

Abstract
Genotyping of clinical Mycobacterium tuberculosis complex (MTBC) strains has become a standard tool for epidemiological tracing and for the investigation of the local and global strain population structure. Of special importance is the analysis of the expansion of multidrug (MDR) and extensively drug-resistant (XDR) strains. Classical genotyping and, more recently, whole-genome sequencing have revealed that the strains of the MTBC are more diverse than previously anticipated. Globally, several phylogenetic lineages can be distinguished whose geographical distribution is markedly variable. Strains of particular (sub)lineages, such as Beijing, seem to be more virulent and associated with enhanced resistance levels and fitness, likely fueling their spread in certain world regions. The upcoming generalization of whole-genome sequencing approaches will expectedly provide more comprehensive insights into the molecular and epidemiological mechanisms involved and lead to better diagnostic and therapeutic tools.
Genomic technologies have revealed that strains of tuberculosis-causing bacteria are more diverse than previously anticipated. Several lineages (e.g., Beijing) are more virulent and drug-resistant than others.
With an estimated 100 million new infections, 8.5 million new notified cases, and 1.5 million deaths yearly, tuberculosis (TB) is still a vivid, major public health problem globally (Dye and Williams 2010; Diel et al. 2013).
Among well-known socioepidemiological factors favoring the spread of the disease are bad living conditions (e.g., poverty, malnutrition), war, and displacement, as well as the widespread breakdown of social and TB control systems. Especially in countries of sub-Saharan Africa, the situation is aggravated by high coinfection rates with HIV (Corbett et al. 2003; Dye and Williams 2010). On the pathogen’s side, drug-resistant and multidrug-resistant (MDR; resistance at least to isoniazid [INH] and rifampicin [RMP]) Mycobacterium tuberculosis strains have emerged worldwide and pose a serious threat for TB control (Dye and Williams 2010; Gandhi et al. 2010). High rates of resistant and MDR TB have especially been reported in former Eastern USSR republics such as Azerbaijan and Uzbekistan (Dye and Williams 2010; Gandhi et al. 2010). Even more worrisome is the emergence of a barely treatable form of TB—namely, extensively drug-resistant TB (XDR TB), which is defined as MDR plus additional resistance to any fluoroquinolone and at least one of three injectable drugs (i.e., amikacin, kanamycin, or capreomycin). The detection and prevention of the spread of these most dangerous forms of TB is particularly important.
A successful control of the disease is based on several components that range from rapid and accurate diagnostics and effective therapy to the detection of recent transmission chains and outbreaks. Precise knowledge of the actual epidemiological situation is crucial for the optimization of local and global TB control measures and the early detection of changes in dynamics (e.g., the enhanced spread of MDR strains in particular regions [Niemann et al. 2010]). For example, recent studies show that, in addition to ineffective therapy, recent transmission is a key factor fostering the MDR epidemic (Pardini et al. 2009; Niemann et al. 2010).
For disclosing such transmission events, as well as longitudinal outbreaks and transmission hot spots, effective genotyping of clinical MTBC isolates at the strain level is the key. In so-called molecular epidemiological studies, genotyping is used in addition to classical epidemiological approaches, such as contact-tracing investigations, to decipher recent transmission chains and identify epidemiological links (Barnes and Cave 2003). Classical genotyping can be performed using a variety of techniques that interrogate different classes of genetic markers and generate either strain-specific banding patterns (IS6110 DNA fingerprint), bar code–like signals (spoligotyping), or numerical patterns (24 locus-MIRU-VNTR typing) (Schurch and van Soolingen 2012). However, thanks to rapid improvements in next-generation sequencing (NGS) technologies, comparison of nearly complete genome sequences of M. tuberculosis strains is progressively becoming the ultimate approach of choice, providing the maximal resolution power especially needed for resolving complex outbreak situations (Roetzer et al. 2013; Walker et al. 2013a). Epidemiological inferences are made based on the logical assumption that isolates from patients from the same clonal transmission chain have identical genotyping results/identical or nearly identical genome sequences, whereas isolates from unrelated patients are nonclonal and have different genotyping results/significantly different genome sequences. At population level, strains with identical/nearly identical molecular results are grouped in clusters that are indicative for possible cases of recent transmission (reviewed in Barnes and Cave 2003; Schurch and van Soolingen 2012).
In addition, M. tuberculosis genotyping and, more recently, whole-genome sequencing have provided fascinating new insights into the population structure and long-term evolution of the pathogen and the relationship between the strain genetic background, the human host populations, and disease characteristics (Hirsh et al. 2004; Gagneux et al. 2006a; Wirth et al. 2008; Comas et al. 2013). A number of studies showed that although the bulk of the pathogen’s population is highly clonal (Sreevatsan et al. 1997; Supply et al. 2003; Hirsh et al. 2004), several main phylogenetic lineages coexist with a marked differential geographic distribution, suggesting close coevolution with the human host (Hershberg et al. 2008; Wirth et al. 2008; Comas et al. 2013; Supply et al. 2013). Moreover, increasing evidence suggests that the underlying genetic diversity has a significant impact on the pathogenicity and immunogenicity of individual strains (reviewed in Gagneux and Small 2007). This is thus changing the classical paradigm of TB, in which host (e.g., immune status) and environmental factors (e.g., duration and intensity of exposure to infection) were contemplated as major/sole factors driving the outcome of the infection, whereas the pathogen’s diversity was considered unimportant (Gagneux and Small 2007).
In the following sections, molecular epidemiology using classical genotyping and whole-genome sequencing will be considered first, as this discipline intrinsically focuses on limited genetic variation and short-term evolutionary scales, with usual time spans ranging from months to years. The second part will consider the diversity and evolution of the pathogen at larger evolutionary and global population scales.
MOLECULAR EPIDEMIOLOGY BY CLASSICAL GENOTYPING
In molecular epidemiology, research or public health questions range from the analysis of exogenous reinfection (e.g., in hospitals) to the analysis of particular outbreaks and population-based longitudinal epidemiological studies (Small et al. 1993a,b, 1994; Aznar et al. 1995; Barnes et al. 1996; Golub et al. 2001; Diel et al. 2002; Allix-Beguec et al. 2008c). Because the timescale considered is relatively short and before the advent of NGS, molecular epidemiology has focused on the analysis of changes driven by specific, highly polymorphic genetic markers.
IS6110 DNA fingerprinting has been the standard genotyping technique for more than a decade (van Embden et al. 1993). The technique is based on the detection of the insertion element IS6110, which is present in variable copy numbers and positions in the genomes of clinical isolates (McAdam et al. 1990; Cave et al. 1991; Mazurek et al. 1991). Thus, IS6110 DNA fingerprinting generates highly variable banding patterns that provide high-resolution power for distinguishing different strains (Fig. 1). The use of a common, dedicated analysis software (e.g., Bionumerics), the creation of large genotyping databases (Heersma et al. 1998), and the international agreement on standardization (van Embden et al. 1993) led to worldwide application of this approach for molecular epidemiological analysis.
Figure 1.

IS6110 DNA fingerprints of 19 Mycobacterium tuberculosis complex strains. PvuII-digested chromosomal DNA was separated on a 1% agarose gel, transferred via vacuum blot on a nylon membrane, and hybridized with a 245-bp internal PCR fragment of IS6110. Lanes 1 and 19, reference strain Mt.14323; lanes 2–18, diverse clinical M. tuberculosis complex isolates.
However, because IS6110 DNA fingerprinting—relying on Southern blot analysis—is labor intensive and requires large amounts of DNA purified from well-grown cultures, different alternative techniques based on polymerase chain reaction (PCR) have been evaluated.
The first PCR-based method that has reached global application is spoligotyping. This technique is based on the reverse blot detection of 43 variable spacer sequences in the direct repeat (DR) region that is present in virtually all MTBC isolates (Kamerbeek et al. 1997). As the presence or absence of these spacer sequences is variable among strains, spoligotyping is generating variable bar code–like patterns that can be used for discrimination of clinical isolates (Fig. 2) (Kamerbeek et al. 1997). However, compared with IS6110 DNA fingerprinting and 24-locus MIRU-VNTR typing (see below), spoligotyping has a clearly much lower-resolution power for discriminating MTBC isolates. Thus, in molecular epidemiological studies, it should be used in combination with another, high-resolution method (Barnes and Cave 2003; Prodinger 2007).
Figure 2.

Spoligotyping patterns of 13 Mycobacterium tuberculosis complex strains. Lanes 1–11, clinical M. tuberculosis complex isolates; 12, no DNA; 13, Mycobacterium bovis BCG; 14, M. tuberculosis H37Rv.
The second PCR-based method that has reached worldwide application is MIRU-VNTR typing, which is significantly more effective. This method is based on the analysis of variable numbers of tandem repeats (VNTR) of genetic elements including mycobacterial interspersed repetitive units (MIRUs), which are mostly intergenic. After identification and use of different markers (Supply et al. 1997; Frothingham and Meeker-O’Connell 1998; Smittipat and Palittapongarnpim 2000; Supply et al. 2000; Mazars et al. 2001; Le Fleche et al. 2002; Roring et al. 2002; Skuce et al. 2002; Warren et al. 2004), a 24-locus-based format has been internationally standardized for typing of M. tuberculosis (Supply et al. 2006). The sizes of the amplicons obtained from the 24 markers are determined after electrophoretic analysis on agarose gels or on multicapillary DNA sequencers, after individual or multiplexed amplification of the target loci, respectively. The use of a DNA sequencer allows for faster, semiautomated analysis (amplicon sizing and allele calling) of the generated data (Supply et al. 2006). Importantly, regardless of the version used, the final typing result is the same and consists of a portable 24-number genotype, corresponding to the number of repeat units found in each locus. This portability allowed the development of large databases and versatile web-based tools for online analysis of MIRU-VNTR and spoligotyping data, including a universal nomenclature system, such as the MIRU-VNTRPlus (Allix-Beguec et al. 2008b; Weniger et al. 2010) and the SIT-VIT databases (Demay et al. 2012).
The rate of genetic drift of MIRU-VNTR markers in M. tuberculosis strains in actual transmission chains and within infected patients has been carefully calibrated by analyzing hundreds of isolates from genetically diverse clusters with proven epidemiological links and sets of clonal serial isolates (Savine et al. 2002; Supply et al. 2006), as well as clonal isolates from an extended longitudinal outbreak spanning 15 years (Allix-Beguec et al. 2014). As only scarce single-locus variations were observed among such clonal isolates, a cluster suggestive of clonal transmission is defined based on strict conservation of the 24 alleles among clinical isolates. Such cluster analysis based on 24 loci, optionally combined with spoligotyping, was shown to provide a close to similar predictive value as IS6110 fingerprinting for tracing TB transmission in several Western Europe countries or settings (e.g., Oelemann et al. 2007; Allix-Beguec et al. 2008a; Bidovec-Stojkovic et al. 2011; Roetzer et al. 2011; de Beer et al. 2013) (Fig. 3).
Figure 3.
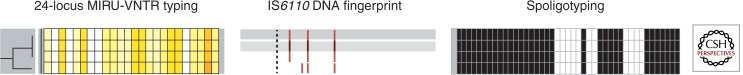
Analysis of four Mycobacterium tuberculosis complex strains with IS6110 DNA fingerprinting, 24-locus MIRU-VNTR typing, and spoligotyping. Strains are ordered in a tree based on the similarity of their 24-locus MIRU-VNTR typing results. MIRU-VNTR alleles are color-coded. Note the concordant one-locus change by MIRU-VNTR typing and the one-band change by IS6110 fingerprinting, and the conserved spoligotypes among the four isolates, illustrating the higher-resolution power of the two former versus the latter methods.
However, 24-locus MIRU-VNTR typing lacks resolution power for accurately discriminating closely related clones, which often compose populations of Beijing strains predominating in some world regions. A consensus set of four additional, hypervariable MIRU-VNTR loci has been recently defined that, when used in complement to standard 24-locus-based MIRU-VNTR typing, substantially improves the resolution power on Beijing strain populations and still retains sufficient clonal stability to follow epidemic spread of a particular strain over time (Allix-Beguec et al. 2014).
MOLECULAR EPIDEMIOLOGY BY WHOLE-GENOME SEQUENCING
Although classical genotyping methods target highly variable genetic elements, they only interrogate a small fraction of the genome. Therefore, they can intrinsically not capture microevolution potentially occurring in other genomic regions. In contrast, NGS-based approaches give access to nearly complete genome sequences. The extended use of such approaches for research and epidemiological control has become possible through the rapid improvements and quickly increasing affordability of NGS technologies. So-called benchtop NGS systems (e.g., Illumina MiSeq) can be integrated in a routine workflow, with a throughput adapted to a normal microbiological laboratory (Koser et al. 2012a; Walker et al. 2013b). In addition, commercially available easy-to-use kits for DNA library preparation and sample multiplexing now require only low amounts of genomic DNA, which limits the time needed for culturing the slow growing mycobacteria.
Several studies showed that, in accordance with the wider spectrum of analysis to detect genetic variation, whole-genome-based approaches provide more resolution power than classical genotyping (e.g., 24-locus MIRU-VNTR typing and IS6110 fingerprinting) (Niemann et al. 2009; Schurch et al. 2010; Roetzer et al. 2013; Walker et al. 2013a). In particular, whole-genome sequence analysis allows for a more precise distinction of isolates belonging to a specific recent transmission chain from other, closely but not directly related isolates, which is especially useful to resolve complex outbreak situations (Gardy et al. 2011; Roetzer et al. 2013; Walker et al. 2013a). In such situations, the phylogenetic trees based on the obtained genome-wide single-nucleotide polymorphism (SNP) data of the isolates correlate much better with the available epidemiological data and the spatiotemporal distribution patterns of the corresponding TB cases, compared with trees based on classical typing data (Roetzer et al. 2013; Walker et al. 2013a) (Fig. 4). In addition to a better time-dependent signal, the higher resolution offered by whole-genome-based analysis also provides other valuable information. For example, the presence of a particularly contagious case (i.e., a superspreader), leading to multiple secondary cases, can be detected by the observation of star-like topologies in the phylogenetic tree, where clonal variants only differing by a few SNPs and corresponding to secondary cases branch directly from a central node, representing the same single source case. Furthermore, the identification of vacant nodes in the tree topology is predictive of undiagnosed cases in the population. The unidirectional accumulation of SNPs allows a more clear-cut association of new cases to particular previous cases in a longitudinal outbreak, thus allowing for more targeted contact tracing investigations (Roetzer et al. 2013; Walker et al. 2013a). This is of special utility for the precise determination of the source cases for patient isolates of particular importance such as MDR isolates.
Figure 4.

Discrimination of 86 outbreak strains based on IS6110 DNA fingerprint patterns, 24-locus MIRU-VNTR typing, and 85 single-nucleotide polymorphisms (SNPs) detected by next-generation sequencing (NGS) (Roetzer et al. 2013). IS6110 band positions were normalized to enable mutual comparison of all isolate fingerprints. The strains are ordered in UPGMA (unweighted pair group method with arithmetic mean)—a tree based on the similarity of the IS6110 DNA fingerprint patterns (A) or concatenated SNP sequences (B). MIRU-VNTR alleles are color-coded with yellow shading, with cutoffs ranging from 0 (white) to 35 (red) repeat units. Single-locus differences in 24-locus MIRU-VNTR genotypes are indicated by asterisks. Concatenated SNP sequences are likewise color-coded. Note the largely clustered isolates by IS6110 fingerprinting and MIRU-VNTR typing, contrasting with the resolution into smaller clusters or singletons by NGS-based SNP analysis.
To identify and delineate recent transmission chains based on whole-genome sequences, a key parameter to calibrate is the level of genome-wide variation that occurs in M. tuberculosis strains within and between infected individuals over time. In different studies, the observed levels of divergence between such longitudinal isolates collected from chronically infected patients or from epidemiologically linked cases virtually never exceeded three to five SNPs, which thus defines a cutoff range for predicting recent transmission (Schurch et al. 2010; Roetzer et al. 2013; Walker et al. 2013a). The calculated mean rate of change in DNA sequence was approximately 0.5 SNPs per genome per year, providing thus a quantitative estimate of the short-term evolution rate of M. tuberculosis in the human host population (Roetzer et al. 2013; Walker et al. 2013a). Interestingly, this rate nicely matches the mutation rate estimated for M. tuberculosis in a macaque infection model (Ford et al. 2011).
Compared with classical genotyping, another major advantage of whole-genome sequencing is that it simultaneously provides direct and valuable information for predicting drug resistance, as well as highly precise phylogenetic identification (Koser et al. 2013). This combination of such diagnostic and epidemiological information in a single assay is a great benefit, especially for M. tuberculosis, for which early detection of drug resistance is important. Analysis based on whole-genome sequencing goes far beyond conventional molecular tests, focusing on known selected mutations in hot spot regions of genes involved in resistance to first and second line antituberculous drugs. In principle, whole-genome sequencing captures most, if not all, the gene sequences determining the so-called M. tuberculosis resistome, which thus allows interrogation of the entire spectrum of known mutations associated with drug resistance (Koser et al. 2012b; Roetzer et al. 2013). Moreover, whole-genome sequencing at high depth enables us to detect the emergence and coexistence of different drug-resistance-conferring mutations before selection and fixation of a final mutant, in possible combination with compensatory mutations (Sun et al. 2012; Merker et al. 2013). Such detection is of clinical relevance as the coexistence of wild-type and mutant subpopulations resulting in heteroresistance might confound the current phenotypic and molecular drug-resistance tests (Sun et al. 2012; Merker et al. 2013), as well as conclusions on transmission or secondary acquisition of drug resistance.
Although NGS-based molecular tracing of M. tuberculosis has a clearly vast potential, its generalized use is still hampered by some remaining challenges. Despite rapid cost decreases, NGS analyses still remains too costly for many microbial/mycobacteriological laboratories. In addition, the bioinformatics analysis and interpretation of the data require adapted computer and data storage infrastructure. In contrast to classical genotyping, technical and analytical modalities (e.g., the precise delimitation of the genome sequence that is taken into account in the analysis; the minimal sequence coverage) have not yet been standardized with the consequence that data sets generated by different laboratories are not yet directly comparable. Universal databases are not yet available (e.g., for multicentric longitudinal epidemiological studies and surveillance). For optimal prediction of drug resistance, although some databasing tools are already available such as TBDReaMDB (Sandgren et al. 2009), more comprehensive and well-curated databases are needed to better and more quantitatively correlate genetic data with drug susceptibility phenotypes. These challenges are being addressed by large international consortia (e.g., http://patho-ngen-trace.eu/), to accelerate the implementation of NGS-based epidemiology and diagnostics.
GLOBAL DIVERSITY AND EVOLUTION OF M. tuberculosis
A substantial part of what we know today about the diversity of the MTBC initially stems from molecular epidemiological surveys, which explored the pathogen’s population structure in various countries for contextual molecular epidemiological interpretations. The use of IS6110 fingerprinting, MIRU-VNTR typing, and spoligotyping lead to the recognition of distinct families or groups of closely related strains and genotypes predominating in different world regions. One of the first and most emblematic examples is the report of strains with particular, closely related IS6110 fingerprints prevalent in East Asian countries, and particularly in the region of Beijing, China, leading to their designation as Beijing strains (van Soolingen et al. 1995). Other groups of strains were also recognized in other geographic regions, also by other genetic markers, and were therefore assumed to represent different clonal lineages (e.g., Kremer et al. 1999; Sola et al. 2001; Supply et al. 2001). As a result, databases that compile spoligotyping (Brudey et al. 2006; Demay et al. 2012) and 24-locus MIRU-VNTR typing data collected worldwide (i.e., MIRU-VNTRPlus [Allix-Beguec et al. 2008b; Weniger et al. 2010]) currently offer maybe the best qualitative view on the global diversity of the MTBC population. As of early 2014, SITVIT counts more than 7000 different spoligotypes (http://www.pasteur-guadeloupe.fr:8081/SITVIT_ONLINE/description.jsp#), and MIRUVNTRplus more than 14,000 different MIRU-VNTR types (based on the 15 most discriminatory loci; http://www.miru-vntrplus.org/MIRU/types15.faces). SpolDB/SITVIT data provide a vision of the geographic distribution of the main spoligotype signatures (defined by the presence/absence of some of the 43 spacers interrogated by spoligotyping) (Filliol et al. 2003; Brudey et al. 2006; Demay et al. 2012), known to reflect to a certain degree the main clonal lineages defined by more orthodox phylogenetic markers (see below). However, although analysis of MIRU-VNTR typing data complemented by detection of spoligotype signatures is of high predictive value for classifying isolates into lineages (Cardoso Oelemann et al. 2011), spoligotyping and, to a lesser extent, MIRU-VNTR typing data both show homoplasy, which makes them not optimal for phylogenetic reconstruction (Comas et al. 2009).
Therefore, the phylogenetic structure of the MTBC described below has essentially been established by exploitation of classical phylogenetic markers (i.e., sequence-based [Sreevatsan et al. 1997; Baker et al. 2004; Gutierrez et al. 2005; Hershberg et al. 2008] and comparative genomics-based [Cole et al. 1998; Brosch et al. 2002; Fleischmann et al. 2002; Gutacker et al. 2002; Garnier et al. 2003; Hirsh et al. 2004; Filliol et al. 2006; Gagneux et al. 2006a; Gutacker et al. 2006; Comas et al. 2010, 2013a; Homolka et al. 2012; Supply et al. 2013]), including analysis of SNPs, regions of genomic differences (RDs)/large sequence polymorphisms (LSPs), and whole-genome sequences.
Compared with other bacteria, the MTBC population is genetically highly compact and essentially clonal with no significant evidence of interstrain horizontal gene transfer and/or recombination (Supply et al. 2003; Hirsh et al. 2004; Comas et al. 2013) (except for Mycobacterium canettii, see below). A recent whole-genome phylogeny based on 259 strains that represent the global diversity of the MTBC identified around 34.000 polymorphic sites with pairwise distances of no more than approximately 2000 SNPs separating any MTBC strain (Comas et al. 2013). This compactness, as well as the vertical inheritance of most RDs, LSPs, and SNPs (with very low levels of homoplastic variants), indicates that this whole population results from recent clonal expansion, probably from a few or a single common ancestor(s) (Sreevatsan et al. 1997; Brosch et al. 2002; Hirsh et al. 2004). Despite the relative limitation in genetic diversity of classical MTBC strains, seven main lineages of human strains (including M. tuberculosis sensu stricto, in lineages 1–4 and 7, as well as M. africanum in lineages 5 and 6), as well as one branch of animal strains, can be clearly distinguished (Fig. 5) (Hirsh et al. 2004; Gagneux et al. 2006a; Comas et al. 2013).
Figure 5.

Whole-genome-based phylogeny of 219 isolates of all major MTBC lineages according to Comas et al. (2013). Major lineages are depicted by specific colors. (Image courtesy of Inaki Comas.)
Consistent with the differential geographic distribution of the different genotype groups or families defined by classical genotyping markers (see above), the different human-adapted lineages are remarkably associated with the particular, sympatric human population groups that carry them. This association is apparent both at a global level, when analyzing isolates from patients living on different continents, and often at a local level as well, when analyzing isolates from residents of a particular cosmopolitan setting originating from different countries (Hirsh et al. 2004; Gagneux et al. 2006a). In the latter case, this association remains visible even when TB transmission takes place in the country of residence, outside the geographic region of origin. This could reflect the non–mutually exclusive contribution of social factors limiting contacts between different host population groups in the studied settings and biological factors including adaptation of M. tuberculosis lineages to different host populations (Hirsh et al. 2004; Gagneux et al. 2006a). The role of social factors is suggested (e.g., a contrario by the observation of a weaker association between lineages and host populations in situations or places favoring everyday contacts between human population groups [Varghese et al. 2013]). The hypothesis of biological adaptation is likewise supported by the observation that the association between a pathogen’s lineages and host population groups appears disrupted in immunodeficient patients (Gagneux et al. 2006a; Fenner et al. 2013).
The notion of host-adapted genotypes is also suggested by the structuration of the animal strain branch that correlates with host ranges of the different corresponding strains. Although classical Mycobacterium bovis strains can be found in a range of mammalian species (cow, badger, wild boar, deer, lion, etc.), the strains of other variant MTBC members show a clearly restricted and specific host range, such as Mycobacterium microti (vole), Mycobacterium caprae (goat), Mycobacterium pinnipedii (seal), and Mycobacterium orygis (oryx). Therefore, and because of the conservation of >99% at the nucleotide level among the genomes of the animal and human strains (higher than the genetic threshold usually considered for defining different bacterial species), the different MTBC members (and lineages) may be regarded as a series of so-called ecotypes, occupying distinct niches, instead of different species (Smith et al. 2006, 2009).
The association and possible adaptation of pathogen lineages to particular host populations suggest parallel evolution between the pathogen and its host. For the human strain lineages, such coevolution is supported by several lines of evidence. For example, the observation that the most deeply rooted lineages (lineages 5 and 6) are found exclusively in Africa suggests an African origin for the MTBC (Comas et al. 2013). Similarities that exist between the phylogeny and the geographic distribution of the MTBC strains and that of the main human mitochondrial haplogroups suggest parallel divergence between the pathogen and its host (Comas et al. 2013). Of note, such similarities were not so clear when an MTBC phylogeny was compared with that of human Y chromosome data; however, the latter comparison did not take into account the frequencies of geographic distribution of the MTBC strains to define strain phylogeographical lineages (Pepperell et al. 2013). Also in favor of parallel evolution is the strikingly matching demographic changes, detected by analysis of both MTBC genotyping and whole-genome sequence data, suggesting expansion of the pathogen population coinciding with the human population densification/expansion at the Neolithic period or at the industrialization period two or three centuries ago (Wirth et al. 2008; Comas et al. 2013; Pepperell et al. 2013). Furthermore, recent tests of alternative models using MTBC whole-genome sequence data with initial points calibrated against several key dates in human evolution suggested a striking correlation between dating of splits of specific MTBC lineages and estimated dates of major migratory movements (i.e., lineage 1 alias Indo-Oceanic and the early dispersal around the Indian Ocean; lineage 2-4 including Eurasian lineages vs. the later dispersal throughout Eurasia). Interestingly, the long-term genome genome-wide evolution rate estimated posteriorly on this basis was much lower than the short-term mutation rate of 0.5 SNP per year and per genome based on longitudinal isolates from outbreak or chronically infected patients (Comas et al. 2013) (see above). Although it was proposed that M. tuberculosis was mostly under relaxed purifying selection (Hershberg et al. 2008), this difference between short-term and long-term rates might reflect the increased effect over time of fixation or removal of SNPs by natural selection (Namouchi et al. 2012; Comas et al. 2013). In accordance with this idea, the proportion of nonsynonymous versus synonymous mutations was found to be much lower in M. canettii than among classical MTBC members, which is compatible with an even longer time during which the evolutionary early-branching M. canettii was exposed to purifying selection (Supply et al. 2013).
An African origin for the MTBC is also supported by genotyping (van Soolingen et al. 1997; Fabre et al. 2004; Gutierrez et al. 2005) and whole-genome sequence data (Supply et al. 2013) indicating that M. canettii represents the earliest evolutionary branch of the tubercle bacilli. M. canettii, also named smooth tubercle bacilli (STB) and/or Mycobacterium prototuberculosis, regroups special TB clinical isolates, which have a distinctive smooth colony phenotype on culture media. In contrast to classical MTBC members, M. canettii strains are highly recombinogenic (i.e., nonclonal) and show much higher numbers of SNPs in their genomes (up to approximately 60,000 SNPs). Strikingly, most of the M. canettii isolates have been obtained from patients living or having lived in East Africa. Together with the earlier branching of M. canettii, this geographic restriction thus also points to an African origin of the MTBC. The fact that the strains from these early branching TB bacilli have all been isolated from human cases argues against the hypothesis of a zoonotic origin of the MTBC, consistently with the evolutionary scenarios based on analysis of LSPs or MIRU-VNTR markers (Brosch et al. 2002; Wirth et al. 2008). These data also suggest a coexistence between the pathogen and its human host that predated the clonal emergence of the classical members of the MTBC (Gutierrez et al. 2005; Brisse et al. 2006; Supply et al. 2013).
The hypothesis of a stable longitudinal host–pathogen coevolution that resulted in MTBC lineage-specific pathobiological characteristics (Homolka et al. 2010; Brites and Gagneux 2012) is further supported by the identification of human immunity-related gene variants that appear to protect from infection with a particular MTBC genotype (e.g., Intemann et al. 2009; Thye et al. 2011).
PATHOBIOLOGICAL IMPACT OF THE GENOMIC DIVERSITY
Although the genetic diversity of the MTBC is relatively low, one key finding is that roughly two-thirds of the SNPs found in clinical isolates are nonsynonymous and thus potentially have functional consequences (Sreevatsan et al. 1997; Hershberg et al. 2008). There is increasing evidence that this genomic diversity may influence important pathobiological properties, such as the transmissibility and virulence of the MTBC strains, as well as the immune responses and clinical pictures that they evoke (reviewed in Gagneux and Small 2007). Such an influence has been indicated by field studies looking at epidemiological or clinical correlates of transmissibility and virulence of strains from different lineages (e.g., de Jong et al. 2006, 2008; Kong et al. 2007; Caws et al. 2008; Rakotosamimanana et al. 2010) or by detecting lineage-specific differences in virulence-related features, mycobacterial transcriptomic responses, or cytokine induction patterns in animal or cellular infection models (Lopez et al. 2003; Homolka et al. 2010; Portevin et al. 2011; Reiling et al. 2013).
One of the most marked differences in pathogenicity among MTBC strains was observed for M. canettii isolates, which consistently are the most divergent and early branching tubercle bacilli compared with the rest of the population. In mouse infection models, M. canettii isolates showed lower virulence/persistence compared with M. tuberculosis. These findings led to the conclusion that the MTBC emerged from an ancestral M. canettii–like pool of mycobacteria by gain of as yet unknown persistence and virulence mechanisms, which favored its subsequent clonal radiation worldwide (Supply et al. 2013).
Specific pathobiological properties of the Beijing/East Asian lineage (alias lineage 2) are also intensively studied. Indeed, a number of reports suggest their selective spread in certain regions of the world, frequently (but not always) in association with drug resistance (e.g., Cowley et al. 2008; Niemann et al. 2010; reviewed in Glynn et al. 2002; Parwati et al. 2010). This spread has been proposed to result from several potential selective advantages. Among different possible causes, conflicting evidence based on mouse infection models and molecular epidemiological studies exists for a potential role of Bacillus Calmette–Guérin (BCG) vaccination, which has been hypothesized to be less protective against Beijing strains and thus induce an escape favoring the selective spread of these strains (Lopez et al. 2003; Tsenova et al. 2005; Jeon et al. 2008; Ordway et al. 2011). Higher virulence and, for example, an elicited immune response skewed toward a less protective Th1 response have also been indicated for some Beijing strains investigated using animal infection models (Lopez et al. 2003; Dormans et al. 2004; Manca et al. 2004; Reed et al. 2004). However, recent data suggested differences in pathogenicity and other properties such as transmissibility at Beijing sublineage level (Kato-Maeda et al. 2010; Kato-Maeda et al. 2012), thus suggesting more complex associations between strain genetic background and phenotypes than previously thought.
Still relatively little is known about the genetic polymorphisms that could explain these (sub)lineage-specific phenotypic differences. For example, the production of a phenolic glycolipid by an intact pks15-1 gene in some Beijing strains has been shown to modulate the early host cytokine response in vitro, which may explain the induction of a suboptimal Th1 response mentioned above, but this production does not in itself confer increased virulence (Sinsimer et al. 2008). Beijing strains classified as “modern” also constitutively up-regulate the DosR regulon, involved in adaption for survival during periods of nonreplicating persistence in vitro. Such up-regulation was proposed to provide selective advantages in conditions encountered during chronic or latent phases of infection (Reed et al. 2007). A frameshift mutation has been identified in these Beijing strains, in the gene encoding the DosT sensor kinase putatively interacting with DosR in a corresponding two-component regulatory system. However, this mutation is not directly responsible for the constitutive up-regulation of the DosR regulon (Fallow et al. 2010). Along the same lines, a massive gene duplication event spanning 350 kb and including the dosR region was detected among some but not all sublineages of modern Beijing strains, which can thus not in itself explain the constitutive up-regulation of the DosR regulon by the modern Beijing strains (Domenech et al. 2010).
Increasing knowledge is being gained regarding the molecular mechanisms underlying the association between drug resistance and strain genetic background. There is some evidence, however strongly debated (Werngren and Hoffner 2003; Werngren 2013), for a higher mutation rate of strains of the East Asian/Beijing (sub)lineage compared with the strains, leading to increased rate of drug resistance acquisition in vitro, as well as a predicted higher probability of multidrug resistance in TB patients infected with these strains (de Steenwinkel et al. 2012; Ford et al. 2013). Although specific SNPs have been identified in Beijing strains in genes involved in DNA replication, recombination, and repair, none of these polymorphisms has yet been shown to confer increased mutability in M. tuberculosis (Mestre et al. 2011). Another factor that likely plays a major role is the interplay between the fitness cost associated to drug resistance mutations, acquisition of compensatory mutations, and, more generally, influence of epistasis, defined as the phenotypic interaction between mutations affecting different regions of a same protein or different gene products involved in a same or linked metabolic pathway(s) (Muller et al. 2013). For example, high-fitness-cost mutations in katG, the 16S rRNA, or rpoB, causing resistance to isoniazid, aminoglycosides, or rifampicin, respectively, have been found to be associated with compensatory mutations in the regulatory region of ahpC (Gagneux et al. 2006b; Hazbon et al. 2006), other regions of the 16S rRNA (Shcherbakov et al. 2010), or rpoA or rpoC (Casali et al. 2012; Comas et al. 2012). More than 30% of the MDR clinical isolates from Abkhazia/Georgia, Uzbekistan, and Kazakhstan, which are the countries with the highest incidence of MDR TB worldwide, were found to harbor mutations in rpoA or rpoC putatively compensating for rpoB mutations (Comas et al. 2012). Such configurations of mutations were especially found in Beijing strains that predominate in these regions, and are correlated with high molecular clustering of the corresponding isolates, thus suggesting increased transmissibility as a result of drug resistance associated to an increased fitness. Such epistatic interaction may plausibly be influenced by the genetic background of specific (sub)lineages or strains, which could offer more or less favorable arrays of preexisting mutations for compensating the cost of an emerging drug resistance mutation (Muller et al. 2013). As mentioned above, coemergence of drug resistance and compensatory mutations have also been observed by deep genome sequencing of M. tuberculosis populations isolated from TB patients during development of drug resistance (Sun et al. 2012; Merker et al. 2013).
CONCLUDING REMARKS
Recently accumulated data have contributed to change previous dogmas on the diversity of the pathogen and on the origin of the disease and have provided as well fascinating, new insights into the parallel evolution of M. tuberculosis and its human host. At the same time, these data also indicate that complex interactions probably underpin the links between the strain genetic background, drug resistance, and other features of biomedical interest. Integrative approaches coupling whole-genome sequencing to transcriptomic analysis, epidemiological analysis, and biological characterization are expected to provide the power needed to identify the determinants involved.
Footnotes
Editors: Stefan H.E. Kaufmann, Eric J. Rubin, and Alimuddin Zumla
Additional Perspectives on Tuberculosis available at www.perspectivesinmedicine.org
REFERENCES
- Allix-Beguec C, Fauville-Dufaux M, Supply P 2008a. Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 46: 1398–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allix-Beguec C, Harmsen D, Weniger T, Supply P, Niemann S 2008b. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J Clin Microbiol 46: 2692–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allix-Beguec C, Supply P, Wanlin M, Bifani P, Fauville- Dufaux M 2008c. Standardised PCR-based molecular epidemiology of tuberculosis. Eur Respir J 31: 1077–1084. [DOI] [PubMed] [Google Scholar]
- Allix-Beguec C, Wahl C, Hanekom M, Nikolayevskyy V, Drobniewski F, Maeda S, Campos-Herrero I, Mokrousov I, Niemann S, Kontsevaya I, et al. 2014. Proposal of a consensus set of hypervariable mycobacterial interspersed repetitive-unit-variable-number tandem-repeat loci for subtyping of Mycobacterium tuberculosis Beijing isolates. J Clin Microbiol 52: 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar J, Safi H, Romero J, Alejo A, Gracia A, Palomares JC 1995. Nosocomial transmission of tuberculosis infection in pediatrics wards. Pediatr Infect Dis J 14: 44–48. [DOI] [PubMed] [Google Scholar]
- Baker L, Brown T, Maiden MC, Drobniewski F 2004. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis. Emerg Infect Dis 10: 1568–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PF, Cave MD 2003. Molecular epidemiology of tuberculosis. N Engl J Med 349: 1149–1156. [DOI] [PubMed] [Google Scholar]
- Barnes PF, el-Hajj H, Preston-Martin S, Cave MD, Jones BE, Otaya M, Pogoda J, Eisenach KD 1996. Transmission of tuberculosis among the urban homeless. JAMA 275: 305–307. [PubMed] [Google Scholar]
- Bidovec-Stojkovic U, Zolnir-Dovc M, Supply P 2011. One year nationwide evaluation of 24-locus MIRU-VNTR genotyping on Slovenian Mycobacterium tuberculosis isolates. Respir Med 105: S67–S73. [DOI] [PubMed] [Google Scholar]
- Brisse S, Supply P, Brosch R, Vincent V, Gutierrez MC 2006. ‘A re-evaluation of M. prototuberculosis’: Continuing the debate. PLoS Pathog 2: e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D, Gagneux S 2012. Old and new selective pressures on Mycobacterium tuberculosis. Infect Genet Evol 12: 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, et al. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci 99: 3684–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuno L, Arora J, Baumanis V, et al. 2006. Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso Oelemann M, Gomes HM, Willery E, Possuelo L, Batista Lima KV, Allix-Beguec C, Locht C, Goguet de la Salmoniere YO, Gutierrez MC, Suffys P, et al. 2011. The forest behind the tree: Phylogenetic exploration of a dominant Mycobacterium tuberculosis strain lineage from a high tuberculosis burden country. PLoS ONE 6: e18256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali N, Nikolayevskyy V, Balabanova Y, Ignatyeva O, Kontsevaya I, Harris SR, Bentley SD, Parkhill J, Nejentsev S, Hoffner SE, et al. 2012. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res 22: 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave MD, Eisenach KD, McDermott PF, Bates JH, Crawford JT 1991. IS6110: Conservation of sequence in the Mycobacterium tuberculosis complex and its utilization in DNA fingerprinting. Mol Cell Probes 5: 73–80. [DOI] [PubMed] [Google Scholar]
- Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NT, Thuong NT, Stepniewska K, Huyen MN, Bang ND, Loc TH, et al. 2008. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog 4: e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. [DOI] [PubMed] [Google Scholar]
- Comas I, Homolka S, Niemann S, Gagneux S 2009. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE 4: e7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet 42: 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44: 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, et al. 2013. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet 45: 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, Dye C 2003. The growing burden of tuberculosis: Global trends and interactions with the HIV epidemic. Arch Intern Med 163: 1009–1021. [DOI] [PubMed] [Google Scholar]
- Cowley D, Govender D, February B, Wolfe M, Steyn L, Evans J, Wilkinson RJ, Nicol MP 2008. Recent and rapid emergence of W-Beijing strains of Mycobacterium tuberculosis in Cape Town, South Africa. Clin Infect Dis 47: 1252–1259. [DOI] [PubMed] [Google Scholar]
- de Beer JL, van Ingen J, de Vries G, Erkens C, Sebek M, Mulder A, Sloot R, van den Brandt AM, Enaimi M, Kremer K, et al. 2013. Comparative study of IS6110 restriction fragment length polymorphism and variable-number tandem-repeat typing of Mycobacterium tuberculosis isolates in the Netherlands, based on a 5-year nationwide survey. J Clin Microbiol 51: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong BC, Hill PC, Brookes RH, Gagneux S, Jeffries DJ, Otu JK, Donkor SA, Fox A, McAdam KP, Small PM, et al. 2006. Mycobacterium africanum elicits an attenuated T cell response to early secreted antigenic target, 6 kDa, in patients with tuberculosis and their household contacts. J Infect Dis 193: 1279–1286. [DOI] [PubMed] [Google Scholar]
- de Jong BC, Hill PC, Aiken A, Awine T, Antonio M, Adetifa IM, Jackson-Sillah DJ, Fox A, Deriemer K, Gagneux S, et al. 2008. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J Infect Dis 198: 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Steenwinkel JE, ten Kate MT, de Knegt GJ, Kremer K, Aarnoutse RE, Boeree MJ, Verbrugh HA, van Soolingen D, Bakker-Woudenberg IA 2012. Drug susceptibility of Mycobacterium tuberculosis Beijing genotype and association with MDR TB. Emerg Infect Dis 18: 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demay C, Liens B, Burguiere T, Hill V, Couvin D, Millet J, Mokrousov I, Sola C, Zozio T, Rastogi N 2012. SITVITWEB—A publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect Genet Evol 12: 755–766. [DOI] [PubMed] [Google Scholar]
- Diel R, Schneider S, Meywald-Walter K, Ruf CM, Rusch-Gerdes S, Niemann S 2002. Epidemiology of tuberculosis in Hamburg, Germany: Long-term population-based analysis applying classical and molecular epidemiological techniques. J Clin Microbiol 40: 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diel R, Loddenkemper R, Zellweger JP, Sotgiu G, D’Ambrosio L, Centis R, van der Werf MJ, Dara M, Detjen A, Gondrie P, et al. 2013. Old ideas to innovate tuberculosis control: Preventive treatment to achieve elimination. Eur Respir J 42: 785–801. [DOI] [PubMed] [Google Scholar]
- Domenech P, Kolly GS, Leon-Solis L, Fallow A, Reed MB 2010. Massive gene duplication event among clinical isolates of the Mycobacterium tuberculosis W/Beijing family. J Bacteriol 192: 4562–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormans J, Burger M, Aguilar D, Hernandez-Pando R, Kremer K, Roholl P, Arend SM, van Soolingen D 2004. Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin Exp Immunol 137: 460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C, Williams BG 2010. The population dynamics and control of tuberculosis. Science 328: 856–861. [DOI] [PubMed] [Google Scholar]
- Fabre M, Koeck JL, Le Fleche P, Simon F, Herve V, Vergnaud G, Pourcel C 2004. High genetic diversity revealed by variable-number tandem repeat genotyping and analysis of hsp65 gene polymorphism in a large collection of “Mycobacterium canettii” strains indicates that the M. tuberculosis complex is a recently emerged clone of “M. canettii”. J Clin Microbiol 42: 3248–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallow A, Domenech P, Reed MB 2010. Strains of the East Asian (W/Beijing) lineage of Mycobacterium tuberculosis are DosS/DosT-DosR two-component regulatory system natural mutants. J Bacteriol 192: 2228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner L, Egger M, Bodmer T, Furrer H, Ballif M, Battegay M, Helbling P, Fehr J, Gsponer T, Rieder HL, et al. 2013. HIV infection disrupts the sympatric host–pathogen relationship in human tuberculosis. PLoS Genet 9: e1003318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol I, Driscoll JR, van Soolingen D, Kreiswirth BN, Kremer K, Valetudie G, Dang DA, Barlow R, Banerjee D, Bifani PJ, et al. 2003. Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. J Clin Microbiol 41: 1963–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, Fyfe J, Garcia-Garcia L, Rastogi N, Sola C, et al. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: Insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol 188: 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, et al. 2002. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol 184: 5479–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, et al. 2011. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet 43: 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, Johnston JC, Gardy J, Lipsitch M, Fortune SM 2013. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet 45: 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frothingham R, Meeker-O’Connell WA 1998. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology 144: 1189–1196. [DOI] [PubMed] [Google Scholar]
- Gagneux S, Small PM 2007. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 7: 328–337. [DOI] [PubMed] [Google Scholar]
- Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, Nicol M, Niemann S, Kremer K, Gutierrez MC, et al. 2006a. Variable host–pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci 103: 2869–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ 2006b. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312: 1944–1946. [DOI] [PubMed] [Google Scholar]
- Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, Bayona J 2010. Multidrug-resistant and extensively drug-resistant tuberculosis: A threat to global control of tuberculosis. Lancet 375: 1830–1843. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, et al. 2011. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med 364: 730–739. [DOI] [PubMed] [Google Scholar]
- Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, Pryor M, Duthoy S, Grondin S, Lacroix C, Monsempe C, et al. 2003. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci 100: 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn JR, Whiteley J, Bifani PJ, Kremer K, van Soolingen D 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: A systematic review. Emerg Infect Dis 8: 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub JE, Cronin WA, Obasanjo OO, Coggin W, Moore K, Pope DS, Thompson D, Sterling TR, Harrington S, Bishai WR, et al. 2001. Transmission of Mycobacterium tuberculosis through casual contact with an infectious case. Arch Intern Med 161: 2254–2258. [DOI] [PubMed] [Google Scholar]
- Gutacker MM, Smoot JC, Migliaccio CA, Ricklefs SM, Hua S, Cousins DV, Graviss EA, Shashkina E, Kreiswirth BN, Musser JM 2002. Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: Resolution of genetic relationships among closely related microbial strains. Genetics 162: 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutacker MM, Mathema B, Soini H, Shashkina E, Kreiswirth BN, Graviss EA, Musser JM 2006. Single-nucleotide polymorphism–based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J Infect Dis 193: 121–128. [DOI] [PubMed] [Google Scholar]
- Gutierrez MC, Brisse S, Brosch R, Fabre M, Omais B, Marmiesse M, Supply P, Vincent V 2005. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog 1: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, Garcia-Garcia L, et al. 2006. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 50: 2640–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heersma HF, Kremer K, van Embden JD 1998. Computer analysis of IS6110 RFLP patterns of Mycobacterium tuberculosis. Methods Mol Biol 101: 395–422. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, Roach JC, Kremer K, Petrov DA, Feldman MW, et al. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol 6: e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM 2004. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci 101: 4871–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka S, Niemann S, Russell DG, Rohde KH 2010. Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: Delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog 6: e1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka S, Projahn M, Feuerriegel S, Ubben T, Diel R, Nubel U, Niemann S 2012. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS ONE 7: e39855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, et al. 2009. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5: e1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon BY, Derrick SC, Lim J, Kolibab K, Dheenadhayalan V, Yang AL, Kreiswirth B, Morris SL 2008. Mycobacterium bovis BCG immunization induces protective immunity against nine different Mycobacterium tuberculosis strains in mice. Infect Immun 76: 5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, et al. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol 35: 907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato-Maeda M, Kim EY, Flores L, Jarlsberg LG, Osmond D, Hopewell PC 2010. Differences among sublineages of the East-Asian lineage of Mycobacterium tuberculosis in genotypic clustering. Int J Tuberc Lung Dis 14: 538–544. [PMC free article] [PubMed] [Google Scholar]
- Kato-Maeda M, Shanley CA, Ackart D, Jarlsberg LG, Shang S, Obregon-Henao A, Harton M, Basaraba RJ, Henao-Tamayo M, Barrozo JC, et al. 2012. Beijing sublineages of Mycobacterium tuberculosis differ in pathogenicity in the guinea pig. Clin Vaccine Immunol 19: 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Cave MD, Zhang L, Foxman B, Marrs CF, Bates JH, Yang ZH 2007. Association between Mycobacterium tuberculosis Beijing/W lineage strain infection and extrathoracic tuberculosis: Insights from epidemiologic and clinical characterization of the three principal genetic groups of M. tuberculosis clinical isolates. J Clin Microbiol 45: 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, et al. 2012a. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog 8: e1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koser CU, Feuerriegel S, Summers DK, Archer JA, Niemann S 2012b. Importance of the genetic diversity within the Mycobacterium tuberculosis complex for the development of novel antibiotics and diagnostic tests of drug resistance. Antimicrob Agents Chemother 56: 6080–6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koser CU, Bryant JM, Becq J, Torok ME, Ellington MJ, Marti-Renom MA, Carmichael AJ, Parkhill J, Smith GP, Peacock SJ 2013. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N Engl J Med 369: 290–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PW, Martin C, Palittapongarnpim P, Plikaytis BB, Riley LW, Yakrus MA, et al. 1999. Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: Interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol 37: 2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Fleche P, Fabre M, Denoeud F, Koeck JL, Vergnaud G 2002. High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol 2: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez B, Aguilar D, Orozco H, Burger M, Espitia C, Ritacco V, Barrera L, Kremer K, Hernandez-Pando R, Huygen K, et al. 2003. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol 133: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Reed MB, Freeman S, Mathema B, Kreiswirth B, Barry CE 3rd, Kaplan G 2004. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect Immun 72: 5511–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, Tibayrenc M, Locht C, Supply P 2001. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci 98: 1901–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek GH, Cave MD, Eisenach KD, Wallace RJ Jr, Bates JH, Crawford JT 1991. Chromosomal DNA fingerprint patterns produced with IS6110 as strain-specific markers for epidemiologic study of tuberculosis. J Clin Microbiol 29: 2030–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdam RA, Hermans PW, van Soolingen D, Zainuddin ZF, Catty D, van Embden JD, Dale JW 1990. Characterization of a Mycobacterium tuberculosis insertion sequence belonging to the IS3 family. Mol Microbiol 4: 1607–1613. [DOI] [PubMed] [Google Scholar]
- Merker M, Kohl TA, Roetzer A, Truebe L, Richter E, Rusch-Gerdes S, Fattorini L, Oggioni MR, Cox H, Varaine F, et al. 2013. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PLoS ONE 8: e82551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre O, Luo T, Dos Vultos T, Kremer K, Murray A, Namouchi A, Jackson C, Rauzier J, Bifani P, Warren R, et al. 2011. Phylogeny of Mycobacterium tuberculosis Beijing strains constructed from polymorphisms in genes involved in DNA replication, recombination and repair. PLoS ONE 6: e16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B, Borrell S, Rose G, Gagneux S 2013. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet 29: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Schock U, Gicquel B, Rocha EP 2012. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res 22: 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, Koser CU, Gagneux S, Plinke C, Homolka S, Bignell H, Carter RJ, Cheetham RK, Cox A, Gormley NA, et al. 2009. Genomic diversity among drug sensitive and multidrug resistant isolates of Mycobacterium tuberculosis with identical DNA fingerprints. PLoS ONE 4: e7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, Diel R, Khechinashvili G, Gegia M, Mdivani N, Tang YW 2010. Mycobacterium tuberculosis Beijing lineage favors the spread of multidrug-resistant tuberculosis in the Republic of Georgia. J Clin Microbiol 48: 3544–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelemann MC, Diel R, Vatin V, Haas W, Rusch-Gerdes S, Locht C, Niemann S, Supply P 2007. Assessment of an optimized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing system combined with spoligotyping for population-based molecular epidemiology studies of tuberculosis. J Clin Microbiol 45: 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway DJ, Shang S, Henao-Tamayo M, Obregon-Henao A, Nold L, Caraway M, Shanley CA, Basaraba RJ, Duncan CG, Orme IM 2011. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin Vaccine Immunol 18: 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardini M, Niemann S, Varaine F, Iona E, Meacci F, Orru G, Yesilkaya H, Jarosz T, Andrew P, Barer M, et al. 2009. Characteristics of drug-resistant tuberculosis in Abkhazia (Georgia), a high-prevalence area in Eastern Europe. Tuberculosis (Edinb) 89: 317–324. [DOI] [PubMed] [Google Scholar]
- Parwati I, van Crevel R, van Soolingen D 2010. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect Dis 10: 103–111. [DOI] [PubMed] [Google Scholar]
- Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, Birren B, Galagan J, Feldman MW 2013. The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathog 9: e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portevin D, Gagneux S, Comas I, Young D 2011. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog 7: e1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodinger WM 2007. Molecular epidemiology of tuberculosis: Toy or tool? A review of the literature and examples from Central Europe. Wien Klin Wochenschr 119: 80–89. [DOI] [PubMed] [Google Scholar]
- Rakotosamimanana N, Raharimanga V, Andriamandimby SF, Soares JL, Doherty TM, Ratsitorahina M, Ramarokoto H, Zumla A, Huggett J, Rook G, et al. 2010. Variation in gamma interferon responses to different infecting strains of Mycobacterium tuberculosis in acid-fast bacillus smear-positive patients and household contacts in Antananarivo, Madagascar. Clin Vaccine Immunol 17: 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, Kaplan G, Barry CE, 3rd. 2004. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431: 84–87. [DOI] [PubMed] [Google Scholar]
- Reed MB, Gagneux S, Deriemer K, Small PM, Barry CE, 3rd. 2007. The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J Bacteriol 189: 2583–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling N, Homolka S, Walter K, Brandenburg J, Niwinski L, Ernst M, Herzmann C, Lange C, Diel R, Ehlers S, et al. 2013. Clade-specific virulence patterns of Mycobacterium tuberculosis complex strains in human primary macrophages and aerogenically infected mice. mBio 30: e00250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roetzer A, Schuback S, Diel R, Gasau F, Ubben T, di Nauta A, Richter E, Rusch-Gerdes S, Niemann S 2011. Evaluation of Mycobacterium tuberculosis typing methods in a 4-year study in Schleswig-Holstein, Northern Germany. J Clin Microbiol 49: 4173–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roetzer A, Diel R, Kohl TA, Ruckert C, Nubel U, Blom J, Wirth T, Jaenicke S, Schuback S, Rusch-Gerdes S, et al. 2013. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med 10: e1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roring S, Scott A, Brittain D, Walker I, Hewinson G, Neill S, Skuce R 2002. Development of variable-number tandem repeat typing of Mycobacterium bovis: Comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J Clin Microbiol 40: 2126–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren A, Strong M, Muthukrishnan P, Weiner BK, Church GM, Murray MB 2009. Tuberculosis drug resistance mutation database. PLoS Med 6: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savine E, Warren RM, van der Spuy GD, Beyers N, van Helden PD, Locht C, Supply P 2002. Stability of variable-number tandem repeats of mycobacterial interspersed repetitive units from 12 loci in serial isolates of Mycobacterium tuberculosis. J Clin Microbiol 40: 4561–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch AC, van Soolingen D 2012. DNA fingerprinting of Mycobacterium tuberculosis: From phage typing to whole-genome sequencing. Infect Genet Evol 12: 602–609. [DOI] [PubMed] [Google Scholar]
- Schurch AC, Kremer K, Kiers A, Daviena O, Boeree MJ, Siezen RJ, Smith NH, van Soolingen D 2010. The tempo and mode of molecular evolution of Mycobacterium tuberculosis at patient-to-patient scale. Infect Genet Evol 10: 108–114. [DOI] [PubMed] [Google Scholar]
- Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Bottger EC 2010. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol 77: 830–840. [DOI] [PubMed] [Google Scholar]
- Sinsimer D, Huet G, Manca C, Tsenova L, Koo MS, Kurepina N, Kana B, Mathema B, Marras SA, Kreiswirth BN, et al. 2008. The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infect Immun 76: 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuce RA, McCorry TP, McCarroll JF, Roring SM, Scott AN, Brittain D, Hughes SL, Hewinson RG, Neill SD 2002. Discrimination of Mycobacterium tuberculosis complex bacteria using novel VNTR-PCR targets. Microbiology 148: 519–528. [DOI] [PubMed] [Google Scholar]
- Small PM, McClenny NB, Singh SP, Schoolnik GK, Tompkins LS, Mickelsen PA 1993a. Molecular strain typing of Mycobacterium tuberculosis to confirm cross-contamination in the mycobacteriology laboratory and modification of procedures to minimize occurrence of false-positive cultures. J Clin Microbiol 31: 1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small PM, Shafer RW, Hopewell PC, Singh SP, Murphy MJ, Desmond E, Sierra MF, Schoolnik GK 1993b. Exogenous reinfection with multidrug-resistant Mycobacterium tuberculosis in patients with advanced HIV infection. N Engl J Med 328: 1137–1144. [DOI] [PubMed] [Google Scholar]
- Small PM, Hopewell PC, Singh SP, Paz A, Parsonnet J, Ruston DC, Schecter GF, Daley CL, Schoolnik GK 1994. The epidemiology of tuberculosis in San Francisco. A population-based study using conventional and molecular methods. N Engl J Med 330: 1703–1709. [DOI] [PubMed] [Google Scholar]
- Smith NH, Gordon SV, de la Rua-Domenech R, Clifton-Hadley RS, Hewinson RG 2006. Bottlenecks and broomsticks: The molecular evolution of Mycobacterium bovis. Nat Rev Microbiol 4: 670–681. [DOI] [PubMed] [Google Scholar]
- Smith NH, Hewinson RG, Kremer K, Brosch R, Gordon SV 2009. Myths and misconceptions: The origin and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 7: 537–544. [DOI] [PubMed] [Google Scholar]
- Smittipat N, Palittapongarnpim P 2000. Identification of possible loci of variable number of tandem repeats in Mycobacterium tuberculosis. Tuber Lung Dis 80: 69–74. [DOI] [PubMed] [Google Scholar]
- Sola C, Filliol I, Legrand E, Mokrousov I, Rastogi N 2001. Mycobacterium tuberculosis phylogeny reconstruction based on combined numerical analysis with IS1081, IS6110, VNTR, and DR-based spoligotyping suggests the existence of two new phylogeographical clades. J Mol Evol 53: 680–689. [DOI] [PubMed] [Google Scholar]
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Musser JM 1997. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci 94: 9869–9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE III, et al. 2012. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis 206: 1724–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Magdalena J, Himpens S, Locht C 1997. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol 26: 991–1003. [DOI] [PubMed] [Google Scholar]
- Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C 2000. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol 36: 762–771. [DOI] [PubMed] [Google Scholar]
- Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C 2001. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol 39: 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Warren RM, Banuls AL, Lesjean S, Van Der Spuy GD, Lewis LA, Tibayrenc M, Van Helden PD, Locht C 2003. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol Microbiol 47: 529–538. [DOI] [PubMed] [Google Scholar]
- Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, Willery E, Savine E, de Haas P, van Deutekom H, Roring S, et al. 2006. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 44: 4498–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, Majlessi L, Criscuolo A, Tap J, Pawlik A, et al. 2013. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet 45: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thye T, Niemann S, Walter K, Homolka S, Intemann CD, Chinbuah MA, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, et al. 2011. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS ONE 6: e20908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenova L, Ellison E, Harbacheuski R, Moreira AL, Kurepina N, Reed MB, Mathema B, Barry CE, 3rd, Kaplan G 2005. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis 192: 98–106. [DOI] [PubMed] [Google Scholar]
- van Embden JD, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicquel B, Hermans P, Martin C, McAdam R, Shinnick TM, et al. 1993. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J Clin Microbiol 31: 406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Soolingen D, Qian L, de Haas PE, Douglas JT, Traore H, Portaels F, Qing HZ, Enkhsaikan D, Nymadawa P, van Embden JD 1995. Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J Clin Microbiol 33: 3234–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Soolingen D, Hoogenboezem T, de Haas PE, Hermans PW, Koedam MA, Teppema KS, Brennan PJ, Besra GS, Portaels F, Top J, et al. 1997. A novel pathogenic taxon of the Mycobacterium tuberculosis complex, Canetti: Characterization of an exceptional isolate from Africa. Int J Syst Bacteriol 47: 1236–1245. [DOI] [PubMed] [Google Scholar]
- Varghese B, Supply P, Allix-Beguec C, Shoukri M, Al-Omari R, Herbawi M, Al-Hajoj S 2013. Admixed phylogenetic distribution of drug resistant Mycobacterium tuberculosis in Saudi Arabia. PLoS ONE 8: e55598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, et al. 2013a. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect Dis 13: 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TM, Monk P, Grace Smith E, Peto TE 2013b. Contact investigations for outbreaks of Mycobacterium tuberculosis: Advances through whole genome sequencing. Clin Microbiol Infect 19: 796–802. [DOI] [PubMed] [Google Scholar]
- Warren RM, Victor TC, Streicher EM, Richardson M, van der Spuy GD, Johnson R, Chihota VN, Locht C, Supply P, van Helden PD 2004. Clonal expansion of a globally disseminated lineage of Mycobacterium tuberculosis with low IS6110 copy numbers. J Clin Microbiol 42: 5774–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weniger T, Krawczyk J, Supply P, Niemann S, Harmsen D 2010. MIRU-VNTRplus: A web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res 38: W326– W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werngren J 2013. Mycobacterium tuberculosis Beijing type mutation frequency. Emerg Infect Dis 19: 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werngren J, Hoffner SE 2003. Drug-susceptible Mycobacterium tuberculosis Beijing genotype does not develop mutation-conferred resistance to rifampin at an elevated rate. J Clin Microbiol 41: 1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth T, Hildebrand F, Allix-Beguec C, Wolbeling F, Kubica T, Kremer K, van Soolingen D, Rusch-Gerdes S, Locht C, Brisse S, et al. 2008. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog 4: e1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]