

Abstract
The majority of human kidney stones are comprised of multiple calcium oxalate monohydrate (COM) crystals encasing a calcium phosphate nucleus. The physiochemical mechanism of nephrolithiasis has not been well determined on the molecular level; this is crucial to the control and prevention of renal stone formation. This work investigates the role of phosphate ions on the formation of calcium oxalate stones; recent work has identified amorphous calcium phosphate (ACP) as a rapidly forming initial precursor to the formation of calcium phosphate minerals in vivo. The effect of phosphate on the nucleation of COM has been investigated using the constant composition (CC) method in combination with scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Our findings indicate COM nucleation is strongly promoted by the presence of phosphate; this occurs at relatively low phosphate concentrations, undersaturated with respect to brushite (dicalcium phosphate dehydrate, DCPD) formation. The results show that ACP plays a crucial role in the nucleation of calcium oxalate stones by promoting the aggregation of amorphous calcium oxalate (ACO) precursors at early induction times. The coaggregations of ACP and ACO precursors induce the multiple-point nucleation of COM. These novel findings expand our knowledge of urinary stone development, providing potential targets for treating the condition at the molecular level.
Short abstract
In solutions that are supersaturated with respect to calcium oxalate, the presence of phosphate results in the initial formation of amorphous calcium phosphate clusters; these clusters were found to promote the aggregation of amorphous calcium oxalate complexes which induce the nucleation and growth of urinary stones.
Introduction
Nephrolithiasis is a significant health problem which has been afflicting humankind for many centuries.1−4 Approximately 85% of human kidney stones are a combination of calcium oxalate monohydrate (COM) crystals with calcium phosphate (Ca–P) phases as a nucleus.5,6 These stones have multiple COM crystals which have aggregated around a Ca–P core forming large clusters.7,8 Although calcium oxalate stone formation is a very common health problem, the mechanism of stone formation is still poorly understood. To better understand how renal stones form in vitro, the role of calcium phosphate on the process of calcium oxalate nucleation and crystal growth must be further investigated. It is accepted that Ca–P phases play a critical role in the formation of large aggregate renal stones and also influence stone size.
Previous studies have suggested that the Ca–P crystal phases, brushite (DCPD, CaHPO4·2H2O) or hydroxyapatite (HAP), may serve as heterogeneous nuclei that initiate the precipitation of calcium oxalate monohydrate (COM).9−14 The influences of these minerals on calcium oxalate mineral formation is challenged by speciation ccalculations based on reported phosphate concentrations in human urine and the relative supersaturation (σ) of the suggested Ca–P phases (Supporting Information, Figure S1). In healthy acidic uric conditions, urine is undersaturated with respect to DCPD mineral formation, and is slightly supersaturated with respect to HAP, with supersaturation less than 5.0. The induction time of HAP nucleation at such concentrations is known to be more than several weeks or longer.15 The formation of DCPD under these conditions is unlikely: calcium and phosphate concentration would need to increase 5–10 times higher than the normal concentration of calcium and phosphate in urine for the solution to be supersaturated with respect to DCPD (Supporting Information, Figure S1c,d). This would indicate that DCPD crystals are difficult to form directly in urine. The supersaturation of Ca–P minerals is relatively low in urine, suggesting that the initial crystal Ca–P phases are difficult to form directly in urine. This may indicate the formation of less soluble lower energy Ca–P phases such as amorphous calcium phosphate, inducing the heterogeneous nucleation of COM. The urinary environment in patients with recurrent calcium nephrolithiasis has been reported to be supersaturated with respect to brushite;16−19 in these people the induction time for nucleation of DCPD is relatively low. The nucleation and crystal growth of DCPD will decrease the calcium concentration in urine and as a result decrease the supersaturation with respect to COM. Thus, the direct formation of DCPD in vivo will suppress the nucleation and growth of COM, which is in conflict with early thinking of DCPD promoting the formation of calcium oxalate stones by heterogeneous nucleation.
Recent investigations have indicated that amorphous cluster aggregation is a key step in slow biomineralization processes.15,20 In a slightly supersaturated Ca–P solution, stable amorphous Ca–P (ACP) complexes can form spontaneously with a small thermodynamic barrier and then coagulate into large amorphous aggregates in the early induction period. Prenuclei of HAP or DCPD could form within the solid amorphous Ca–P complexes via a solid–solid nucleation process. With the small supersaturation in urine, HAP nuclei are difficult to form directly (induction time of more than 1 week); the amorphous CaP complexes could be stable for appreciable times. Many studies indicated that amorphous precursors are also involved in calcium oxalate nucleation processes.12,13 Although attention has been paid to the role of amorphous Ca–P phases in the HAP nucleation, the influence of ACP on COM formation has been ignored.
The existence of amorphous calcium phosphate complexes and their coaggregation with amorphous calcium oxalate may be a key initial step in the mechanism leading to the formation of calcium oxalate (CaOx) stones.20,21 In the present study, we systematically investigated the role of phosphate on the nucleation of COM using a sensitive constant composition (sCC) method that utilizes potentiometric glass electrode and calcium selective electrode monitoring of the calcium ion and hydrogen ion activities (0.001 pH resolution) associated with the nucleation events.15 This method allows the simultaneous monitoring of the species association and complexation in solution, as well as the nucleation and crystal growth of COM. The analysis was augmented by our scanning electron microscopy (SEM) and transmission electron microscopy (TEM) studies performed in parallel. The results unequivocally show that the presence of amorphous calcium phosphate complexes promotes the aggregation of amorphous CaOx precursors, inducing the nucleation of COM crystallites, and the subsequent formation of multicrystal aggregates.
Materials and Methods
The CC method can mimic in vivo biologically stabilized milieu for crystallization.29 Titrant solutions are added to maintain constant reaction solution concentrations during the experiments. A potentiometer is used to control titrant addition from stepper-motor-driven burets. Reaction initiation triggered, potentiometrically, the addition of titrant solutions, which have concentrations calculated based upon the speciation computations using mass balance and electroneutrality expression together with the extended Debye–Hückel equation.
The double constant composition method (DCC) (two CC devices incorporating two different ion selective sensors) is used to simultaneously control two competing crystallization processes at constant driving force.13 For this study, COM and brushite labeled as BA and BC, respectively, share a common B ion (calcium). For the simultaneous growth of BA and BC crystals, B ion (calcium ion specific electrode) and C ion (pH electrode) selective electrodes are used to control BA and BC titrants, respectively.22
COM (BA) phase/controlled by calcium electrode:
Brushite (BC) phase/controlled by glass electrode:
where W and T are the total concentrations in the reaction solutions and titrants, respectively, and Ceff,COM and Ceff,brushite are the effective titrant concentrations with respect to COM and brushite, respectively.
Preparation of Solutions
Calcium chloride dihydrate (CaCl2·2H2O) was purchased from OmniPur (purity ≥ 99.0%, lot no. G07000246A) and potassium dihydrogen phosphate (KH2PO4) was purchased from J.T. Baker (purity ≥ 99.7%, lot no. H08479). Potassium oxalate (K2Ox) was purchased from OmniPur (purity ≥ 99.0%). Other chemical reagents were of analytical grade and purchased from J.T. Baker. Water was deionized and triply distilled before use. Solutions used in this study were prepared fresh, and filtered twice through 50 nm pore size filters (SterliTech, lot no. 37682). The concentration of calcium stock solution was determined by EDTA titration.
COM Nucleation Experiments
In this study, COM nucleation experiments were made at pH 6.5 at 37.0 ± 0.1 °C in magnetically stirred double-walled 250 mL Pyrex vessels at values of the relative supersaturation with respect to COM (σCOM) of 0.745 and 1.02. The relative supersaturation, σ, and supersaturation ratio, S, are given by eq 1:
| 1 |
where v is the number of ions in a formula unit of COM, and IAP and Ksp are the ionic activity and thermodynamic solubility products (Ksp = 2.47 × 10–9 M2 at 37.0 °C), respectively. Solution speciation calculations were made using the extended Debye–Hückel equation proposed by Davies.23,24
Supersaturated reaction solutions were prepared by the slow mixing of sodium chloride (NaCl), potassium dihydrogen phosphate (KH2PO4), potassium oxalate (K2Ox), potassium hydroxide (KOH), and calcium chloride (CaCl2) stock solutions ([Ca2+] = 0.045 M). The solution compositions for different supersaturation experiments are shown in Table 1.
Table 1. Solution Compositions for Different Supersaturation Experiments and Species Concentration at Equilibrium Conditions (pH 6.5, IS = 0.15 M, T = 37.0 °C) Obtained by Species Calculation Program.
| [Ox2–]/M | [Ca2+]/M | [P]/M | σDCPD | σCOM | σHAP | [CaOx]/M | [Ca2Ox]2+/M | |
|---|---|---|---|---|---|---|---|---|
| I | 3.5 × 10–4 | 3.5 × 10–4 | N/A | N/A | 0.745 | N/A | 1.9 × 10–5 | 4.5 × 10–7 |
| II | 3.8 × 10–4 | 3.8 × 10–4 | 8.0 × 10–3 | –0.42 | 0.745 | 2.76 | 1.90 × 10–5 | 4.08 × 10–7 |
| III | 4.71 × 10–4 | 4.71 × 10–4 | 3.7 × 10–2 | 0.10 | 0.745 | 4.41 | 1.90 × 10–5 | 3.19 × 10–7 |
| IV | 4.74 × 10–4 | 4.74 × 10–4 | 3.0 × 10–3 | –0.60 | 1.02 | 2.04 | 3.16 × 10–5 | 9.22 × 10–7 |
CO2 was completely removed by pumping pure, presaturated, N2 gas (>2 h) into both reaction solutions and calcium chloride stock solution prior to mixing. The pH and calcium ion concentration were monitored during crystallization experiments, with a pH electrode (Orion 91-01 ±0.1 mV) and calcium ion selective electrode (Orion 93-20 ±0.1 mV) coupled with a single-junction Ag/AgCl reference electrode (Orion 90-01).
Precipitates were collected from the solution by filtration during the experiments, and samples were dried and sputter-coated with graphite under vacuum before examination by field-emission scanning electron microscopy (SEM; Hitachi SU-70) at 20 keV.
Transmission electron microscopy (TEM) investigations of nucleated particles removed at various time intervals were carried out using a JEOL-2010 TEM at an accelerating voltage of 200 kV.
Results
Recently developed sCC techniques were combined with SEM and TEM to investigate the effect of phosphate on COM nucleation. This technique was used to investigate COM homogeneous nucleation; experiments were performed at a constant supersaturation with respect to COM (σCOM) of 0.745 at pH 6.5 and with 0.15 M ionic strength, at 37 °C.
Phosphate was found to induce COM nucleation. The events of calcium oxalate nucleation were monitored by a calcium selective electrode, in a free drift nucleation experiment (no titrant added); the calcium concentration slowly increased throughout the induction time. This indicates the release of free calcium ions into solution at this point in the nucleation process; after this small detectable release of the calcium, the concentration decreases as COM nuclei form and crystal growth occurs (Figure 1b).
Figure 1.

CC crystallization plots in a mixed solution of calcium oxalate (σCOM = 0.745) with and without phosphate at initial pH 6.5, ionic strength 0.15 M. (a) Volume of titrant addition plotted against time in control experiments ([Ca2+] = [Ox2–] = 3.5 × 10–4 M, without phosphate). (b) Calcium concentration change in free drift nucleation system ([Ca2+] = [Ox2–] = 3.5 × 10–4 M, without phosphate). (c) Titrant addition curve for phosphate containing system ([P] = 8.0 × 10–3 M, [Ca2+] = [Ox2–] = 3.81 × 10–4 M, σDCPD = −0.42, and σHAP =2.76). (d) pH and calcium concentration changes for phosphate containing system ([P] = 8.0 × 10–3 M, [Ca2+] = [Ox2–] = 3.81 × 10–4 M, σDCPD = −0.42, and σHAP =2.76). In the presence of phosphate, even the solution is undersaturated with respect to DCPD, and the nucleation of COM was strongly promoted by shortening the induction time from ∼1150 to ∼590 min.
Constant composition reactions were also carried out. In the presence of phosphate COM nucleated in approximately half the time as reactions without phosphate (Figure 1a,c). The addition of phosphate to these reactions at relatively low concentrations ([P] = 8.00 × 10–3 M, [Ca2+] = [Ox2–] = 3.81 × 10–4 M), concentrations undersaturated with respect to the formation of DCPD (σDCPD = −0.412), decreased the induction time for COM nucleation by nearly half to 590 min under (Figure 1c). The rate of titrant addition in sCC reactions was based on the potential change of the calcium selective electrode. When the results of the reactions in the presence of phosphate are compared with the control (Figure 1c and 1a), it is clear the rate of titrant addition in COM nucleation reactions in the presence of phosphate (0.57 mL/h) was considerably less than that of the control system (6.6 mL/h). This decrease indicates that COM crystal growth is retarded in the presence of phosphate.
At phosphate concentrations undersaturated with respect to the formation of DCPD, the calcium concentration slowly increased during the initial stages of the COM induction period. At the same time, the pH decreased in the induction period (Figure 1d); this increase is likely related to the formation of amorphous calcium phosphate as previously described by Nancollas et.al.15
At higher phosphate concentration ([P] = 3.71 × 10–2 M, [Ca2+] = [Ox2–] = 4.71 × 10–4 M), conditions supersaturated with respect to DCPD (σDCPD = 0.10), the induction time was greatly reduced to only 60 min. The formation of DCPD crystals was detected, promoting COM nucleation and decreasing the induction time for COM formation to 60 min from 1150 min in reactions without phosphate (Figure 2).
Figure 2.
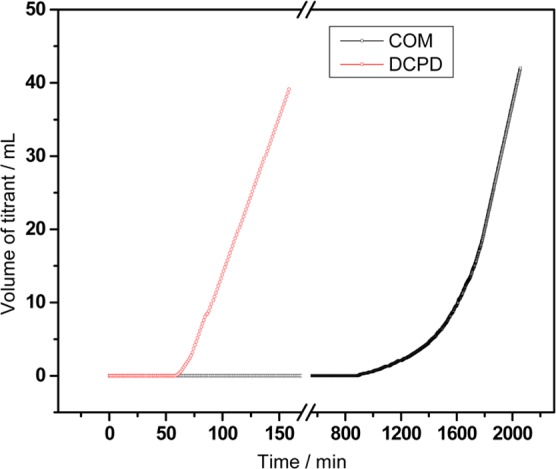
Double constant composition plots for COM nucleation (σCOM = 0.745) in the presence of higher amount of phosphate ([P] = 3.71 × 10–2 M, [Ca2+] = [Ox2–] = 4.71 × 10–4 M) at pH 6.5, ionic strength 0.15 M. Solution is supersaturated with respect to DCPD (σDCPD = 0.10) and HAP (σHAP = 4.41).
In addition to sCC experiments, COM free drift nucleation experiments were carried out to further investigate the effect of phosphate on the nucleation of COM. The free drift nucleation experiments were done at a higher supersaturation with respect to calcium oxalate formation which decreased the induction time (σCOM = 1.02) at pH 6.5. These experiments were initiated by the addition of calcium solution to the reaction solution. Changes in calcium concentration and pH were monitored throughout the reactions. The presence of phosphate in the reaction solution ([P] = 3.0 × 10–3 M) at concentrations undersaturated with respect to DCPD (σDCPD = −0.6) resulted in a rapid increase in pH followed by a slower decrease (Figure 3, stage I). In the CO2 free system, the pH change is a result of the varying equilibria of phosphate and oxalate species. A similar discussion in our previous work regarding the relatively slower dehydration rate of calcium ions compared to that of phosphate ions suggests the final addition of calcium to the reaction solution resulted in the calcium-deficient cluster formation. These clusters are not stable: the rapid release of excess HPO42– results in a sharp increase in pH following calcium addition.15 As this occurs, calcium ions associate with the HPO42– which results in the observed decrease in calcium concentration at this stage. The initial pH and calcium concentration changes confirmed the formation of amorphous calcium phosphate cluster in the calcium oxalate nucleation system. At the same time, calcium oxalate complex formation also contributed to the decrease in calcium concentration observed during this stage (Figure 3, stage I). Following stage I the pH decreased slightly and the calcium concentration increased (Figure 3, stage II); this corresponds to the formation of COM at this point in the induction period. The induction time or COM nucleation at σCOM = 1.02 in the presence of phosphate ([P] = 3.0 × 10–3 M) was 130 min, decreased from 430 min in control experiments (Supporting Information, Figure S2); these conditions are undersaturated in DCPD. At stage III, the calcium concentration and pH remain constant; this is likely related to a solid phase transition from calcium oxalate precursors to calcium oxalate prenuclei. Following the 120 min stabilization in pH and calcium concentration, the calcium concentration decreased. This is the start of stage IV, which is associated with the COM crystal growth (Figure 3, stage IV).
Figure 3.
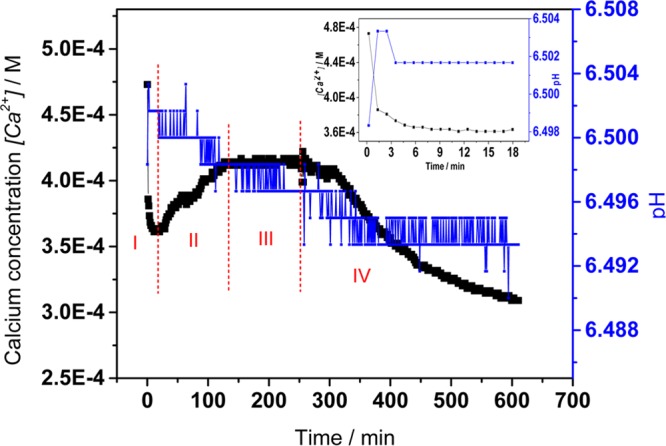
Calcium and pH changes during nucleation of calcium oxalate (σCOM = 1.02) in the presence of phosphate ([P] = 3.0 × 10–3 M, [Ca2+] = [Ox2–] = 4.74 × 10–4 M, σDCPD = −0.60, and σHAP = 2.045) at pH 6.5, ionic strength 0.15 M. The inset highlights the decrease in calcium concentration during the first 20 min of the reaction.
The precipitates were harvested at various time intervals and investigated by high-resolution transmission electron microscopy (TEM), selected area electron diffraction (SAED), and scanning electron microscopy (SEM). In the absence of phosphate large aggregates up to 100 nm were detected, in addition to smaller Ca–Ox particles with size less than 5 nm diameter. The slow addition of calcium solution for about 60 min (Figure 4a), followed by aging for 250 min, produced some larger aggregates (up to 500 nm) with porous features. SAED indicated the samples were amorphous in nature (Figure 4b). The largest porous aggregates formed (larger than 1000 nm) 1000 min after the addition of calcium to the reaction solution; at the same time some denser aggregates with crystal-like shape were detected (Figure 4c). After the induction time (1200 min, Figure 4d), small crystal nuclei were observed by TEM and the previous large porous amorphous aggregates were not detected by SAED. This would indicate that the large amorphous Ca–Ox aggregates dissolved after nuclei formation and crystal growth. These findings are supported by the dissolution–reprecipitation25 and solution-mediated transformation26 mechanisms. The COM crystal was observed by SEM (Figure 4e). The TEM results showed that the COM nucleation took place via an amorphous phase pathway, in which large porous amorphous particles were first formed by aggregation of small Ca–Ox clusters in the induction time, and then the denser nuclei formed and induced the desolation of amorphous precursors.
Figure 4.

Phase and morphology evolution of calcium oxalate precipitates during the induction periods in the absence of phosphate with supersaturation of σCOM = 0.745 at initial pH 6.5 and ionic strength 0.15 M. (a–d) TEM micrographs and SAED pattern of precipitates at the induction period; (e) SEM micrograph of COM crystal at end of experiment. Scale bar: (a) 20, (b) 50, (c) 100, (d) 50, and (e) 400 nm.
The morphology evolution was investigated by SEM. After the slow (5 min) addition of calcium solution to the reaction solution, separated particles about 30 nm in size were detected (Figure 5a). At 250 min after the addition of calcium large aggregates were formed. The Ca/P ratio detected by energy dispersive X-ray spectroscopy of these aggregates was Ca/P 3.6 (Figure 5b); this relatively high Ca/P ratio suggests the formation of coaggregates of ACP and Ca–Ox complexes early in the induction periods. At 500 min (induction time was 590 min), larger aggregates (over 2 μm) formed, which contained nuclei of COM (Figure 5c). After 1600 min plate-like COM crystal aggregates formed (Figure 5d).
Figure 5.

SEM micrographs depicting the morphology evolution of calcium oxalate precipitates during the induction period in the presence of phosphate ([P] = 8.0 × 10–3 M, [Ca2+] = [Ox2–] = 3.81 × 10–4 M, σDCPD = −0.42, and σHAP = 2.76) with supersaturation degree of σCOM = 0.745 at initial pH 6.5 and ionic strength 0.15 M. The induction time was 590 min.
TEM micrographs clearly show the coaggregating structure formed by small Ca–Ox clusters (less than 5 nm) surrounding the large amorphous calcium phosphate complex spheres (about 60 nm) after the addition of calcium to the reaction solution (Figure 6a); 60 min after the addition of the calcium solution, larger aggregates of ACP spheres were observed and these formed larger coaggregates (Figure 6b). The SAED pattern showed that these large coaggregates were amorphous. High energy electron bombardment during TEM caused the adsorbed Ca−Ox clusters on the surface of ACP spheres to became larger. These results indicated that the existence of ACP promoted the aggregation of small Ca–Ox clusters on the surface, an important step to control nucleation of COM.
Figure 6.
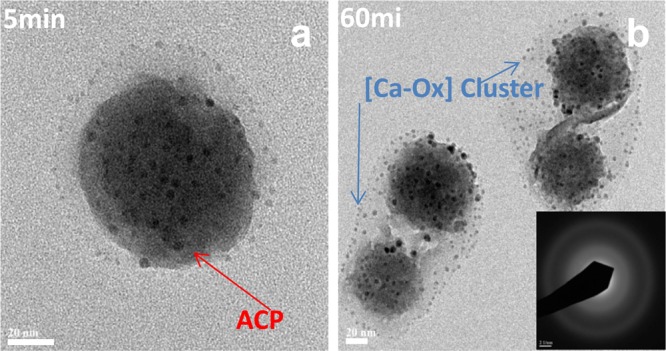
TEM micrographs and SAED pattern of aggregates between large amorphous calcium phosphate and smaller peripheral amorphous Ca–Ox precursors at early induction period of COM nucleation in the presence of phosphate (8.0 × 10–3 M) with undersaturated DCPD (σDCPD = −0.42). Scale bar: 20 nm.
Discussion
This work has demonstrated that, in the presence of phosphate, even in undersaturated solutions with respect to Ca–P phases, the nucleation of COM can be strongly promoted. This work further demonstrates that the COM nuclei form via amorphous Ca–Ox precursor clusters, which coaggregate with ACP to form large amorphous particles before nucleus formation in the presence of phosphate.
In a supersaturated solution of calcium oxalate, many Ca–Ox species will form due to ion dehydration and association. Speciation calculations indicate that there are mainly two calcium oxalate species, the uncharged [CaOx] complex and the positive [Ca2Ox]2+ complex (Table 1), and of the two the former predominated. Kinetic effects due to the faster dehydration rate of Ox when compared with calcium, the Ox-deficient species, [Ca2Ox·H2O]2+, result in the formation of these species which transform to the thermodynamically more stable [CaOx] complex following the release of extra calcium ions. This was observed as a slow increase of calcium concentration during the induction period. In the absence of phosphate, these Ca–Ox complexes will aggregate to small particles of COM which induce the formation of COM nuclei.
Previous studies on hydroxyapatite nucleation have shown that a relatively stable amorphous cluster with the composition of [Ca-(HPO4)1+x·nH2O]2x− forms initially in Ca–P systems. These amorphous clusters self-assemble to large ACP-I phases early in the induction period of HAP nucleation.15,27 In the presence of phosphate, the existence of negative Ca–P complexes, [Ca-(HPO4)1+x·nH2O]2x−, will promote the aggregation of positive Ca–Ox clusters, [Ca2Ox·H2O]2+ by electrostatic attraction and form the ACP–ACO coaggregating structure as shown in TEM images. At the same time, some calcium ions released from slow condensation of [Ca2Ox·H2O]2+ clusters will complex with more HPO42– and induce a further hydrolysis of H2PO4–, resulting in a slow pH decrease during the induction period.
The coaggregation effect from ACP promotes the formation of large calcium oxalate complexes, which promote the formation of COM prenuclei increasing to their critical size, and forming stable nuclei. The numerous Ca–Ox clusters aggregating around the ACP sphere provide multiple sites for the following nucleation and growth, resulting in multiple COM crystals encapsulating the calcium phosphate phase. This is similar to a Ca–Ox renal stone. The agglomerate structure of multiple COM crystals aggregating around the calcium phosphate phase results in a simulated kidney stone which is too large to pass the urinary tract.
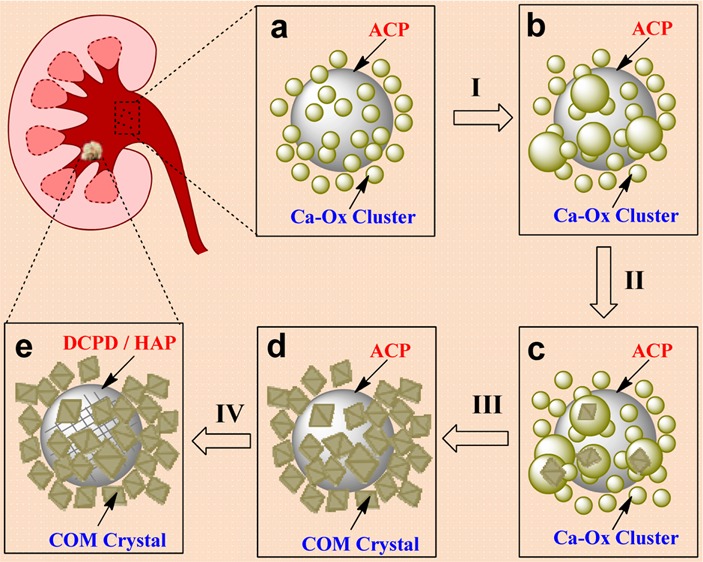
Based on the above results, a new mechanism for calcium renal stone formation has been proposed (Figure 7). An amorphous calcium oxalate (ACO) cluster containing phosphate is formed, driven by the electrostatic interaction of ACP and ACO. The result is an amorphous coaggregate with ACO surrounding an ACP sphere (Figure 7a). The ACO clusters self-assemble to larger size ACO particles on the ACP surface (Figure 7b, process I). The numerous large ACO particles provide the ideal site for COM prenuclei formation during the process of ACO phase condensation (Figure 7c, process II). As the size of prenuclei reaches a critical size, the COM nuclei will stabilize and induce the crystal growth (process III). At the same time, the small ACO particles will be dissolved. Multiple nuclei grow on the ACP surface and finally form a structure of numerous COM crystals encapsulating an ACP sphere (Figure 7d). Due to the coaggregating structure, the size of the whole agglomerate quickly increases, making it difficult to expel from the body, and a renal stone has formed. The confined ACP phase will transform to Ca–P crystal, such as DCPD or HAP, via a slow confined solid phase transition process (Figure 7e, process IV). The coaggregation of the amorphous precursors ACP and ACO, followed by the nucleation of COM surrounding a large ACP cluster, provides a possible initial step in the mechanism leading to the formation of renal stones.
Figure 7.

Schematic illustration of the nucleation mechanism for COM in the presence of phosphate at biological conditions. (a) ACP–ACO coaggregates form as small amorphous Ca–Ox clusters aggregate around large ACP spheres early in the induction period, (b) ACP surface promotes ACO aggregation, (c) multiple prenuclei form within ACO clusters, (d) a composite of multiple COM crystals forms, and (e) a kidney stone containing Ca–P crystals is formed by the confined phase transition of ACP.
The new mechanism of stone formation via coaggregation of amorphous precursors is relevant to the current investigation of the mechanism of nephrolithiasis and is an interesting comparison with the previous mechanism of heterogeneous nucleation induced by DCPD.5,28,29 The condition for DCPD supersaturation is unlikely in urine, which needs a relatively high calcium or phosphate concentration in the acidic urine environment. However, the formation and growth of DCPD will consume the calcium ion and decrease the supersaturation level of COM.
The new physicochemical mechanism for kidney stone formation highlights the important role of phosphate, and the formation of calcium phosphate nidus. Modern renal stone treatment involving proteins, amino acids, and organic crystal-growth inhibitors such as citric acid typically ignore the side effects of promoting amorphous precursor aggregation.30−32 Addressing the effects of coaggregation may lead to more effective renal stone treatments.
Conclusions
In solutions that are supersaturated with respect to calcium oxalate, the presence of phosphate results in the initial formation of amorphous calcium phosphate clusters. These clusters were found to promote the aggregation of amorphous calcium oxalate complexes which induce the nucleation and growth of urinary stones. These revelations both expand our knowledge of urinary stone development and provide potential targets for treating the condition at the molecular level. We expect that the results will inspire novel approaches to our understanding of kidney stone formation mechanisms, which will lead to innovative therapeutic interventions to improve the health of patients with kidney diseases.
Acknowledgments
The presented work was supported by National Institute of Dental and Craniofacial Research (NIDCR) Grant DE003223 to G.H.N.
Supporting Information Available
Effects of pH, calcium concentration, and phosphate concentration on supersaturation of COM (σCOM), DCPD (σDCPD), and HAP (σHAP) with oxalate concentration [Ox] = 8.0 × 10–5 M and ionic strength of 0.15 M. Volume of titrant addition and change of calcium concentration curves for COM crystallization with supersaturation of σCOM = 1.02 without phosphate at pH 6.5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† B.X. and T.J.H. made equal contributions.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Evan A.; Lingeman J.; Coe F. L.; Worcester E. Kidney Int. 2006, 69, 1313–1318. [DOI] [PubMed] [Google Scholar]
- Tang R.; Nancollas G. H.; Giocondi J. L.; Hoyer J. R.; Orme C. A. Kidney Int. 2006, 70, 71–78. [DOI] [PubMed] [Google Scholar]
- Sakhaee K. Kidney Int. 2009, 75, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushinsky D. A.; Asplin J. R.; Grynpas M. D.; Evan A. P.; Parker W. R.; Alexander K. M.; Coe F. L. Kidney Int. 2002, 61, 975–987. [DOI] [PubMed] [Google Scholar]
- Tiselius H. G.; Lindback B.; Fornander A. M.; Nilsson M. A. Urol. Res. 2009, 37, 181–192. [DOI] [PubMed] [Google Scholar]
- Reginato A. J.; Kurnik B. Semin. Arthritis Rheum. 1989, 18, 198–224. [DOI] [PubMed] [Google Scholar]
- Herring L. C. J. Urol. 1962, 88, 545–562. [DOI] [PubMed] [Google Scholar]
- Prien E. L.; Prien E. L. Am. J. Med. 1968, 45, 654–672. [DOI] [PubMed] [Google Scholar]
- Pak C. Y. C. J. Clin. Invest. 1969, 48, 1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C. Y. C.; Eanes E. D.; Ruskin B. Proc. Natl. Acad. Sci.U.S.A. 1971, 68, 1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan A. P.; Lingemon J. E.; Coe F. L.; Worcester E. M. Semin. Nephrol. 2008, 28, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. R.; Glenton P. A. J. Urol. 1995, 153, 811–817. [PubMed] [Google Scholar]
- Ebrahimpour A.; Perez L.; Nancollas G. H. Langmuir 1991, 7, 577–583. [Google Scholar]
- Lee T.; Lin Y.C. Cryst. Growth Des. 2011, 11, 2973–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B.; Halter T. J.; Borah B. M.; Nancollas G. H. Cryst. Growth Des. 2014, 14, 1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C. Y. C.; Hayashi Y.; Finlayson B.; Chu S. J. Lab. Clin. Med. 1977, 89, 891–901. [PubMed] [Google Scholar]
- Marangella M.; Vitale C.; Bagnis C.; Bruno M.; Ramello A. Nephron 1999, 81, 38–44. [DOI] [PubMed] [Google Scholar]
- Opalko F. J.; Adair J. H.; Khan S. R. J. Cryst. Growth 1997, 181, 410–417. [Google Scholar]
- Bushinsky D. A.; Parker W. R.; Asplin J. R. Kidney Int. 2000, 57, 550–560. [DOI] [PubMed] [Google Scholar]
- Gower L. B. Chem. Rev. 2008, 108, 4551–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer D.; Voelkel A.; Coelfen H. Science 2008, 322, 1819–1822. [DOI] [PubMed] [Google Scholar]
- Tomson M. B.; Nancollas G. H. Science 1978, 200, 1059–1060. [DOI] [PubMed] [Google Scholar]
- Yang X. D.; Xie B.; Wang L.; Qin Y.; Henneman Z. J.; Nancollas G. H. CrystEngComm 2011, 13, 1153–1158. [Google Scholar]
- Wang L. J.; Tang R.; Bonstein T.; Bush P.; Nancollas G. H. J. Dent. Res. 2006, 85, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eanes E. D.; Termine J. D.; Nylen M. U. Calcif. Tissue Res. 1973, 12, 143–158. [DOI] [PubMed] [Google Scholar]
- Boskey A. L.; Posner A. S. J. Phys. Chem. 1973, 77, 2313–2317. [Google Scholar]
- Habraken W. J.; Tao J.; Brylka L. J.; Friedrich H.; Bertinetti L.; Schenk A. S.; Verch A.; Dmitrovic V.; Bomans P. H.; Frederik P. M.; Laven J.; van der Schoot P.; Aichmayer B.; de With G.; DeYoreo J. J.; Sommerdijk N. A. Nat. Commun. 2013, 4, 1507–1518. [DOI] [PubMed] [Google Scholar]
- Khan S. R. J. Urol. 1997, 157, 376–383. [PubMed] [Google Scholar]
- Pak C. Y. C. J. Cryst. Growth 1981, 53, 202–208. [Google Scholar]
- Pak C. Y. C.; Rodgers K.; Poindexter J. R.; Sakhaee K. J. Urol. 2008, 180, 1532–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S. R.; Wierzbicki A.; Salter E. A.; Zepeda S.; Orme C. A.; Hoyer J. R.; Nancollas G. H.; Cody A. M.; DeYoreo J. J. J. Am. Chem. Soc. 2005, 127, 9036–9044. [DOI] [PubMed] [Google Scholar]
- Fleisch H. Kidney Int. 1978, 13, 361–371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.