

Abstract
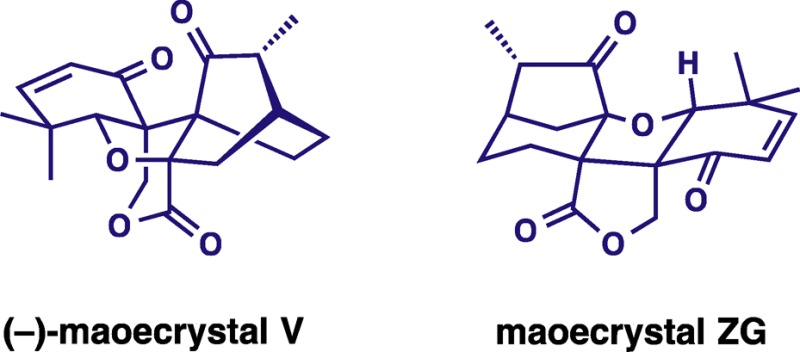
The evolution of a program directed at the enantioselective total synthesis of maoecrystal V, a highly modified ent-kauranoid, is described. An early stage chiral auxiliary-directed asymmetric C–H functionalization for the construction of a key benzofuran intermediate enabled the first asymmetric synthesis of the natural enantiomer of maoecrystal V, confirming the assigned stereochemistry. A divergent course of the central intramolecular Diels–Alder reaction, which is dependent on the nature of the dienophile, initially led to the development of an unanticipated and previously unknown isomer of maoecrystal V, which we named maoecrystal ZG. In light of the reported selective and potent cytotoxic activity of maoecrystal V, the cytotoxic properties of maoecrystal ZG were also investigated.
Introduction
The first investigations of diterpenoids from plants in the Isodon genus date back over 100 years ago to 1910, when Yagi isolated a crystalline principle plectranthin from “enmei-so”, a medication from the leaves of I. japonica and I. trichocarpa that have been used for the treatment of gastrointestinal disorders in Japan for centuries.1 In the Henan province in China, the leaves of I. rubescnes are still used by residents for the treatment of respiratory and gastrointestinal bacterial infections, inflammation, and cancer. Enmein was the first compound isolated from I. japonica in 1958, and its structure was established in 1968 by X-ray crystallography.2 Since then, over 600 individual diterpenoids from Isodon plants, displaying a broad spectrum of biological activity, have been characterized.3 Whereas the largest group is defined by the classic ent-kaurane carbon skeleton (Figure 1), another sizable structural type is 6,7-seco-ent-kauranes, examples of which include compounds like scuponeatin N, trichorabdals, longirabdolactone, and longikaurin. Related maoecrystals L4 and O5 were both isolated from I. eriocalyx.
Figure 1.

General structures and selected members of ent-kaurane diterpenoids from Isodon plants.
More recently discovered constituents of I. eriocalyx, maoecrystals V6 and Z,7 are special members of the ent-kaurane diterpenoid family because of unprecedented, highly rearranged polycyclic ring systems found in their molecular structures. The structure of maoecrystal Z combines a trans-fused 6,5,6-carbotricyclic framework with a bridging six-membered lactone and has been classified as a 6,7:8,15-di-seco-7,20-olide-6,8-cyclo-ent-kaurane. Maoecrystal V is characterized by an intricate blend of bicyclo[2.2.2]octan-2-one, δ-valerolactone, and cyclohexene subunits with a strained central oxolane ring. Both compounds have been found to display notable cytotoxic activity. The potencies of maoecrystal Z were comparable across the three human cell lines examined: K562 leukemia (IC50 2.9 μg/mL), MCF7 breast (IC50 1.6 μg/mL), and A2780 ovarian (IC50 1.5 μg/mL). Remarkably, maoecrystal V showed high selectivity against the HeLa cell line (IC50 2.9 μg/mL), with the activities for K562, A549 lung carcinoma, and BGC-823 adenocarcinoma lower by at least 6 orders of magnitude.
The total syntheses of only a few ent-kauranoids have been accomplished. Some of the pioneering achievements have been disclosed by the Mander group who reported a total synthesis of 15-deoxyeffusin almost three decades ago.8 The same group accessed longirabdolactone from giberellic acid by semisynthesis.9 Steviol and isosteviol are other ent-kauranes with a classic polycyclic skeleton that stimulated early interest as synthesis targets. The first synthesis by Mori in 197010 was followed by concise total syntheses by Ziegler,11 Snider,12 and Baran13 probing distinct strategies. Isolation of structurally unique maoecrystal V, and subsequently maoecrystal Z, is one of the recent events that reinvigorated interest in the synthesis of ent-kauranoid natural products. Reisman and co-workers developed and successfully executed a concise unified strategy for the synthesis of (−)-maoecrystal Z, (−)-trichorabdal A, and (−)-longikaurin E.14 Earlier this year, total syntheses of scuponeatin N were reported by Zhai15 and Thomson.16 Maoecrystal V, with its intricate structure distinct from other ent-kauranes, proved to be an especially compelling synthesis target, and many research groups reported different strategies for its synthesis.17,18 Presently, the total synthesis of maoecrystal V in racemic form has been completed by the groups of Yang,19 Danishefsky,20 and ours.21
Herein, we provide a full account of the enantioselective total synthesis of (−)-maoecrystal V22 and of its non-natural isomer maoecrystal ZG. The enantiodetermining step of the synthesis is a Rh-catalyzed C–H functionalization forming a dihydrobenzofuran intermediate ii (Scheme 1). We discovered that the course of the synthesis is critically dependent on the choice of dienophile partner in the central intramolecular Diels–Alder (IMDA) cycloaddition of intermediate iii forging the bicyclo[2.2.2]octan-2-one subunit. Our first effort unexpectedly delivered maoecrystal ZG, a structural isomer of the original target, which provided an opportunity to explore its antiproliferative properties. Compound 2 appears to have an unprecedented ring system. Modification of the dienophile in iii allowed for the ultimate completion of the first enantioselective total synthesis of maoecrystal V.
Scheme 1. Abbreviated Presentation of the Total Synthesis of (−)-Maoecrystal V (1) and Maoecrystal ZG (2).

Results and Discussion
Synthesis Plan
A central factor that influenced the design of the synthesis was the realization that the assembly of the central tetrahydrofuran ring, highlighted in Scheme 2, at first a seemingly simple operation, was going to present a substantial challenge. This notion was validated by the growing number of reports on the progress toward the total synthesis of 1 from several groups that halted at this stage, after successfully addressing other challenges such as the installation of the quaternary centers.17 The challenge was ascribed to the well-recognized strain associated with the five-membered heterocycle in 1. Guided in part by these observations, our goal was to preserve the oxolane ring for as long as reasonable during the progressive bond disconnection process. The cyclohexenone subunit was envisioned to arise from either an aldol cyclocondensation of a methyl ketone–aldehyde precursor or ring closing metathesis. Subsequent simplification was aimed at the C16 methyl group and the lactone ring. The six-membered lactone was to be constructed by a 6-exo radical cyclization of an acyl radical onto the enol ether. This risky blueprint was faced with potential complications due to (1) possible instability of the enol ether, (2) lack of precedent and therefore limited confidence in the reliability of radical cyclizations onto enol ethers within complex substrates, and (3) the need for the 6-exo cyclization to occur contrary to the electronic preferences of the enol ether. After installation of the tether at the C20 hydroxy group, the bicyclo[2.2.2]octan-2-one retron was disassembled via an IMDA process with a synthetic equivalent of ethylene. In addition to enabling the IMDA reaction, the function of the tether was to deliver the dienophile from the correct face of the cyclohexadeinone ring system. After further analysis, which involved ester enolate hydroxymethylation and phenol oxidation, dihydrobenzofuran vii was identified as an early synthesis target, thus fulfilling our goal to preserve the central oxolane ring during the course of the synthesis. Essential to the plan was the prospect of using the recent C–H functionalization technologies advanced by the groups of Davies and Yu to effectively access dihydrobenzofuran vii from precursors like viii.23,26,28,30
Scheme 2. Synthesis Plan.

The main variables intentionally left open to experimental feedback were the identity of groups X, Y, and Z (Scheme 2). For group Y, the preferred options were either CH2 or a protected hydroxyl group (aldehyde was deemed too reactive). We initially selected the methylene group because it promised a more direct elaboration to the cyclohexenone via RCM, recognizing its potential reactivity during the radical cyclization onto the enol ether. The nature of the tether (group X) was to be guided by two parameters: the ease of installation and then removal. The main purpose of group Z in diazo precursor viii was to support effective C–H functionalization, in terms of both chemical efficiency and enantioselectivity.
Development of the Enantioselective Synthesis of Benzofuran Intermediates by Rh-Catalyzed C–H Functionalization
Catalytic Asymmetric Approaches
Catalytic enantioselective synthesis of dihydrobenzofurans via intramolecular C–H insertions in the presence of chiral rhodium catalysts has been developed by Davies24,26 and Hashimoto.25 These findings provided the basis for the development of the synthesis of (−)-maoecrystal V by the aforementioned strategy, in which the early construction of a dihydrobenzofuran intermediate would be the enantiodetermining stage of the synthesis. Although the majority of examples involve insertions into benzylic ether C–H bonds, a few examples with aliphatic ethers were also reported. Perhaps the most relevant precedent is illustrated in Scheme 3a.24
Scheme 3.

The synthesis of the first substrate for the enantioselective C–H functionalization studies began by O-alkylation of sesamol with readily accessible neopentylic alcohol 6 under Mitsunobu conditions (Scheme 3b). The resulting ether was advanced through directed ortho-metalation followed by acylation with methyl chloroxoacetate via an arylzinc intermediate. The diazo ester (9) was accessed in two steps from 8 by tosylhydrazone formation and subsequent fragmentation under basic conditions in the presence of DBU.
The initial screening focused on identifying the preferred catalyst/solvent system at ambient temperature (Table 1). The library of catalysts used in this study is depicted in Figure 2. With Rh2(S-PTTL)4 catalyst, the best diastereoselectivity and enantioselectivity were observed in dichloromethane (DCM), although moderately higher yields were obtained in hexane and toluene (entries 3, 4). Extensive catalyst screening in DCM was disappointingly unproductive, and generally did not lead to improvement in stereoselectivity, although a somewhat higher yield of 42% was observed with Rh2(S-TCPTTL)4 (entry 6).
Table 1. Enantioselective C–H Insertion with Diazo Ester 9a.
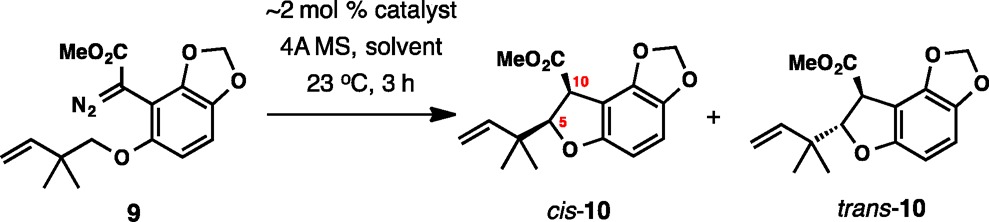
| entry | catalyst | solvent | yield (%)a | cis:trans | ee, cis (%) | ee, trans (%) |
|---|---|---|---|---|---|---|
| 1 | Rh2(S-PTTL)4 | CH2Cl2 | 25 | 9.1:1 | 70 | 50 |
| 2 | Rh2(S-PTTL)4 | EtOAc | trace | |||
| 3 | Rh2(S-PTTL)4 | hexane | 34 | 6.7:1 | 51 | |
| 4 | Rh2(R-PTTL)4 | PhMe | 47 | 5.9:1 | −60 | −33 |
| 5 | Rh2(S-NTTL)4 | CH2Cl2 | 27 | 9.1:1 | 54 | |
| 6 | Rh2(S-TCPTTL)4 | CH2Cl2 | 42 | 30 | 18 | |
| 7 | Rh2(R-BPTV)4 | CH2Cl2 | trace | |||
| 8 | Rh2(S-PTTL)4 | CH2Cl2c | 53 | 10.0:1 | 60 | |
| 9 | Rh2(S-PTAD)4 | CH2Cl2c | 22 | 3.7:1 | 48 | |
| 10 | Rh2(R-BTPCP)4 | CH2Cl2c | NR | |||
| 11 | Rh2(R-BPTV)4 | CH2Cl2c | trace | |||
| 12 | Rh2(S-biTISP)2 | CH2Cl2c | trace | 1:1 | ||
| 13 | Rh2(S-TCPTAD)4 | CH2Cl2c | 24 | 3:1 | 8 |
Diastereomer ratios and enantiomeric excess were determined by HPLC analysis.
Combined isolated yield of cis-10 and trans-10.
At reflux.
Figure 2.

Chiral rhodium catalysts used in asymmetric C–H functionalization of 9, 11, and 22.
Further incremental progress was reached when the temperature of the reaction was increased to reflux in DCM. The Rh2(S-PTTL)4 catalyst again had the best yield, delivering a 53% combined yield of the two diastereomers with a 10:1 ratio, in which the major isomer was formed in 60% ee (entry 8). Additional screening under these conditions (reflux in DCM) led to no further improvement and Rh2(S-PTTL)4 was found to be the optimal catalyst system for a potential application in total synthesis. Importantly, the high diastereoselectivity of 10:1 was critical to maintaining the original enantiomer ratio, because the catalyst controls the absolute stereochemistry at C10 rather than C5 (maoecrystal V numbering).24 Position C10 is epimerizable and is in fact inverted later during synthesis with a net result of erosion of the ee.
Overall, although the results in entry 8 were found to fall short of those desired, they provided a reasonable starting point for a potential application in a total synthesis setting. In particular, a 53% yield and the observed 10:1 diastereoselectivity would minimize the impact of enantioselectivity erosion in subsequent transformations. Although the enantioselectivity at 60% was not very high, there was a reasonable expectation that it could be further upgraded during the course of the synthesis.
Another reaction that was extensively studied with a view for a potential application in the enantioselective total synthesis of maoecrystal V is depicted in Table 2. The substrate (11) was prepared analogously to 9.21,27 Here, the nature of the ester substituent was also varied (Me or t-Bu). In general, higher chemical yields of products cis-12 and trans-12 were observed with these substrates. One important conclusion from this study is that replacing the terminal alkene in 9 with p-methoxybenzyloxy resulted in a loss of diastereoselectivity across the range of catalysts in several solvents. Although rather high enantioselectivity of up to 80% could be achieved with catalysts Rh2(S-PTTL)4 and Rh2(S-BPTV)4, the low diastereoselectivity of ∼1:1 observed for 11 (R = Me) negated the ability to control stereochemistry at C5 (entries 1 and 11). In fact, with any of the catalysts examined, the critically important diastereoselectivity never reached the levels observed for diazo ester 9. Although the tert-butyl ester (11, R = t-Bu) generally displayed higher diastereoselectivity, the highest level among the studied reactions (3.8:1) was recorded for the methyl ester with catalyst Rh2(S-BTPCP)4 (13). However, enantioselectivity was low for both cis-12 (ee 36%) and trans-12 (ee 11%).
Table 2. Enantioselective C–H Insertion with Diazo Esters 11a.
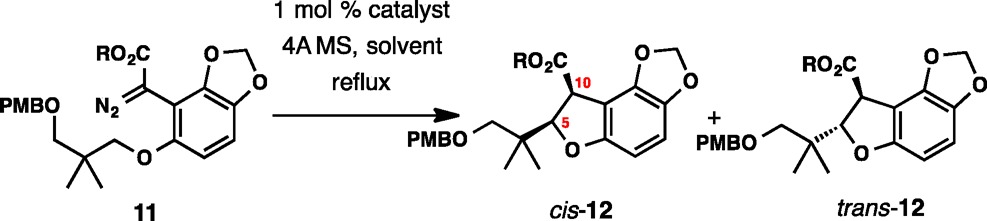
| entry | R | catalyst | solvent | yield %b | cis:trans | ee, cis (%) | ee, trans (%) |
|---|---|---|---|---|---|---|---|
| 1 | Me | Rh2(S-PTTL)4 | DMB | 71 | 1.4:1 | 45 | 80 |
| 2 | Me | Rh2(S-PTTL)4 | CHCl3 | 63 | 1.6:1 | 49 | 67 |
| 3 | Me | Rh2(S-PTTL)4 | c-C6H12 | 69 | 1.6:1 | 41 | 77 |
| 4 | t-Bu | Rh2(S-PTTL)4 | DMB | 55 | 3.0:1 | <5 | 66 |
| 5 | t-Bu | Rh2(S-PTTL)4 | CHCl3 | 28 | 3.0:1 | <5 | 57 |
| 6 | t-Bu | Rh2(S-PTTL)4 | c-C6H12 | 70 | 3.5:1 | 23 | 59 |
| 7 | Me | Rh2(S-PTA)4 | c-C6H12 | 79 | 1.0:1 | 18 | 57 |
| 8 | Me | Rh2(R-PTV)4 | c-C6H12 | 76 | 1:1.1 | ∼42 | ∼78 |
| 9 | Me | Rh2(S-NTTL)4 | c-C6H12 | 74 | 2.2:1 | 29 | 56 |
| 10 | Me | Rh2(S-PTAD)4 | c-C6H12 | 79 | 1.4:1 | 41 | 54 |
| 11 | Me | Rh2(R-BPTV)4 | c-C6H12 | 76 | 1:1.1 | ∼43 | ∼80 |
| 12 | Me | Rh2(S-biTISP)4 | c-C6H12 | 78 | 1:1.4 | ∼10 | 38 |
| 13 | Me | Rh2(S-BTPCP)4 | c-C6H12 | 65 | 3.8:1 | ∼36 | ∼11 |
| 14 | Me | Rh2(R-DPCP)4 | c-C6H12 | 74 | 1:1.1 | 19 | ∼18 |
| 15 | Me | Rh2(S-DOSP)4 | c-C6H12 | 71 | 1.0:1 | <5 | 7 |
| 16 | Me | Rh2(S-BNP)4 | c-C6H12 | 73 | 1.9:1 | ∼19 | ∼45 |
| 17 | t-Bu | Rh2(S-BTPCP)4 | c-C6H12 | 77 | 2.8:1 | 42 | 63 |
| 18 | t-Bu | Rh2(S-biTISP)2 | c-C6H12 | 77 | 2.5:1 | ∼46 | 46 |
| 19 | t-Bu | Rh2(S-BNP)4 | c-C6H12 | 83 | 3.3:1 | 13 | 24 |
Diastereomer ratios determined by 1H NMR analysis of crude reaction mixture. Enantiomeric excesses were determined by HPLC analysis.
Combined isolated yield of cis-12 and trans-12.
Davies and Yu recently reported an elegant enantioselective approach to 2,3-dihydrobenzofurans based on consecutive C–C and C–O bond-forming C–H functionalization reactions,28 which we also explored for our purposes (Scheme 4). The desired transformation was complicated by the need to use challenging neopentylic ether 13 as the substrate whereas the precedent existed only for benzylic ethers. A preliminary evaluation of the first step of the process was carried out with ether 13 or 14 and Rh2(OAc)4 in dichloromethane.29 No conversion was observed in this reaction, confirming that 13 is indeed a challenging substrate. To explore the C–O bond formation, substrate 15b was prepared by aldol reaction between aldehyde 16 and ester methyl 2-(benzo[d][1,3]dioxol-4-yl)acetate (99% yield, dr 12:1). After a partial separation of the syn and anti aldol products, the Pd-catalyzed oxidative cyclization to 2,3-dyhydrobenzofuran was studied. Under the reported conditions,28,30 the pure syn-15b showed low reactivity, and only oxidation to keto ester 17 was observed. However, a mixture of syn-15b and anti-15b (∼2:1) afforded trans-2,3-dihydrobenzofuran 24, ketone 17, and isomerically pure syn-15b.
Scheme 4.

These outcomes suggest that, unlike syn-15b, anti-15b can potentially serve as a useful substrate for the synthesis of 2,3-dihydrobenzofuran 24 via oxidative C–H functionalization, provided it is accessible in an enantio- and diastereoselective fashion.
An approach based on a trans-annular cyclization was also assessed (Scheme 5). Nine-membered diazo lactone 19 was prepared from keto ester 18 in four steps. No CH insertion product 20 was observed upon treatment with Rh2(OAc)4, and only the corresponding keto lactone was isolated along with the recovered starting material.
Scheme 5.

In summary, among the catalytic asymmetric methods, the current results suggest that only the conversion of 9 to cis-10 in the presence of Rh2(S-PTTL)4 at reflux in DCM (53% yield, 10:1 dr, 60% ee, Table 1, entry 8) could be considered for a potential application in the enantioselective total synthesis of 1.
Auxiliary-Based Approaches
Chiral auxiliaries attached at the ester carbonyl group have been shown to be effective instruments for controlling stereochemistry in C–H functionalization reactions of diazo esters, with several applications in total synthesis.31 Our initial foray into the auxiliary-based methods began with menthol as the stereodirecting group, which was found to be ineffective. Significantly improved stereocontrol was achieved when pyrrolidine amides from lactic and mandelic acids were investigated. The preparation of the substrates from keto ester 18 was accomplished in four steps as illustrated in Scheme 6.
Scheme 6.

Diastereoselective C–H insertions with lactamide 22a and mandelamide 22b have been investigated with a selected group of catalysts under different reaction conditions. Although only the cis-23a and cis-23b are shown in Table 3, up to four diastereomeric products are produced in the reaction. To simplify analysis, the mixture of diastereomers was subjected to a rather facile methanolysis (CH3ONa, CH3OH, 23 °C, 5 min). Doing so also accomplishes complete isomerization at C10, giving only the trans-substituted product (24) in pure form, the enantiomeric excess of which was then measured by HPLC. The initial screening with Rh2(R-DOSP)4 catalyst revealed that CH2Cl2 was the optimal solvent, although the reactivity was lower in toluene and, especially, in cyclohexane (entries 4 and 8–10), whereas the use of acetonitrile resulted in decomposition. The diastereoselectivity was low in dichloroethane (entry 7). The catalysts Rh2(S-PTAD)4, Rh2(R-PTAD)4, and Rh2(pfb)4 were also unreactive (entries 1, 2, and 6). The highest yields and diastereoselectivities in the formation of 23a were first observed with both Rh2(S-DOSP)4 and Rh2(R-DOSP)4 (entries 3 and 4). After methanolysis, the same enantiomer of 24 was produced from both catalysts in 62 and 70% ee, respectively. The use of the simple achiral catalyst Rh2(OAc)4 led to comparable results, giving 24 in 65% ee in good yield (entry 5). Further improvements were achieved with mandelamide substrate 22b. Although the enantiomeric purity of the final methanolysis product 24 was somewhat lower with Rh2(S-DOSP)4 and Rh2(R-DOSP)4 (entries 11 and 12), dirhodium tetraacetate provided 24 in 76% ee.
Table 3. Lactamide and Mandelamide as Chiral Auxiliaries .
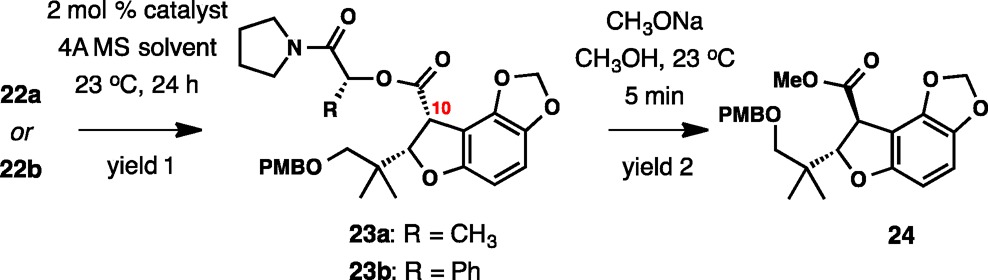
| entry | starting material | catalyst | solvent | yield 1 (%)a | yield 2 (%)b | ee %e |
|---|---|---|---|---|---|---|
| 1 | 22a | Rh2(S-PTAD)4 | CH2Cl2 | <10 | ||
| 2 | 22a | Rh2(R-PTAD)4 | CH2Cl2 | <10 | ||
| 3 | 22a | Rh2(S-DOSP)4 | CH2Cl2 | 63 | 83 | 62 |
| 4 | 22a | Rh2(R-DOSP)4 | CH2Cl2 | 74 | 86 | 70 |
| 5 | 22a | Rh2(OAc)4 | CH2Cl2 | 84 | 84 | 65 |
| 6 | 22a | Rh2(O2CC4F9)4 | CH2Cl2 | 0 | ||
| 7 | 22a | Rh2(R-DOSP)4 | (CH2Cl)2 | 72 | ||
| 8 | 22a | Rh2(R-DOSP)4 | PhMe | 61 | ||
| 9 | 22a | Rh2(R-DOSP)4 | c-C6H12 | <10 | ||
| 10 | 22a | Rh2(R-DOSP)4 | CH3CN | 0 | ||
| 11 | 23b | Rh2(S-DOSP)4 | CH2Cl2 | 84 | 89 | 59 |
| 12 | 23b | Rh2(R-DOSP)4 | CH2Cl2 | 57 | 77 | 52 |
| 13 | 23b | Rh2(OAc)4 | CH2Cl2 | 72 | 85c | 76 |
| 14 | 23b | Rh2(OAc)4 | CH2Cl2 | 61d | 84 | 84 |
Isolated yield.
Combined isolated yield.
Isolated yield, complete isomerization to the trans isomer observed.
Major diastereomer isolated in pure form by column chromatography.
Determined by HPLC.
The optimal enantiomeric purity of 84% ee was obtained from 22b with Rh2(OAc)4 after a careful chromatographic purification of the major diastereomer, which prevented erosion of enantiomeric purity upon methanolysis (entry 14). Overall, this result provided a practical entry point to enantioenriched 2,3-dihydrobenzofuran 24 for application in the total synthesis of maoecrystal V.32
Investigation of the IMDA Reaction
As illustrated in Schemes 1 and 2, a key reaction en route to 1 is the formation of the bicyclo[2.2.2]octan-2-one subunit by an IMDA addition.33 We intended to pursue a tether-based delivery of ethylene as a dienophile in the IMDA reaction. The range of tether groups that we considered for this purpose is indicated in Figure 3. Among constraints placed on the tether groups for the total synthesis applications are (1) efficient installation, (2) capacity to enable a high-yielding IMDA process, and (3) efficient removal, ideally solved in one step. We previously described preliminary studies that targeted compounds 25a–c.34 Although they have been very informative in validating the IMDA approach, the third constraint was not fulfilled as the tether removal from compounds 25a–c required several steps at best and was deemed inefficient.
Figure 3.
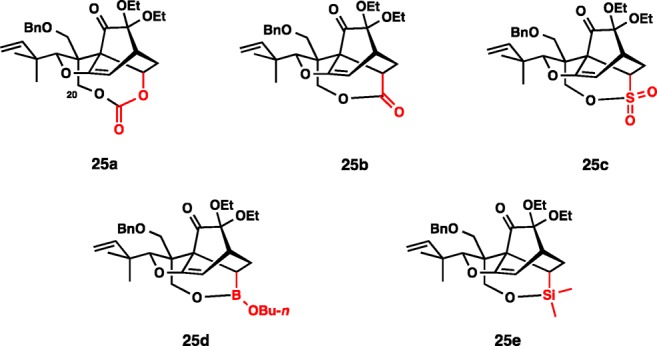
Potential products of the key IMDA reaction considered for the total synthesis of maoecrystal V.
The butyl boronate (25d) and dimethylsilyl tethers (25e) appeared to be more promising. We were especially intrigued by the borinate tether because it has limited application in the context of total synthesis and promised the development of new chemistry during the course of its removal.35 With the goal of exploring the boronate tether, the synthesis of the substrate (29) was initiated by the benzyloxymethylation of trans-10 (Scheme 7). Within our previous experience, the enolate alkylation was consistently best performed through the intermediacy of the zincate enolate.36 Alkylation product 26 was produced in 95% yield as a single isomer. Reduction of the methyl ester with LiAlH4 was followed by elaboration of the dioxolane ring in 27 by the action of methylmagnesium bromide at 80 °C in benzene, which delivered 2-ethoxy phenol 28 in 91% yield over the 2 steps. A rapid oxidation of the phenol with PhI(O2CCF3)2 in ethanol (15 min, 94% yield) provided the substrate for the IMDA reaction.
Scheme 7.

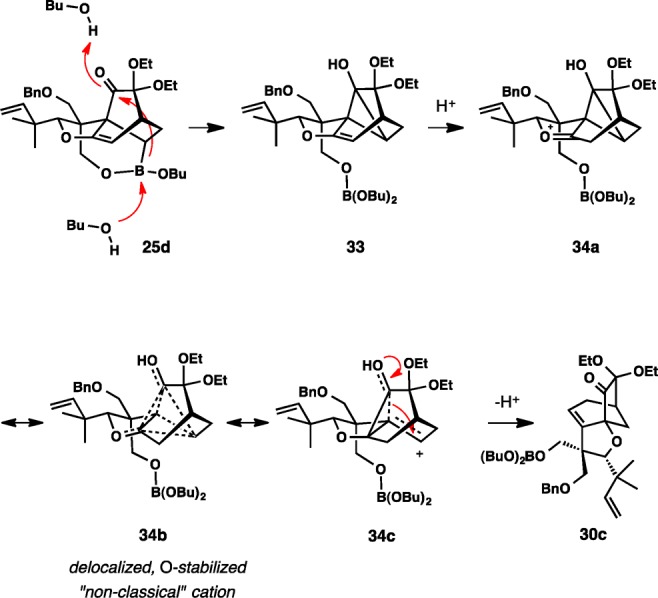
Vinylboronate 30a was generated from 29 and commercially available vinylboronic acid dibutyl ester in situ through a facile exchange reaction. Upon heating at 130 °C in o-dichlorobenzene, a new product was formed in 72% yield in a rather clean reaction. Contrary to our expectations, the product was not the direct IMDA addition product 25d but appeared to lack the boronic ester group. Based on NMR studies of the product, including NOE and homodecoupling experiments, our initial hypothesis was that we fortuitously obtained protodeboronation product 32 (Scheme 7), which is well suited for our synthesis plans. However, the true identity of the product has only been conclusively deduced after completion of the total synthesis, which revealed that the product (31) contained the rearranged bicyclo[3.2.1]octan-7-one ring system (vide infra).
Several reaction pathways can be considered for the formation of 31 from 30a. Although details are not clear at present, our current hypothesis favors the simplest path and involves the initial formation of [4+2] cycloadduct 25d. Proton transfer to enol ether 25d forms a stabilized oxycarbenium ion 30b. The 1,2-shift of the acyl group, which should be further facilitated by the properly aligned carbon—boron bond, leads directly to deborylation product 30c. After hydrolytic workup, the final product 31 with a free hydroxyl is isolated. An alternative, more complex pathway is depicted in Scheme 8. Here, the first step is an admittedly speculative and, as far as we know, unprecedented formation of cyclopropanol intermediate 33 from the initial [4+2] cycloadduct 25d. This apparent intramolecular nucleophilic attack of an alkylboronate on the carbonyl group is possibly aided by a secondary overlap between the πC—O and σ*C—B bonds. The resulting enol ether in 33 should be particularly susceptible to protonation because of the strong stabilizing effect of the adjacent cyclopropanol substituent, which should give a highly stabilized nonclassical carbocation represented by resonance forms 34a, 34b, and 34c. Its deprotonation affords 30c directly.37
Scheme 8.
Evaluation of the silicon tether began with silylation of intermediate 29 with dimethylvinylchlorosilane (Scheme 9a). The IMDA reaction of silyl ether 35 took place smoothly upon heating at 120 °C for 6 h, giving the intended product in an excellent 96% yield.
Scheme 9.

An intermolecular variant of Diels–Alder reaction with two dienophiles, nitroethylene and vinyl phenyl sulfide,38 was also examined for the construction of the bicyclo[2.2.2]octan-2-one ring system of maoecrystal V (Scheme 9b). If successful, the nitro or phenylthio group could be removed by reduction under radical conditions.39
The principal lessons that we drew from these studies are (1) the IMDA reactions with the boron and silicon tethers were the most valuable in meeting our synthetic needs, albeit giving drastically different outcomes, and (2) intermolecular Diels–Alder reactions with nitroethylene and vinyl phenyl sulfide as ethylene equivalents did not appear to be promising.
Total Synthesis of Maoecrystal ZG (2)
Even with some uncertainty whether compound 32 was the product of the reaction between cyclohexadienone 29 and dibutyl vinylboronate, the synthesis was advanced as delineated in scheme 10. In retrospect, it became clear that the actual product was in fact 31. Formation of the lactone by radical cyclization was set up by the efficient conversion of 31 to phenyl selenocarbonate 36 in 94% yield. Lactone 37 formed readily upon treatment of 36 with tri-n-butyltin hydride in the presence of AIBN at reflux in benzene, requiring little optimization (53% yield). Minor reduction to the corresponding formate was also observed (∼5%), in addition to other unidentified byproducts. At this stage the gem-ethoxy substituents were removed by reduction with samarium(II) iodide in the presence of methanol (87% yield). Cleavage of the benzyl ether was achieved under forcing conditions upon heating at 50 °C with DDQ, which afforded primary alcohol 39 in 75% yield along with recovered starting material.
Scheme 10.
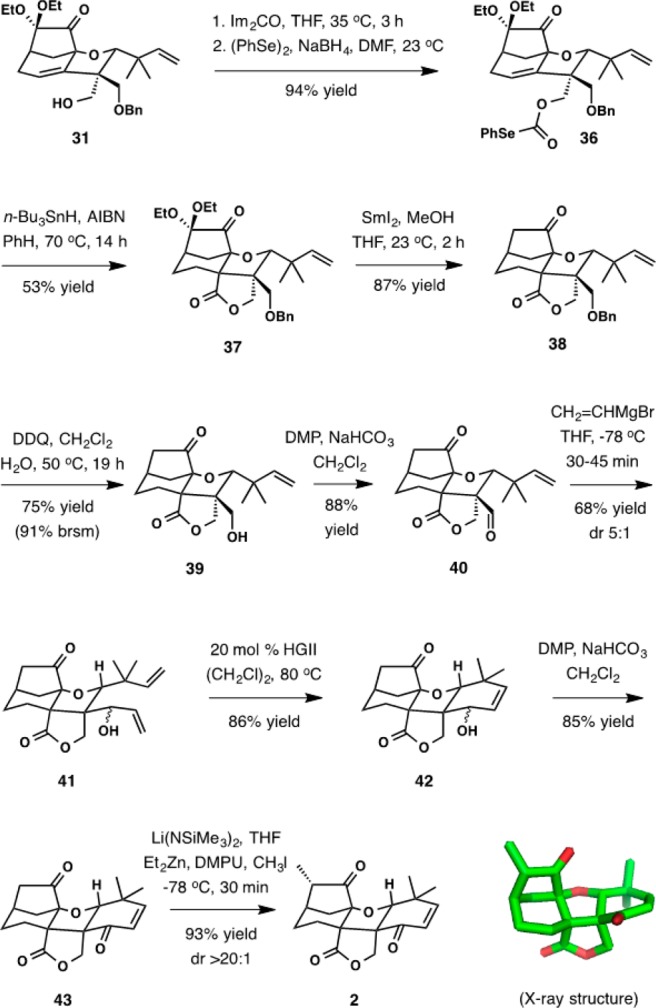
The final phase of the synthesis began with oxidation of 39 to aldehyde 40 with Dess–Martin periodinane (DMP) (88% yield) followed by chemoselective addition of vinylmagnesium bromide to aldehyde in the presence of the lactone and ketone groups (68% yield, dr 5:1). Ring-closing metathesis in the presence of 20 mol % Hoveyda-Grubbs II catalyst (HGII) accomplished the formation of the last ring of the target compound in 86% yield. The resulting product was oxidized to enone 43 with DMP in 85% yield. In the final step, methylation of zincate enolate generated from 43 with LiN(SiMe3)2 and Et2Zn occurred in excellent yield and essentially complete diastereocontrol. NMR data of the final product did not match those published for maoecrystal V, which prompted X-ray crystallographic analysis of the product. A single crystal suitable for analysis was produced by crystallization of the compound from ethyl acetate and hexane. The XRD analysis revealed the fascinating, unexpected structure 2, which we refer to as maoecrystal ZG. Its origin was traced to 31, the product of the unusual reaction between cyclohexadienone 29 and dibutyl vinylboronate, and allowed for unambiguous assignment of its structure.
Although falling short of completing the total synthesis of 1, this effort resulted in several important contributions. First, the efficient conversion of cyclohexadienone 29 to 31 in the presence of vinylboronic esters is of fundamental interest and provides an effective entry to functionalized bicyclo[3.2.1]octan-6-one ring systems. Second, maoecrystal ZG, an isomer of 1, appears to be a new natural-product-like chemical entity. Its pentacyclic 6,7-dinor-5,8;9a,20-diepoxy-ent-kaurane skeleton appears to have no precedent among natural products or other known organic compounds. Given its fairly efficient synthesis in 18 straightforward steps and ∼4% overall yield from sesamol, over 10 mg was produced after the first effort, an amount sufficient for a preliminary biological evaluation of 2.
Enatioselective Total Synthesis of (−)-Maoecrystal V (1)
Given the unforeseen creation of 2, the approach to maoecrystal V had to be reevaluated. The success was eventually achieved through the use of the silyl tether in the key IMDA reaction (cf. 25e, Scheme 9). The initial incursion entailed desilylation of 25e, which turned out to be challenging. During the course of our work we found it to be advantageous to carry out the removal of the gem-ethoxy substituents and enolate methylation first. In the event, treatment of 25e with SmI2 and subsequent exposure of the zincate enolate from 44 to CH3I provided intermediate 45 (Scheme 11). Its elaboration to selenocarbonate 47 was followed by radical cyclization upon treatment with n-Bu3SnH and azobis(cyclohexanecarbonitrile) (ABCN) at 80 °C in benzene. Surprisingly, eight-membered lactone 48 was the major product, which was produced in addition to ∼10% of formate 49. None of the expected six-membered lactone formed by cyclization of the acyl radical onto the enol ether was observed.
Scheme 11.

After exploring alternatives, a decision was made to set the synthesis back to p-methoxybenzyl ether 24 that lacks the terminal olefin competing in the radical cyclization (Scheme 12). Starting with enantioenriched 24 (84% ee), the synthesis followed the same blueprint through the IMDA reaction. The lithium enolate generated from 24 was successively treated with Et2Zn and BnOCH2Cl/DMPU, affording 50 in 20:1 dr. Reduction of the methyl ester, dioxolane cleavage, phenol oxidation, and silylation with vinyldimethylchlorosilane provided the precursor of the IMDA reaction 54. In accord with the previous experience, its clean conversion to [4+2] adduct 55 was achieved after heating in toluene at 110 °C for 24 h, which was followed by reductive removal of the two geminal ethoxy groups with samarium(II) iodide.
Scheme 12.

Desilylation of 56 and related [4+2] cycloadducts was complicated by competing formation of rearranged products b and c (Table 4). The reaction was studied in greater detail to optimize the yield of the requisite product a. The initial attempts focused on screening solvents using n-Bu4NF as the reagent (entries 1–11). These studies revealed that the ratio of the three products was significantly influenced by the choice of solvent. Low reactivity was observed in protic solvents. Among the polar aprotic solvents, DMPU emerged as the optimal medium to maximize the fraction of the requisite product a (entry 8). Tetrabutylammonium triphenyldifluorosilicate, a milder desilylation reagent, strongly favored rearranged product b (entry 13), or showed no reactivity in tert-butanol (entry 14), as did cesium fluoride (entry 12). These data indicate that a balance between the pH and reactivity is needed to achieve the desired outcome in the desilylation reaction.
Table 4.
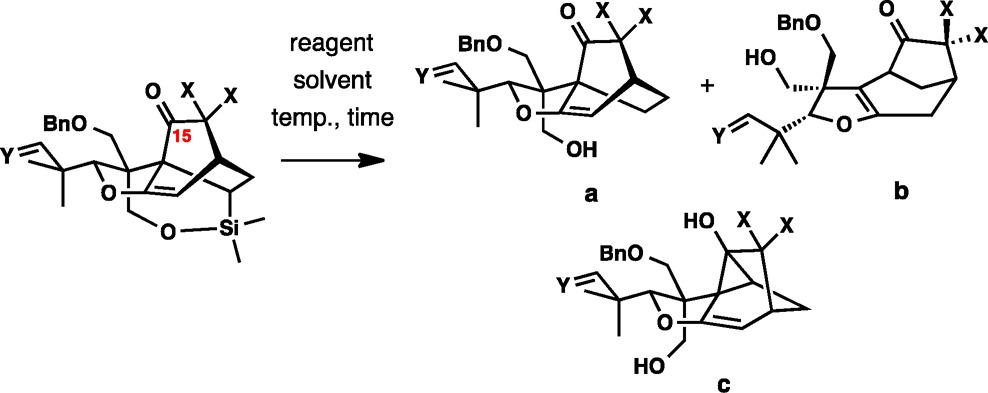
| entry | X | Y | reagent solvent | temp (°C), time (h) | a:b:ca (yield, a) |
|---|---|---|---|---|---|
| 1 | H | H, OBn | Bu4NF, DMSO | 0, 0.2 | 1.0:1.8:1 (20%) |
| 2 | H | H, OBn | Bu4NF, DMSO | 23, 0.2 | 0.9:1.5:1 |
| 3 | H | H, OBn | Bu4NF, DMF | –20, 2 | 1.5:1.3:1 |
| 4 | H | H, OBn | Bu4NF, DMF | 0, 0.5 | 2.3:1.8:1 |
| 5 | H | H, OBn | Bu4NF, DMF | 23, 0.3 | 1.9:1.6:1 |
| 6 | H | H, OBn | Bu4NF, DMF, (slow addition) | 23, 3.2 | 1.6:1.3:1 |
| 7 | H | H, OBn | Bu4NF, NMP | 0, 1.0 | 1.4:1.7:1 |
| 8 | H | H, OBn | Bu4NF, DMPU | 0, 1.0 | 3.2:1.8:1 |
| 9 | H | H, OBn | Bu4NF, DMA | 0, 0.5 | 2.4:1.7:1 |
| 10 | H | H, OBn | Bu4NF, DMSO–t-BuOH (4:1) | 75, 1 | 0.3:1.2:1 |
| 11 | H | H, OBn | Bu4NF, AcOH, THF | 0, 1.0 | no reaction |
| 12 | H | H, OBn | CsF, t-BuOH | 70, 15 | no reaction |
| 13 | H | H, OBn | Bu4NPh3SiF2, DMSO | 50, 36 | 0:20:1 |
| 14 | H | H, OBn | Bu4NPh3SiF2, t-BuOH | 50, 15 | no reaction |
| 15 | OEt | CH2 | Bu4NF, DMF | 23, 0.2 | 0.8:5.1:1 (7%) |
| 16 | OEt | CH2 | Bu4NF, THF | 23, 0.5 | no reaction |
| 17 | OEt | CH2 | Bu4NF, MeCN | 23, 0.2 | 0:3.4:1 |
| 18 | H | CH2 | Bu4NF, DMF | 23, 0.5 | 1.1:3.6:1 |
| 19 | H | H, OPMB | Bu4NF, DMA | 0, 1.0 | 2.9:2.2:1 |
| 20 | H | H, OPMB | Bu4NF, DMPU | 0, 1.0 | 3.2:1.9:1 40–50% |
Determined by NMR analysis of the crude mixture of products. Isolated yield of a is given in parentheses.
We also found that carrying out desilylation immediately after the IMDA reaction before removal of the gem-ethoxy substituents (X = OEt) was less efficient than postremoval desilyation (entries 15–18, cf. entries 15 and 18). The presence of the ethoxy groups favored the formation of rearranged byproduct to a greater extent.
Finally, desilylation of the selected total synthesis intermediate 56 (X = H, Y = H, OPMB, entry 20, Table 4) was best accomplished upon treatment with tetrabutylammonium fluoride at 0 °C for 1 h, which afforded the desired product in 50% yield. It is noteworthy that the desilylation was greatly activated by the C15 carbonyl group. The reactivity of the corresponding C15 alcohol was substantially lower, requiring heating at 80 °C for 12 h for the reaction to reach completion. No improvement in the chemical yield was observed with the reduced substrate.
There are some noteworthy similarities in the results of the desilylation reaction in Table 4 and the proposed formation of 30c depicted in Scheme 8. In particular, the structures of cyclopropanol byproduct c in Table 4 and intermediate 33 (Scheme 8) are very similar. Both appear to be unstable and readily converted to distinct bicyclo[3.2.1]octan-6-one derivatives b (Table 4) or 30c (Scheme 8), respectively, although the reasons for the divergent reactivity of the cyclopropanol intermediates are not clear.
The next immediate goal after desilylation of 56 was introduction of the six-membered lactone by radical cyclization onto enol ether, with regioselectivity opposite to its electronic preferences (Scheme 13). This is a relatively uncommon transformation with only a few examples reported in the literature.40 In our case, only one regiochemical outcome is possible due to structural constraints within the starting material. In the current substrate, selenocarbonate 58, the 4-methoxybenzyloxy substituent at C3 replaces the terminal double bond to avoid competitive cyclization onto eight-membered lactone, the reactivity mode overwhelmingly favored for selenocarbonate 47 (Scheme 11).
Scheme 13.
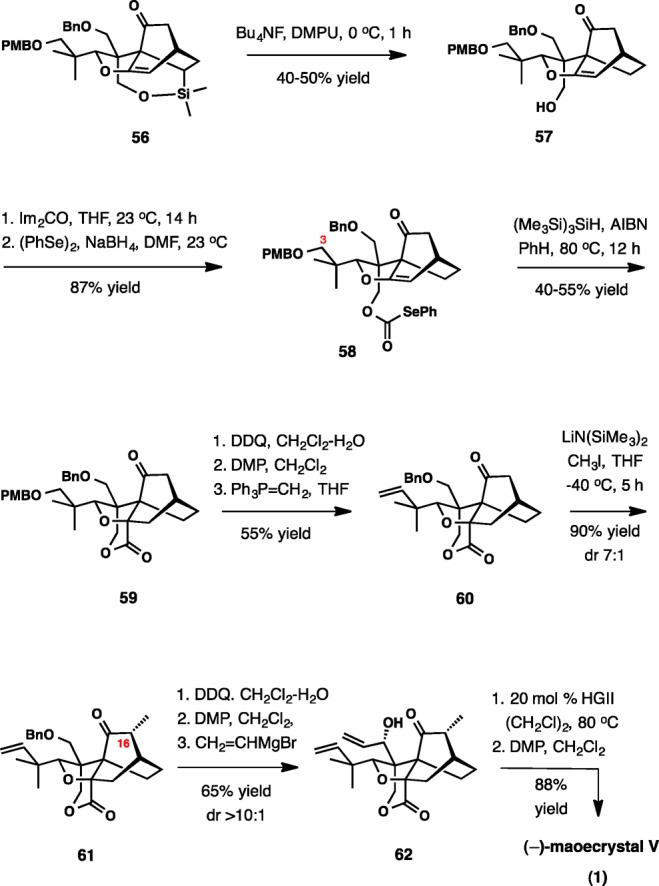
In analogy to selenocarbonate 47, the initial attempts at the radical cyclization of 58 utilized the n-Bu3SnN/AIBN reagent system under various conditions. The results were disappointing. Although the acyl radical was clearly generated, the rate of the 6-exo-cyclization onto the enol ether was very low in comparison to its reduction to the corresponding formate ester, which occurred in up to 84% yield. To reduce the rate of hydrogen abstraction by the acyl radical, and thus favor the cyclization, we investigated tris(trimethylsilyl)silane as a replacement for n-Bu3SnH.41 An immediate improvement was observed, apparently due to the longer lifetime of the acyl radical. Under optimized reaction conditions, the isolated yields of the desired lactone 59 were in the range of 40 to 55%. Notably, the main side product in this reaction results from decarboxylative fragmentation of the acyl radical rather than its reduction to the formate.21
The final phase of the total synthesis began with a three-step conversion of 59 to alkene 60 by oxidative removal of the PMB ether with DDQ, oxidation of the primary alcohol with Dess–Martin periodinane (DMP), and Wittig methylenation (55% yield over three steps). Diastereoselective installation of the methyl group at C16 has been problematic in the previously reported total syntheses of 1.19,20 With ketone 60 as the substrate, α-methylation at C16 was accomplished with 7:1 diastereoselectivity in excellent yield. If this alkylation reaction is postponed until the last step after installation of the cyclohexenone, a 1:1 mixture of diastereomers is produced. Also, when LDA is used in place of LiN(SiMe3)2 for enolate generation, substantially lower yields of 61 were observed due to unidentified side reactions.
Oxidative cleavage of the benzyl ether (61) was accomplished in the presence of DDQ in aqueous dichloromethane at 50 °C for 12 h. Oxidation with DMP followed by the addition of vinylmagnesium bromide in the presence of CeCl3 afforded allylic alcohol 62 as a single diastereomer in 65% overall yield after the three steps. Ring-closing metathesis (20 mol % Hoveyda-Grubbs II catalyst, 1,2-dichloroethane, 80 °C, 3 h) afforded a mixture of the expected allylic alcohol along with the oxidation product, (−)-maoecrystal V. A full oxidation of the mixture with DMP completed the enantioselective synthesis of 1, affording 10 mg of the natural products: [α]D25 −101.1° (c 0.3, CH3OH); lit.6 [α]D25 −92.9° (c 0.7, CH3OH).
Investigation of Antiproliferative Properties of Maoecrystal ZG (2)
Maoecrystal V (1) has shown potent and selective cytotoxic activity against the HeLa cell line, with activity for K562, A549 lung carcinoma, and BGC-823 adenocarcinoma reduced by at least 6 orders of magnitude. Because maoecrystal ZG (2) and the artificial structural isomer of 1 retain the electrophilic cyclohexenone moiety of the natural product and apparently represent a novel chemical entity, we explored its cytotoxic properties with the assistance of the NCI-60 DPT human tumor cell line screen. Somewhat remarkably, 2 showed virtually no growth inhibition at 10 μm concentration across the entire cell panel, underscoring the remarkable cytotoxicity profile of maoecrystal V.
Conclusion
The first enantioselective synthesis of (−)-maoecrystal V was enabled by a stereoselective Rh-catalyzed C–H functionalization as the enantiodetermining step. This reaction provided the key 2,3-dihydrobenzofuran precursor in 84% ee. Catalytic asymmetric and auxiliary-enabled approaches have been investigated. Although both proved promising, the auxiliary-based approach was ultimately used because it performed best in the presence of functional groups necessary for the total synthesis application. The synthesis required 26 linear steps from sesamol (three steps longer than our synthesis of racemic 1), affording (−)-maoecrystal V in ∼0.21% overall yield.
An unexpected course of the IMDA reaction with a tethered vinylboronate led to the initial synthesis of an isomeric product, which we refer to as maoecrystal ZG (2). This compound has a rearranged pentacyclic 6,7-dinor-5,8;9a,20-diepoxy-ent-kaurane skeleton that apparently has no precedent in the realm of natural products, or in fact of any known organic compounds. Evaluation of its cytotoxic activity in the NCI 60 cell panel revealed no growth inhibition across the entire panel, underscoring the unique and selective activity of maoecrystal V itself, which displays selective nanomolar activity against the HeLa cell line.
The expected outcome of the central IMDA reaction using a silicon tether provided the bicyclo[2.2.2]octan-2-one ring system, thus enabling the successful completion of the enantioselective total synthesis of (−)-maoecrystal V, which confirmed the assignment of its absolute stereochemistry.
Acknowledgments
In memory of Chase Mabe. We thank the Developmental Therapeutics Program at NCI for performing the in vitro antitumor screen of 2. A.Z. thanks the NSF (CHE-0836757) and NIGMS (R01 GM077379) for supporting this research. H.M.L.D., D.M.G, and H.W. were supported by NSF under the CCI for Selective C–H Functionalization, CHE-1205646. NMR instrumentation at UC Santa Barbara was supported by the NIH #1S10OD012077-01A1. We thank Dr. Hongjun Zhou (UCSB) for assistance with NMR spectroscopy, and Dr. Guang Wu (UCSB) for assistance with X-ray crystallography.
Supporting Information Available
Detailed experimental procedures, characterization data, copies of 1H and 13C NMR spectra, HPLC traces, and a crystallographic CIF file. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China.
Author Contributions
⊥ A.M. and P.L. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yagi S. J. Kyoto Med. Assoc. 1910, 7, 30. [Google Scholar]
- a Litake Y.; Natsume M. Tetrahedron Lett. 1964, 1259. [Google Scholar]; b Litake Y.; Natsume M. Acta Crystallogr. 1966, 20, 197. [Google Scholar]
- Sun H.-D.; Huang S.-X.; Han Q.-B. Nat. Prod. Rep. 2006, 23, 673. [DOI] [PubMed] [Google Scholar]
- Shen X.; Isogai A.; Furihata K.; Lin Z.; Sun H. D.; Suzuki A. Phytochemistry 1994, 35, 820–821. [Google Scholar]
- Wang J.; Zhao Q. S.; Lin Z. W.; Sun H. D. Acta Bot. Yunnanica 1997, 19, 191. [Google Scholar]
- Li S. H.; Wang J.; Niu X. M.; Shen Y. H.; Zhang H. J.; Sun H. D.; Li M. L.; Tian Q. E.; Lu Y.; Peng C.; Zheng Q. T. Org. Lett. 2004, 6, 4327. [DOI] [PubMed] [Google Scholar]
- Han Q.-B.; Cheung S.; Tai J.; Qiao C.-F.; Song J.-Z.; Tso T.-F.; Sun H.-D.; Xu H.-X. Org. Lett. 2006, 8, 4727. [DOI] [PubMed] [Google Scholar]
- a Kenny M. J.; Mander L. N.; Sethi S. P. Tetrahedron Lett. 1986, 27, 3923. [Google Scholar]; b Kenny M. J.; Mander L. N.; Sethi S. P. Tetrahedron Lett. 1986, 27, 3927. [Google Scholar]
- Adamson G.; Mander L. N. Aust. J. Chem. 2003, 56, 805. [Google Scholar]
- a Mori K.; Nakahara M.; Matsui M. Tetrahedron Lett. 1970, 11, 2411. [Google Scholar]; b Mori K.; Nakahara Y.; Matsui M. Tetrahedron 1972, 28, 3217. [Google Scholar]
- Ziegler F. E.; Kloek J. A. Tetrahedron 1977, 33, 373. [Google Scholar]
- Snider B. B.; Kiselgof J. Y.; Foxman B. M. J. Org. Chem. 1998, 63, 7945. [Google Scholar]
- Cherney E. C.; Green J. C.; Baran P. S. Angew. Chem., Int. Ed. 2013, 52, 9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cha J. Y.; Yeoman J. T. S.; Reisman S. E. J. Am. Chem. Soc. 2011, 133, 14964. [DOI] [PubMed] [Google Scholar]; b Yeoman J. T. S.; Mak V. W.; Reisman S. E. J. Am. Chem. Soc. 2013, 135, 11764. [DOI] [PubMed] [Google Scholar]; c Yeoman J. T. S.; Cha J. Y.; Mak V. W.; Reisman S. E. Tetrahedron 2014, 70, 4070. [Google Scholar]
- Pan Z.; Zheng C.; Wang H.; Chen Y.; Li Y.; Cheng B.; Zhai H. Org. Lett. 2014, 16, 216. [DOI] [PubMed] [Google Scholar]
- Moritz B. J.; Mack D. J.; Tong L.; Thomson R. J. Angew. Chem., Int. Ed. 2014, 53, 2988. [DOI] [PubMed] [Google Scholar]
- Synthetic studies toward maoecrystal V:; a Gong J.; Lin G.; Li C. C.; Yang Z. Org. Lett. 2009, 11, 4770. [DOI] [PubMed] [Google Scholar]; b Krawczuk P. J.; Schone N.; Baran P. S. Org. Lett. 2009, 11, 4774. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Peng F.; Yu M.; Danishefsky S. J. Tetrahedron Lett. 2009, 50, 6586. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nicolaou K. C.; Dong L.; Deng L. J.; Talbot A. C.; Chen D. Y. K. Chem. Commun. 2010, 46, 70. [DOI] [PubMed] [Google Scholar]; e Lazarski K. E.; Hu D. X.; Sterm C. L.; Thomson R. J. Org. Lett. 2010, 12, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Singh V.; Bhalerao P.; Mobin S. M. Tetrahedron Lett. 2010, 51, 3337. [Google Scholar]; g Baitinger I.; Mayer P.; Trauner D. Org. Lett. 2010, 12, 5656. [DOI] [PubMed] [Google Scholar]; h Dong L.; Deng L.; Lim Y. H.; Leugn G. Y. C.; Chen D. Y. K. Chem.—Eur. J. 2011, 17, 5778. [DOI] [PubMed] [Google Scholar]; i Peng F.; Danishefsky S. J. Tetrahedron Lett. 2011, 52, 2104. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Lazarski K. E.; Akpinar B.; Thomson R. J. Tetrahedron Lett. 2013, 54, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Carberry P.; Viernes D. R.; Choi L. B.; Fegley M. W.; Chisholm J. D. Tetrahedron Lett. 2013, 54, 1734. [Google Scholar]; l Zhang Y.; Gong J.; Yang Z. Chem. Rec. 2014, 14, 606. [DOI] [PubMed] [Google Scholar]
- For a recent review on the total synthesis of Isodon diterpenes, see:Lazarski K. E.; Moritz B. J.; Thomson R. J. Angew. Chem., Int. Ed. 2014, 53, 10588. [DOI] [PubMed] [Google Scholar]
- Gong J.; Lin G.; Sun W.; Li C.-C.; Yang Z. J. Am. Chem. Soc. 2010, 132, 16745. [DOI] [PubMed] [Google Scholar]
- Peng F.; Danishefsky S. J. J. Am. Chem. Soc. 2012, 134, 18860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Gu Z.; Zakarian A. J. Am. Chem. Soc. 2013, 135, 14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson and co-workers have concurrently completed the enantioselective total synthesis of (−)-maoecrystal V that appears in the accompanying publication:; Zheng C.; Dubovyk I.; Lazarski K. E.; Thomson R. J. J. Am. Chem. Soc. 2014, 10.1021/ja5109694. [DOI] [PubMed] [Google Scholar]
- Recent reviews and applications:; a Gutekunst W. R.; Baran P. S. J. Org. Chem. 2014, 79, 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yamaguchi J.; Yamaguchi A. D.; Itami K. Angew. Chem., Int. Ed. 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]; c Ueda K.; Amaike K.; Maceiczyk R. M.; Itami K.; Yamaguchi K. J. Am. Chem. Soc. 2014, 136, 13226. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Grazini M. V. A.; Aouad E. Org. Lett. 2001, 3, 1475. [DOI] [PubMed] [Google Scholar]
- Saito H.; Oishi H.; Kitagaki S.; Nakamura S.; Anada M.; Hashimoto S. Org. Lett. 2002, 4, 3887. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Manning J. R. Nature 2008, 451, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- Wang H.; Li G.; Engle K. M.; Yu J.-Q.; Davies H. M. L. J. Am. Chem. Soc. 2013, 135, 6774. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Hedley S. J.; Bohall B. R. J. Org. Chem. 2005, 70, 10737–10742. [DOI] [PubMed] [Google Scholar]
- Wang X.; Lu Y.; Dai H.-X.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 12203–12205. [DOI] [PubMed] [Google Scholar]
- a Kurosawa W.; Kan T.; Fukuyama T. J. Am. Chem. Soc. 2003, 125, 8112. [DOI] [PubMed] [Google Scholar]; b Wang D.-H.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The absolute stereochemistry of 24 was determined as described in the Supporting Information.
- For reviews on the intramolecular Diels–Alder reaction, see:; a Roush W. R.Intramolecular Diels–Alder Reactions. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Eds.; Pergamon Press: New York, 1991; Vol. 5, pp 513–550. [Google Scholar]; b Takao K.-i.; Munakata R.; Tadano K.-i. Chem. Rev. 2005, 105, 4779. [DOI] [PubMed] [Google Scholar]; c Tadano K.-i. Eur. J. Org. Chem. 2009, 4381. [Google Scholar]; d Juhl M.; Tanner D. Chem. Soc. Rev. 2009, 38, 2983. [DOI] [PubMed] [Google Scholar]; Selected recent applications in synthesis:; e Xiao Q.; Young K.; Zakarian A. Org. Lett. 2013, 15, 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tortosa M.; Yakelis N. A.; Roush W. R. J. Am. Chem. Soc. 2008, 130, 2722. [DOI] [PubMed] [Google Scholar]; g Shvartsbart A.; Smith A. B. III. J. Am. Chem. Soc. 2014, 136, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z.; Zakarian A. Org. Lett. 2011, 13, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey R. A.; Thadani A. N.; Lough A. J. J. Am. Chem. Soc. 1999, 121, 450. [Google Scholar]
- a Morita Y.; Suzuki M.; Noyori R. J. Org. Chem. 1989, 54, 1785. [Google Scholar]; Recent applications:; b Ilardi E. A.; Isaacman M. J.; Qin Y. C.; Shelly S. A.; Zakarian A. Tetrahedron 2009, 65, 3261. [Google Scholar]; c Iimira S.; Overman L. E.; Paulini R.; Zakarian A. J. Am. Chem. Soc. 2006, 128, 13095. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fisher E. L.; Wilkerson-Hill S. M.; Sarpong R. J. Am. Chem. Soc. 2012, 134, 9946. [DOI] [PubMed] [Google Scholar]
- Selected examples of discussion on nonclassical carbocation, see:; a Scholz F.; Himmel D.; Heinemann F. W.; Schleyer P. v. R.; Meyer K.; Krossing I. Science 2013, 341, 62. [DOI] [PubMed] [Google Scholar]; b Winstein S.; Trifan D. S. J. Am. Chem. Soc. 1949, 71, 2953. [Google Scholar]
- Nitroethylene as the dienophile, see:; a Corey E. J.; Myers A. G. J. Am. Chem. Soc. 1985, 107, 5574. [Google Scholar]; b Sacher J. R.; Weinreb S. M. Org. Lett. 2012, 14, 2172. [DOI] [PubMed] [Google Scholar]; Phenyl vinyl sulfide as the dienophile, see:; c Gao S.-Y.; Chittimalla S. K.; Chuang G. J.; Liao C. C. J. Org. Chem. 2009, 74, 1632. [DOI] [PubMed] [Google Scholar]; d Kalogiros C.; Hadjiarapoglou L. P. Tetrahedron 2011, 67, 3216. [Google Scholar]
- For removal of nitro group under radical conditions, see:; a Ono N.; Miyake H.; Tamura R.; Kaji A. Tetrahedron Lett. 1981, 22, 1705. [Google Scholar]; b Jakubec P.; Cockfield D. M.; Dixon D. J. J. Am. Chem. Soc. 2009, 131, 16632. [DOI] [PubMed] [Google Scholar]; For removal of phenylthio group under radical radical, see:; c Cohen T.; Ouellette D.; Pushpananda K.; Senaratne A.; Yu L.-C. Tetrahedron Lett. 1981, 22, 3377. [Google Scholar]; d Geum S.; Lee H.-Y. Org. Lett. 2014, 16, 2466. [DOI] [PubMed] [Google Scholar]
- a Davies K. A.; Wulff J. E. Org. Lett. 2011, 13, 5552. [DOI] [PubMed] [Google Scholar]; Also see:; b Quirante J.; Escolano C.; Diaba F.; Bonjoch J. J. Chem. Soc., Perkin Trans. 1 1999, 1157. [Google Scholar]
- a Kanabus-Kaminska J. M.; Hawari J. A.; Griller D.; Chatgilialoglu C. J. Am. Chem. Soc. 1987, 109, 5267. [Google Scholar]; b Chatgilialoglu C.; Griller D.; Lesage M. J. Org. Chem. 1988, 53, 3642. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.