

Abstract
Klinefelter syndrome and Y-chromosomal microdeletion analyses were once the only two genetic tests offered to infertile men. Analyses of aurora kinase C (AURKC) and DPY19L2 are now recommended for patients presenting macrozoospermia and globozoospermia, respectively, two rare forms of teratozoospermia particularly frequent among North African men. We carried out genetic analyses on Algerian patients, to evaluate the prevalence of these syndromes in this population and to compare it with the expected frequency of Klinefelter syndrome and Y-microdeletions. We carried out a retrospective study on 599 consecutive patients consulting for couple infertility at the assisted reproduction unit of the Ibn Rochd Clinique, Constantine, Algeria. Abnormal sperm parameters were observed in 404 men. Fourteen and seven men had typical macrozoospermia and globozoospermia profiles, respectively. Molecular diagnosis was carried out for these patients, for the AURKC and DPY19L2 genes. Eleven men with macrozoospermia had a homozygous AURKC mutation (79%), corresponding to 2.7% of all patients with abnormal spermograms. All the men with globozoospermia studied (n = 5), corresponding to 1.2% of all infertile men, presented a homozygous DPY19L2 deletion. By comparison, we would expect 1.6% of the patients in this cohort to have Klinefelter syndrome and 0.23% to have Y-microdeletion. Our findings thus indicate that AURKC mutations are more frequent than Klinefelter syndrome and constitute the leading genetic cause of infertility in North African men. Furthermore, we estimate that AURKC and DPY19L2 molecular defects are 10 and 5 times more frequent, respectively, than Y-microdeletions.
Keywords: aurora kinase C, DPY19L2, globozoospermia, intracytoplasmic sperm injection, infertility, macrozoospermia
INTRODUCTION
At least 70 million couples worldwide have infertility problems.1 Male infertility has diverse, often multifactorial causes, resulting in quantitative and/or qualitative sperm defects in 61% of cases.2 A large proportion of male infertility cases are caused by genetic defects, but few genes have been formally shown to be associated with spermatogenic defects since the discovery of microdeletions in the 1970s.3 Aurora kinase C (AURKC)4 and DPY19L25 are the two principal genes implicated in sperm defects. They have been found to be mutated in large numbers of unrelated individuals presenting macrozoospermia and globozoospermia, respectively. The abnormal spermatozoa of men with these conditions display typical morphological abnormalities (Figure 1).
Figure 1.
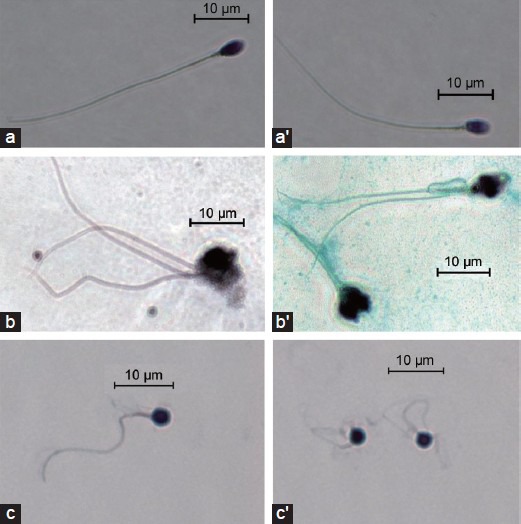
Photographs of representative spermatozoa from (a, a’) a normal donor, (b, b’) a patient with macrozoospermia (with a homozygous aurora kinase C c.144delC mutation) and (c, c’) a patient with globozoospermia (with a homozygous DPY19L2 deletion).
Patients with macrozoospermia produce ejaculates containing mostly large-headed multiflagellar polyploid spermatozoa. The c.144delC deletion has been identified in most North African patients with this condition.6 Further studies led to the identification of rare familial mutations7 and of a nonsense mutation, p.Y248*, in both European and North African men.8 An overall carriage frequency for these defects of 1/50 has been established for individuals from the general population of the Maghreb.6 We have shown that the large-headed spermatozoa of AURKC-deficient patients are tetraploid, indicating that meiosis cannot be completed without a functional AURKC protein.6 This confirms that men with homozygous AURKC mutations are unlikely to be able to father children. A positive genetic diagnosis thus provides a formal contraindication for in vitro fertilization by intracytoplasmic sperm injection (ICSI).6
Globozoospermia is another severe sperm defect leading both to primary infertility and to poor fertilization and embryo growth following ICSI. This phenotype is characterized by the production of small, round-headed spermatozoa with no acrosome. Mutations of SPATA169 and PICK110 have been identified in individuals with a family history of the globozoospermia, suggesting a role of these genes in the phenotype, although they cannot be the main cause of it. In a previous study, a recurrent homozygous deletion of DPY19L2 was found in a large majority of globozoospermia patients, indicating that DPY19L2 deletions were the main cause of the globozoospermia.5 These recurrent deletions were subsequently shown to have occurred by nonallelic homologous recombination (NAHR), due to the presence of repeated sequences on either side of the gene.11 Point mutations were also identified in affected men,12 further confirming the pathogenicity of DPY19L2 mutation. We have shown that DPY19L2 is a transmembrane protein required for the anchoring of the acrosome to the nucleus and that, in its absence, the acrosome is lost during the last stages of spermiogenesis.13 Unlike macrozoospermatozoa, globozoospermatozoa is not associated with any gross chromosomal abnormality, and in vitro fertilization by ICSI was expected to be an effective treatment strategy for these patients. However, fertilization, embryo cleavage and pregnancy rates following ICSI have consistently been disappointingly low for these patients.9
Macrozoospermia and globozoospermia are rare, recurrent forms of teratozoospermia more frequently found in subjects from North Africa than in men from elsewhere. The frequencies of these phenotypes and their etiologies have yet to be determined for the Algerian population. We report here the first genetic analysis of Algerian men with severe forms of teratozoospermia, focusing in particular on men with macro and globozoospermia.
MATERIALS AND METHODS
Semen analysis
Sperm was collected by masturbation after 3 days of abstinence. Spermiograms were performed at the Ibn Rochd Clinique, Constantine, Algeria in accordance with World Health Organization guidelines,14 during the routine biological examination of the patients. David's classification was applied to spermocytograms.15
The variables recorded and analyzed in this study were ejaculate volume (ml), sperm concentration (×106 ml−1), total sperm count (×106), percentage of progressive sperm cells (a + b), sperm viability (%) and percentage of morphologically normal spermatozoa. We determined the percentage of morphologically abnormal spermatozoa (irregular, large-headed, multiple heads, round-headed) by analyzing 100 spermatozoa for each patient.
Sperm abnormalities were defined as followed: (1) asthenospermia (A): samples with a motility (a + b) <50%; (2) oligospermia (O): samples with a concentration <20 million spermatozoa ml−1; (3) astheno-teratospermia (AT): samples with motility <50% and <30% spermatozoa with a normal morphology; (4) oligo-astheno-teratospermia (OAT): samples with a concentration <20 million spermatozoa ml−1, a motility <50% and with <30% of spermatozoa displaying a normal morphology; (5) azoospermia (AZ): an absence of spermatozoa; (6) hypospermia or hyperspermia (H): sperm samples with a volume <2 ml and >6 ml, respectively.
Patient information
In total, 599 men consulting at the assisted reproduction unit of the Ibn Rochd Clinique, Constantine, Algeria, were included in this study between January 2011 and February 2012. All consulted for a period of infertility lasting at least 1 year. Y-microdeletion analysis was not carried out for all patients, so it was not possible to establish the frequency of this molecular defect for this cohort.
We identified 21 patients presenting with the most severe morphological abnormalities: fourteen patients with macrozoospermia and seven with globozoospermia. These patients had a normal somatic karyotype.
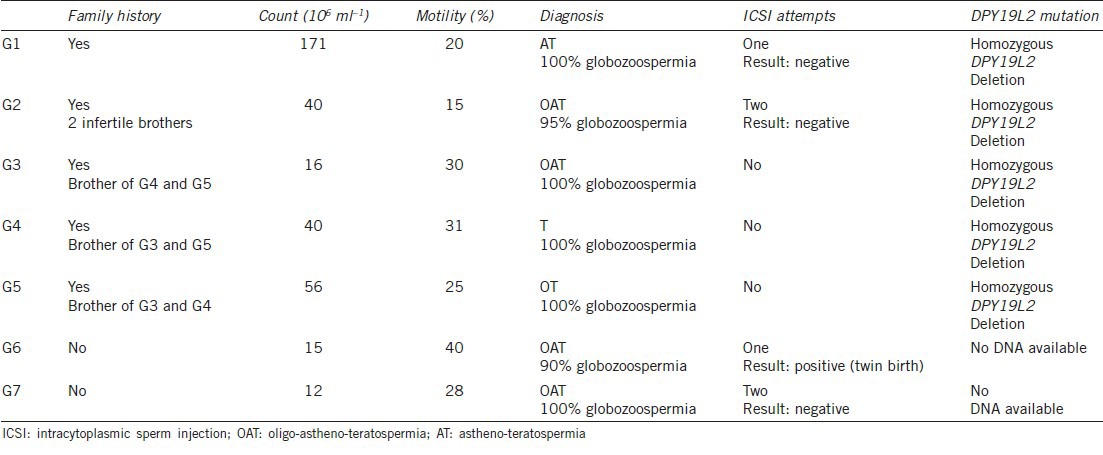
We analyzed the AURKC and DPY19L2 genes for all patients with macrozoospermia and for five of the seven men with globozoospermia. The main clinical characteristics of the patients are shown in Tables 1 and 2. The testosterone, luteinizing hormone and follicle-stimulating hormone concentrations of the patients tested were in the normal range.
Table 1.
Sperm characteristics and genotype of macrozoospermic men
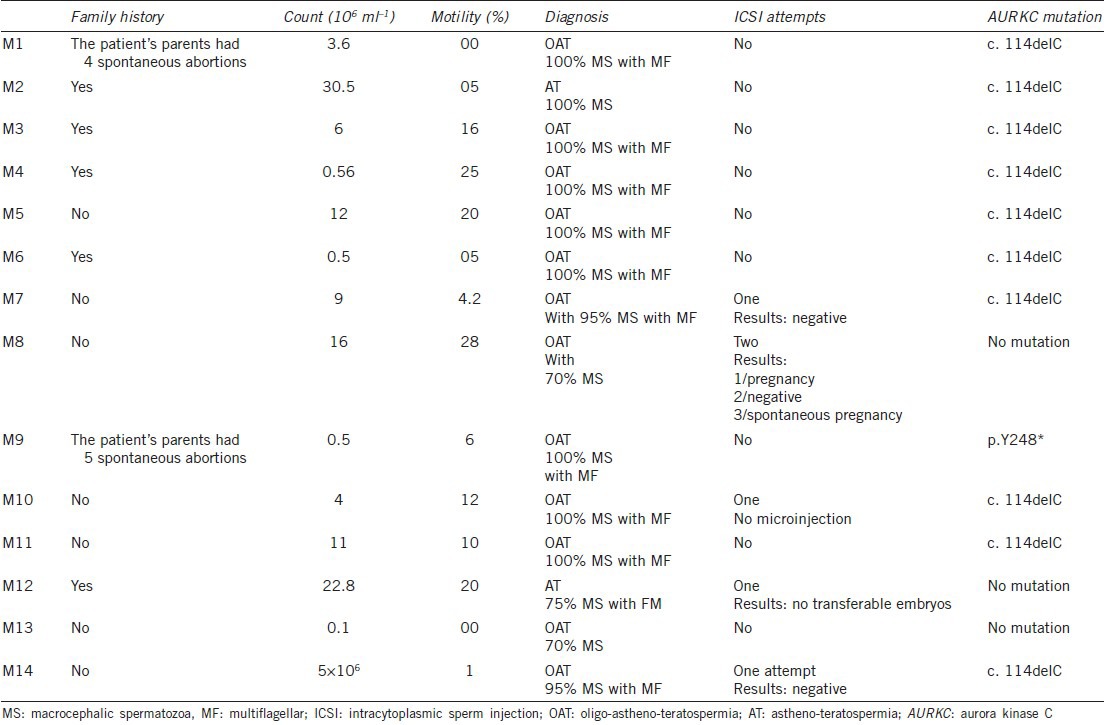
Table 2.
Sperm characteristics and genotype of globozoospermic men
The study was approved by the Local Ethics Committee. Each patient was informed of the purpose of the study and signed a consent form. All national laws and regulations were respected.
DNA extraction
Genomic DNA was extracted from peripheral blood leukocytes with a guanidium chloride extraction procedure, or from saliva with the Oragene DNA self-collection kit (DNA Genotech, Canada), according to the manufacturer's instructions.
Polymerase chain reaction and sequencing analyses
The seven AURKC exons and their intron boundaries were amplified as previously described.4 All analyses were carried out with BigDye Terminator version 3.1 sequencing kits and an ABI PRISM 3130 genetic analyzer (Applied Biosystems, Foster City, CA, USA). The primers and protocols used were as described elsewhere.4 Example of the results obtained is shown in Figure 2.
Figure 2.
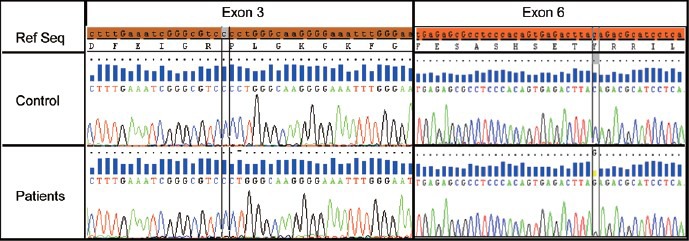
Genetic diagnosis of aurora kinase C (AURKC) exons 3 and 6. Electropherogram showing part of AURKC exons 3 and 6 from a control subject (lane 2) and a patient with a homozygous c.144delC deletion of exon 3 (lane 3, column 2) and another patient with a homozygous p.Y248* mutation of exon 6 (lane 3, column 3). The reference nucleotide sequence and its translation are indicated at the top of the figure.
Multiplex ligation-dependent probe amplification analysis
For this study, three synthetic multiplex ligation-dependent probe amplification (MLPA) probes specific to exons 1, 17 and 22 of DPY19L2 were used, as previously described.12 Given the high level of sequence similarity between DPY19L2 and other DPY19L paralogs and pseudogenes, MLPA probes were designed to match specific DPY19L2 single-nucleotide mismatches at the ligation site.16 We also included three MLPA control probes specific to the OCRL1 gene, as control probes for the determination of copy number. Information about the sequences and ligation sites of these control probes can be obtained from the study by Coutton et al.17 A comparison of the heights of the signals obtained for the control and target probes provides information about the number of copies of the target sequence present in the sampled DNA (Figure 3).
Figure 3.
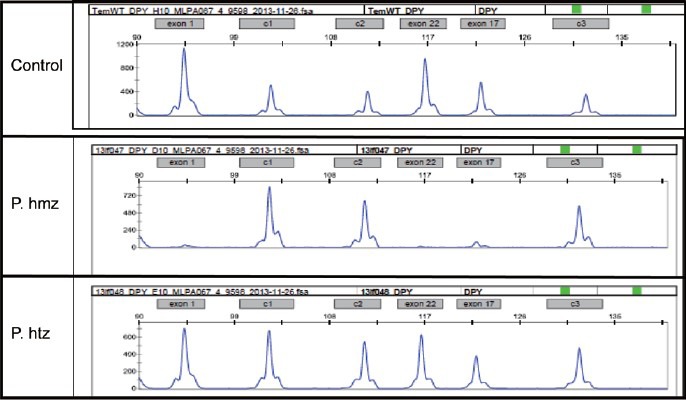
Multiplex ligation-dependent probe amplification (MLPA) analysis of DPY19L2 exons 1, 17 and 22. The probe set contained three control probes for normalization purposes (c1, c2, c3) and three specific DPY19L2 probes (X1, X17 and X22) corresponding to exons 1, 17 and 22 of DPY19L2, respectively. Lane 1: MLPA profile of a normal DNA sample (control). Lane 2: MLPA profile of a patient with a homozygous deletion, showing a total absence of DPY19L2 exon signals. Lane 3: MLPA profile of a patient with a heterozygous deletion, showing a halving of the intensity of the DPY19L2 signals.
RESULTS
Spermogram and spermocytogram
Abnormal spermograms were obtained from 404 (67%) of the 599 men undergoing examinations for infertility. The differential diagnoses were as follows: (1) OAT (n = 104); (2) asthenospermia (n = 83); (3) AT (n = 79); (4) oligozoospermia (n = 76); (5) AZ (n = 46); (6) hypospermia or hyperspermia (n = 16). In total, 183 men presented teratospermia (AT + OAT), corresponding to 45% of the patients with abnormal spermograms or 31% of the men screened for couple infertility.
Fourteen patients presented macrozoospermia and seven presented globozoospermia.
Patients with macrozoospermia
Sperm counts ranged from 0.1 to 30.5 million ml−1, with a mean value of 5.8 million ml−1. All but four of the patients had a low sperm count (<10 million spermatozoa ml−1; Table 1). Mean mobility was low (11.3%), with only four patients displaying normal sperm mobility. Ten patients presented OAT and two had AT.
Aurora kinase C exon 3 was sequenced for all 14 patients. A homozygous c.144delC mutation was found in this exon in 10 patients (Figure 2). The other exons were sequenced for the other four patients. One carried a p.Y248* mutation in exon 6 and no mutation was identified in the other three patients (Figure 2). AURKC mutations, therefore, had a frequency of 78.6% (11/14) in our cohort of patients with macrospermia. The c.144delC variant accounted for 91% of the mutated alleles, with p.Y248* accounting for the rest (9%).
Eight patients with mutations had only large-headed multiflagellar spermatozoa and three displayed macrozoospermia with more than 95% atypical forms. The three men without mutations had better sperm parameters, with only 70%–75% of spermatozoa abnormal.
Three patients with mutations had a family history of male infertility. Sequence analyses for the parents of four patients confirmed that all were heterozygous for the mutations investigated (c.144delC and p.Y248*). All couples presented with primary infertility and two couples reported having suffered several spontaneous abortions, indicating that fertilization and implantation were sometimes possible but that the fetuses were not viable.
Intracytoplasmic sperm injection was attempted before molecular diagnosis for five patients. Three of these patients were homozygous for the c.144delC mutation. In one case (patient M10), no suitable spermatozoa could be identified because 100% of the spermatozoa were abnormal (macrospermatozoa). Patients M7 and M14 displayed 95% macrospermatozoa and a few smaller spermatozoa were identified for injection. In both cases, fertilization rates were low (<50%) and no transferable embryos were obtained. No AURKC mutation could be identified for the other two patients (M12 and M8). No embryos could be transferred for M12, whereas two ICSI procedures were carried out for M8, one of which led to the birth of a healthy child. This couple also subsequently achieved a spontaneous pregnancy.
Patients with globozoospermia
Sperm counts ranged from 12 to 171 million ml−1, with a mean value of 50.8 million ml−1. Mean mobility levels were low (27%). Five patients had OAT and two had AT (Table 2).
All the patients analyzed (n = 5) presented a homozygous deletion of DPY19L2, but three of the subjects analyzed were brothers (Figure 3). Two patients did not give consent for genetic diagnosis. Our findings indicate that all three unrelated subjects presenting with globozoospermia had a DPY19L2 deletion.
DISCUSSION
Frequency of aurora kinase C and DPY19L2 mutations in our cohort and comparison with the estimated frequencies of Klinefelter syndrome and chromosome Y-microdeletions
We screened 599 men presenting at our unit for couple infertility. A male cause for infertility was identified on the basis of an abnormal spermogram for 404 patients. Routine genetic tests (karyotyping and Y-microdeletion analysis) are generally performed only for the most severe cases of teratozoospemia (macrozoospermia and globozoospermia) and for azoospermic men. In our cohort of patients, 46 (11.4%) of the infertile men had AZ. Unfortunatel, karyotyping and Y-microdeletion diagnosis were not carried out systematically. Tüttelmann et al.18 screened a total of 1583 consecutive patients with AZ and found that 14% presented with Klinefelter syndrome and 2% with Y-chromosome microdeletions. These genetic abnormalities occur de novo and we would not, therefore, expect these frequencies to be population-specific. These findings can therefore be extrapolated to our cohort, leading to the prediction of 1.6% of patients with Klinefelter syndrome and 0.23% with Y-microdeletions in our cohort of 404 infertile men. We identified 11 men with a homozygous AURKC mutation (2.7%) and five with a homozygous DPY19L2 deletion (1.2%) in this population. We can therefore conclude that, in Algerian men, and probably more generally in men of North African origin, the most frequent genetic cause of infertility is macrospermia due to AURKC mutation. Furthermore, in infertile North African men, we would expect AURKC and DPY19L2 mutations to be 10 and 5 times more frequent, respectively, than Y-microdeletions.
Frequency of aurora kinase C and DPY19L2 mutations and potential reasons for their high prevalence
The high frequency of AURKC mutations observed here is consistent with the high prevalence of heterozygous c.144delC mutations (1 in 50) in North African men.6 Such a high frequency is surprising for a mutation impairing reproduction when present in the homozygous state. The accumulation of recurrent mutations in the population of North Africa might be accounted for if there was a selective advantage for heterozygous carriers of AURKC mutations. AURKC is involved in the spindle assembly checkpoint (SAC): during meiosis, germ cells with misaligned chromosomes or abnormal microtubule tension that would result in incorrect segregation are blocked in prometaphase by the SAC. Men with heterozygous AURKC mutations may have a faster SAC, allowing meiosis to turn over more rapidly and to result in higher levels of sperm production. This would give these men a slight reproductive advantage, accounting for the high prevalence of the mutated alleles. However, the resulting gametes would be expected to display high rates of aneuploidy, probably resulting in high spontaneous abortion rates. Two of the patients in our cohort reported that their parents had suffered several spontaneous abortions. As a father and the mother are obligate carriers, they would have a high risk of aneuploidy gamete production, as described above.
DPY19L2 deletions were also frequent in our cohort. We have previously shown that these deletions occur by NAHR. New mutations are, therefore, relatively frequent, and we estimated the frequency of de novo deletion at about 1.5 × 10−5 per meiosis, regardless of the origin of the individuals.11 The higher incidence of the globozoospermia observed in North African men is, therefore, probably not due to a higher allelic frequency (as observed for AURKC) but to the higher rate of consanguineous marriages specific to this population, strongly favoring the emergence of recessive traits.
Intracytoplasmic sperm injection treatment for macrozoospermic and globozoospermic men
Careful selection by motile sperm organelle morphology examination was used in a previous study to select the most normal-looking spermatozoa in patients with AURKC c.144delC deletions.19 Only six normal-looking spermatozoa were selected and fluorescence in situ hybridization (FISH) analysis was carried out on these spermatozoa. All six were aneuploid, confirming that ICSI should not be attempted for patients with AURKC mutations, even after very thorough morphological selection.19 In our study, ICSI was attempted for three patients with homozygous deletions. Injection was possible in two cases (M7 and M15), but no transferable embryos were obtained. The three patients with no AURKC mutation presented milder phenotypes, with <75% macrospermatozoa, whereas all the men with mutations had >95% macrospermatozoa). Two patients without AURKC mutations (M8 and M12) underwent ICSI. Injection was possible in each case, and the partner of one of these patients became pregnant. These results confirm the relevance of molecular diagnosis and the feasibility of ICSI for patients without AURKC mutations.
Intracytoplasmic sperm injection is also the only possible option for patients with globozoospermia. No gross chromosomal abnormalities of globozoospermatozoa are observed, but fertilization and pregnancy rates are very low.9,20,21 Recent studies have suggested that the low rates of fertilization observed for globozoospermatozoa result at least partly from the low levels or total absence of phospholipase c-zeta, a protein required for the induction of calcium oscillations triggering oocyte activation.22,23 Artificial oocyte activation (AOA) can be achieved by incubating the oocyte with a calcium ionophore, and the use of this technique seems to improve embryo cleavage and development, although a few pregnancies have been achieved without it.24, However, there has been no evaluation of the effects of this artificial activation technique on the long-term development and health of concept uses. We identified seven patients with a globozoospermia phenotype in our cohort: four unrelated men and three brothers, all with >90% round-headed spermatozoa. A genetic analysis was possible only for the three brothers and two other patients. All had a homozygous DPY19L2 deletion. Several attempts at ICSI were carried out. Fertilization was unsuccessful in all but one case. The exception, patient G6, was not genotyped, and a twin pregnancy resulted from the first attempt at ICSI. This patient had the lowest rate of the globozoospermia among the affected patients, suggesting that he may carry a milder DPY19L2 point mutation or have a mutation of another gene.
GENERAL CONCLUSIONS
We identified 21 men with macrozoospermia or globozoospermia, accounting for 5.2% of the pathological spermograms analyzed (including azoospermic men). Our findings confirm the recurrent nature of the two AURKC mutations and of the DPY19L2 deletion. Macrozoospermia was found to be 2–3 times more frequent than globozoospermia. Our results confirm that ICSI is not appropriate for patients with a homozygous AURKC mutation. As egg donation is not allowed in Algeria, adoption is the only remaining option for these patients. This highlights the importance of AURKC molecular diagnosis for limiting unnecessary ICSI attempts. The prognosis for men without mutations, generally with milder forms of this condition, is better. For these men, we recommend FISH sperm analysis to estimate sperm aneuploidy rate. For patients with globozoospermia, molecular diagnosis does not influence the choice of treatment, but is nevertheless important for the appropriate genetic counseling of couples with this rare form of male infertility. Some centers encourage the use of AOA regardless of the DPY19L2 status. We advocate caution, as the potential long-term effects of AOA have yet to be determined. Our findings highlight the importance of screening for AURKC and DPY19L2 mutations in North African men, as the frequency of mutations of these two genes appears to be higher than that of chromosome Y-microdeletions in this population.
AUTHOR CONTRIBUTIONS
LO, CC and DM carried out and interpreted all the molecular work. LO, AZ, LR, MH, GM, IB, DK, SB and AR supervised data acquisition and management. LO, CA, JF, AR and PFR carried out an analysis and interpreted the data. LO, LR and PFR designed the overall study, supervised all molecular laboratory work, wrote the main draft of the manuscript, had full access to all of the data and take responsibility for the integrity of the data and their accuracy. All the authors revised the manuscript and approved the final version.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
This work was funded in part by GENOPAT 2009, from the French Research Agency (ANR) as part of the “Identification and Characterization of Genes Involved in Infertility (ICG2I)” project.
REFERENCES
- 1.Boivin J, Bunting L, Collins JA, Nygren KG. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22:1506–12. doi: 10.1093/humrep/dem046. [DOI] [PubMed] [Google Scholar]
- 2.Schlosser J, Nakib I, Carré-Pigeon F, Staerman F. Male infertility: definition and pathophysiology. Ann Urol (Paris) 2007;41:127–33. doi: 10.1016/j.anuro.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Tiepolo L, Zuffardi O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet. 1976;34:119–24. doi: 10.1007/BF00278879. [DOI] [PubMed] [Google Scholar]
- 4.Dieterich K, Soto Rifo R, Faure AK, Hennebicq S, Ben Amar B, et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet. 2007;39:661–5. doi: 10.1038/ng2027. [DOI] [PubMed] [Google Scholar]
- 5.Harbuz R, Zouari R, Pierre V, Ben Khelifa M, Kharouf M, et al. A recurrent deletion of DPY19L2 causes infertility in man by blocking sperm head elongation and acrosome formation. Am J Hum Genet. 2011;88:351–61. doi: 10.1016/j.ajhg.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieterich K, Zouari R, Harbuz R, Vialard F, Martinez D, et al. The Aurora Kinase C c.144delC mutation causes meiosis I arrest in men and is frequent in the North African population. Hum Mol Genet. 2009;18:1301–9. doi: 10.1093/hmg/ddp029. [DOI] [PubMed] [Google Scholar]
- 7.Ben Khelifa M, Zouari R, Harbuz R, Halouani L, Arnoult C, et al. A new AURKC mutation causing macrozoospermia: implications for human spermatogenesis and clinical diagnosis. Mol Hum Reprod. 2011;17:762–8. doi: 10.1093/molehr/gar050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben Khelifa M, Coutton C, Blum MG, Abada F, Harbuz R, et al. Identification of a new recurrent aurora kinase C mutation in both European and African men with macrozoospermia. Hum Reprod. 2012;27:3337–46. doi: 10.1093/humrep/des296. [DOI] [PubMed] [Google Scholar]
- 9.Dam AH, Koscinski I, Kremer JA, Moutou C, Jaeger AS, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet. 2007;81:813–20. doi: 10.1086/521314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu G, Shi QW, Lu GX. A newly discovered mutation in PICK1 in a human with globozoospermia. Asian J Androl. 2010;12:556–60. doi: 10.1038/aja.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coutton C, Abada F, Karaouzene T, Sanlaville D, Satre V, et al. Fine characterisation of a recombination hotspot at the DPY19L2 locus and resolution of the paradoxical excess of duplications over deletions in the general population. PLoS Genet. 2013;9:e1003363. doi: 10.1371/journal.pgen.1003363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coutton C, Zouari R, Abada F, Ben Khelifa M, Merdassi G, et al. MLPA and sequence analysis of DPY19L2 reveals point mutations causing globozoospermia. Hum Reprod. 2012;27:2549–58. doi: 10.1093/humrep/des160. [DOI] [PubMed] [Google Scholar]
- 13.Pierre V, Martinez G, Coutton C, Delaroche J, Novella C, et al. DPY19L2 is required for anchoring the acrosome to the nucleus during spermiogenesis and its absence causes globozoospermia in mice. Development. 2012;139:2955–65. doi: 10.1242/dev.077982. [DOI] [PubMed] [Google Scholar]
- 14.World Health Organization. 4th ed. Cambridge, UK: Published on Behalf of the World Health Organization by Cambridge University Press; 1999. WHO Laboratory Manual for the Examination of Human Semen and Sperm-Cervical Mucus Interaction. [Google Scholar]
- 15.Auger J, Eustache F. Standardisation de la classification morphologique des spermatozoïdes humains selon la méthode de David modifiée. Andrologie. 2000;10:358–73. [Google Scholar]
- 16.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coutton C, Monnier N, Rendu J, Lunardi J. Development of a multiplex ligation-dependent probe amplification (MLPA) assay for quantification of the OCRL1 gene. Clin Biochem. 2010;43:609–14. doi: 10.1016/j.clinbiochem.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Tüttelmann F, Werny F, Cooper TG, Kliesch S, Simoni M, et al. Clinical experience with azoospermia: aetiology and chances for spermatozoa detection upon biopsy. Int J Androl. 2011;34:291–8. doi: 10.1111/j.1365-2605.2010.01087.x. [DOI] [PubMed] [Google Scholar]
- 19.Chelli MH, Albert M, Ray PF, Guthauser B, Izard V, et al. Can intracytoplasmic morphologically selected sperm injection be used to select normal-sized sperm heads in infertile patients with macrocephalic sperm head syndrome–. (e1–5).Fertil Steril. 2010;93:1347. doi: 10.1016/j.fertnstert.2008.10.059. [DOI] [PubMed] [Google Scholar]
- 20.Sahu B, Ozturk O, Serhal P. Successful pregnancy in globozoospermia with severe oligoasthenospermia after ICSI. J Obstet Gynaecol. 2010;30:869–70. doi: 10.3109/01443615.2010.515321. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Nagy Z, Joris H, Tournaye H, Devroey P, et al. Successful fertilization and establishment of pregnancies after intracytoplasmic sperm injection in patients with globozoospermia. Hum Reprod. 1995;10:626–9. doi: 10.1093/oxfordjournals.humrep.a136000. [DOI] [PubMed] [Google Scholar]
- 22.Yoon SY, Jellerette T, Salicioni AM, Lee HC, Yoo MS, et al. Human sperm devoid of PLC, zeta 1 fail to induce Ca (2+) release and are unable to initiate the first step of embryo development. J Clin Invest. 2008;118:3671–81. doi: 10.1172/JCI36942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heytens E, Parrington J, Coward K, Young C, Lambrecht S, et al. Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum Reprod. 2009;24:2417–28. doi: 10.1093/humrep/dep207. [DOI] [PubMed] [Google Scholar]
- 24.Kuentz P, Vanden Meerschaut F, Elinati E, Nasr-Esfahani MH, Gurgan T, et al. Assisted oocyte activation overcomes fertilization failure in globozoospermic patients regardless of the DPY19L2 status. Hum Reprod. 2013;28:1054–61. doi: 10.1093/humrep/det005. [DOI] [PubMed] [Google Scholar]