly efficient and photostable photosensitizer based on BODIPY chromophore. J. Am. Chem. Soc. 2005, 127, 12162–12163. [DOI] [PubMed] [Google Scholar]
- Awuah S. G.; You Y. Boron dipyrromethene (BODIPY)-based photosensitizers for photodynamic therapy. RSC Adv. 2012, 2, 11169–11183. [Google Scholar]
- Kamkaew A.; Lim Siang H.; Lee Hong B.; Kiew Lik V.; Chung Lip Y.; Burgess K. BODIPY dyes in photodynamic therapy. Chem. Soc. Rev. 2012, 42, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowt R.; von Bartheld C. S. Sorting of internalized neurotrophins into an endocytic transcytosis pathway via the Golgi system: ultrastructural analysis in retinal ganglion cells. J. Neurosci. 2001, 21, 8915–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M. V. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nature Rev. Neurosci. 2003, 4, 299–309. [DOI] [PubMed] [Google Scholar]
- Pattarawarapan M.; Burgess K. The Molecular Basis of Neurotrophin–Receptor Interactions. J. Med. Chem. 2003, 46, 5277–5291. [DOI] [PubMed] [Google Scholar]
- Louie E.; Chen X. F.; Coomes A.; Ji K.; Tsirka S.; Chen E. I. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2012, 32, 4064–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhecke E.; Adriaenssens E.; Verbeke S.; Meignan S.; Germain E.; Berteaux N.; Nurcombe V.; Le Bourhis X.; Hondermarck H. Brain-Derived Neurotrophic Factor and Neurotrophin-4/5 Are Expressed in Breast Cancer and Can Be Targeted to Inhibit Tumor Cell Survival. Clin. Cancer Res. 2011, 17, 1741–1752. [DOI] [PubMed] [Google Scholar]
- Blasco-Gutierrez M. J.; San Jose-Crespo I. J.; Zozaya-Alvarez E.; Ramos-Sanchez R.; Garcia-Atares N. TrkC: a new predictive marker in breast cancer?. Cancer Invest. 2007, 25, 405–410. [DOI] [PubMed] [Google Scholar]
- Levanti M. B.; Germana A.; Catania S.; Germana G. P.; Gauna-Anasco L.; Vega J. A.; Ciriaco E. Neurotrophin receptor-like proteins in the bovine (Bos taurus) lymphoid organs, with special reference to thymus and spleen. Anat. Histol. Embryol. 2001, 30, 193–198. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Tang Louisa S.; Hu B.; So K.-F.; Yip Henry K. Expression of trkA, trkB, and trkC in injured and regenerating retinal ganglion cells of adult rats. Invest. Ophthalmol. Vis. Sci. 2002, 43, 1954–1964. [PubMed] [Google Scholar]
- Botchkarev V. A.; Metz M.; Botchkareva N. V.; Welker P.; Lommatzsch M.; Renz H.; Paus R. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 act as “epitheliotrophins” in murine skin. Lab. Invest. 1999, 79, 557–572. [PubMed] [Google Scholar]
- Aslakson C. J.; Miller F. R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [PubMed] [Google Scholar]
- Kamkaew A.; Burgess K. Double-Targeting Using a TrkC-Ligand Conjugated To BODIPY-Based ADT Agent. J. Med. Chem. 2013, 56, 7608–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; Farokhzad O. C.; Margalit R.; Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnol. 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- Veenhuizen R.; Oppelaar H.; Ruevekamp M.; Schellens J.; Dalesio O.; Stewart F. Does Tumour Uptake of Foscan Determine PDT Efficacy?. Int. J. Cancer 1997, 73, 236–239. [DOI] [PubMed] [Google Scholar]
- Sasahira T.; Ueda N.; Kurihara M.; Matsushima S.; Ohmori H.; Fujii K.; Bhawal U. K.; Yamamoto K.; Kirita T.; Kuniyasu H. Tropomyosin receptor kinases B and C are tumor progressive and metastatic marker in colorectal carcinoma. Hum. Pathol. 2013, 44, 1098–1106. [DOI] [PubMed] [Google Scholar]
- Solban N.; Rizvi I.; Hasan T. Targeted photodynamic therapy. Lasers Surg. Med. 2006, 38, 522–531. [DOI] [PubMed] [Google Scholar]
- Gomer C. J.; Ferrario A. Tissue distribution and photosensitizing properties of mono-l-aspartyl chlorin e6 in a mouse tumor model. Cancer Res. 1990, 50, 3985–3990. [PubMed] [Google Scholar]
- Boch R.; Canaan A. J.; Cho A.; Dolphin D. D.; Hong L.; Jain A. K.; North J. R.; Richter A. M.; Smits C.; Sternberg E. D. Cellular and antitumor activity of a new diethylene glycol benzoporphyrin derivative (lemuteporfin). Photochem. Photobiol. 2006, 82, 219–224. [DOI] [PubMed] [Google Scholar]
- Koh J.-Y.; Gwag B. J.; Lobner D.; Choi D. W. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science 1995, 268, 573–575. [DOI] [PubMed] [Google Scholar]
- Rao J.; Dragulescu-Andrasi A.; Yao H. Fluorescence imaging in vivo: recent advances. Curr. Opin. Biotechnol. 2007, 18, 17–25. [DOI] [PubMed] [Google Scholar]
- Ballou B.; Ernst L. A.; Waggoner A. S. Fluorescence imaging of tumors in vivo. Curr. Med. Chem. 2005, 12, 795–805. [DOI] [PubMed] [Google Scholar]
- Frangioni J. V. In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol. 2003, 7, 626–634. [DOI] [PubMed] [Google Scholar]
- Sevick-Muraca E. M.; Houston J. P.; Gurfinkel M. Fluorescence-enhanced, near infrared diagnostic imaging with contrast agents. Curr. Opin. Chem. Biol. 2002, 6, 642–650. [DOI] [PubMed] [Google Scholar]
- Chan E.; Mulkerin D.; Rothenberg M.; Holen K. D.; Lockhart A. C.; Thomas J.; Berlin J. A phase I trial of CEP-701 + gemcitabine in patients with advanced adenocarcinoma of the pancreas. Invest. New Drugs 2008, 26, 241–247. [DOI] [PubMed] [Google Scholar]
- Workman P.; Aboagye E. O.; Balkwill F.; Balmain A.; Bruder G.; Chaplin D. J.; Double J. A.; Everitt J.; Farningham D. A. H.; Glennie M. J.; Kelland L. R.; Robinson V.; Stratford I. J.; Tozer G. M.; Watson S.; Wedge S. R.; Eccles S. A. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuvaneswari R.; Gan Y. Y.; Soo K. C.; Olivo M. Targeting EGFR with photodynamic therapy in combination with Erbitux enhances in vivo bladder tumor response. Mol. Cancer 2009, 8, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y.-S.; Chang C.-C.; Chang T.-C.; Yang T.-L.; Young T.-H.; Lou P.-J. Photo-induced antitumor effect of 3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide. Biomed. Res. Int. 2013, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomayko M. M.; Reynolds C. P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.