

Abstract
Histones package and compact DNA by assembling into nucleosome core particles. Most histones are synthesized at S phase for rapid deposition behind replication forks. In addition, the replacement of histones deposited during S phase by variants that can be deposited independently of replication provide the most fundamental level of chromatin differentiation. Alternative mechanisms for depositing different variants can potentially establish and maintain epigenetic states. Variants have also evolved crucial roles in chromosome segregation, transcriptional regulation, DNA repair, and other processes. Investigations into the evolution, structure, and metabolism of histone variants provide a foundation for understanding the participation of chromatin in important cellular processes and in epigenetic memory.
Canonical histones can be replaced by noncanonical variants (e.g., H3 can be replaced by H3.3 at active genes). These may be critical for chromatin-based processes (e.g., transcriptional regulation) and epigenetic memory.
OVERVIEW
Histones package DNA by assembling into nucleosome core particles, whereas the double helix wraps around. Over evolutionary time, histone-fold domain proteins have diversified from archaeal ancestors into the four distinct subunits that comprise the familiar octamer of the eukaryotic nucleosome. Further diversification of histones into variants results in differentiation of chromatin that can have epigenetic consequences. Investigations into the evolution, structure, and metabolism of histone variants provides a foundation for understanding the participation of chromatin in important cellular processes and in epigenetic memory.
Most histones are synthesized at S phase for rapid deposition behind replication forks to fill in gaps resulting from the distribution of preexisting histones. In addition, the replacement of canonical S-phase histones by variants, independent of replication, can potentially differentiate chromatin. The replacement of a canonical histone by a noncanonical variant is a dynamic process that changes the composition of chromatin.
The differentiation of chromatin by a histone variant is especially conspicuous at centromeres, in which the H3 variant, CENP-A, is assembled into specialized nucleosomes that form the foundation for kinetochore assembly. A centromeric H3 (cenH3) counterpart of CENP-A is found in all eukaryotes. In plants and animals, the faithful assembly of cenH3-containing nucleosomes at centromeres does not appear to require centromeric DNA sequences, a spectacular example of epigenetic inheritance. Some cenH3s have evolved adaptively in regions that contact DNA, which suggests that centromeres compete with each other, and cenH3s and other centromere-specific DNA-binding proteins have adapted in response. This process could account for the large size and complexity of centromeres in plants and animals.
Chromatin can also be differentiated outside of centromeres by incorporation of a constitutively expressed form of H3, called H3.3, which is the substrate for replication-independent nucleosome assembly. Replacement with H3.3 occurs at active genes, a dynamic process with potential epigenetic consequences. Differences between H3 and H3.3 in their complement of covalent modifications might underlie changes in the properties of chromatin at actively transcribed loci.
Several H2A variants can also differentiate or regulate chromatin. H2A.X is defined as a variant by a four-amino-acid carboxy-terminal motif whose serine residue is the site for phosphorylation at sites of DNA double-stranded breaks. Phosphorylation of H2A.X is an early event in double-strand break repair, in which it is thought to concentrate components of the repair machinery. H2A.X phosphorylation also marks the inactive XY bivalent during mammalian spermatogenesis and is required for condensation, pairing, and fertility.
H2A.Z is a structurally diverged variant that has long presented an enigma. Studies in yeast have implicated H2A.Z in establishing transcriptional competence and in counteracting heterochromatic silencing. The biochemical complex that replaces H2A with H2A.Z in nucleosomes is an ATP-dependent nucleosome remodeler, providing the first example of a specific function for a member of this diverse class of chromatin-associated machines.
Two vertebrate-specific variants, macroH2A and H2A.B (also called H2A.Bbd), display contrasting features when packaged into nucleosomes in vitro, with macroH2A impeding and H2A.B facilitating transcription. These features are consistent with their localization patterns on the epigenetically inactivated mammalian X chromosome: macroH2A showing enrichment and H2A.B showing depletion.
The emerging view from these studies is that histone variants and the processes that deposit them into nucleosomes provide a primary differentiation of chromatin that might serve as the basis for epigenetic processes.
1. DNA IS PACKAGED BY ARCHITECTURAL PROTEINS IN ALL ORGANISMS
The enormous length of the DNA double helix relative to the size of the chromosome that contains it requires tight packaging, and architectural proteins have evolved for this purpose. The first level of packaging shortens the double helix and protects it from damage while still allowing DNA polymerase to gain full access to each base pair every cell cycle. In addition, these architectural proteins facilitate higher-order folding to further reduce the length of a chromosome. Perhaps because of stringent requirements for packaging DNA, only two structural classes of architectural proteins are found in nearly all cellular life-forms (Talbert and Henikoff 2010): HU proteins that package bacterial DNA, and histones that package eukaryotic DNA. Archaeal DNA is packaged by either HU proteins or histones.
Histones package DNA into nucleosome particles, and this architectural role can account for the fact that histones comprise half of the mass of a eukaryotic chromosome. However, histones have also been found to play diverse roles in gene expression, chromosome segregation, DNA repair, and other basic chromosomal processes in eukaryotes. Specific requirements of these chromosomal processes have led to the evolution of distinct histone variants. The incorporation of a variant histone into a nucleosome represents a potentially profound alteration of chromatin. Indeed, some histone variants are deposited by distinct nucleosome assembly complexes, which suggests that chromatin is diversified, at least in part, by the incorporation and replacement of histone variants.
The four core histones, H2A, H2B, H3, and H4, differ with respect to their propensity to diversify into variants. For example, humans have only one H4 isotype but several H2A paralogs with different properties and functions. Evidently, the different positions of the core histones within the nucleosome particle have subjected them to different evolutionary forces, leading to important diversifications of H2A and H3 but not to H2B and H4 (Fig. 1). The availability of genomic sequences from a wide variety of eukaryotes allows us to conclude that these diversifications have occurred at various times during eukaryotic evolution. However, the evident diversification of an ancestral histone-fold protein into the familiar four core histones must have occurred early in the evolution of the eukaryotic nucleus or perhaps before. By considering these ancient events, we gain insight into the forces that have resulted in subsequent diversification into present-day variants.
Figure 1.

Histone variants. Protein domain structure for the core histones (H3, H4, H2A, and H2B), linker histone H1, and variants of histones H3 and H2A. The histone-fold domain (HFD) is where histone dimerization occurs. Regions of sequence variation in histone variants are indicated in red. WHD, winged-helix domain.
2. EUKARYOTIC CORE HISTONES EVOLVED FROM ARCHAEAL HISTONES
The eukaryotic nucleosome is a complex structure, consisting of an octamer of four core histones wrapped nearly twice by DNA, with histone tails and linker histones mediating a variety of packaging interactions outside the core particle (Arents et al. 1991; Wolffe 1992; Luger et al. 1997). Archaeal nucleosomes are much simpler, and it is evident that they resemble the ancestral particle from which eukaryotic nucleosomes evolved (Malik and Henikoff 2003). An archaeal nucleosome consists of histone-fold domain proteins that lack tails and form a tetrameric particle that is wrapped only once by DNA. The genomes of all ancient archaeal lineages encode histones (Fig. 2), which implies that the eukaryotic nucleosome evolved from an as-yet unidentified archaeal ancestor. The kinship between archaeal and eukaryotic nucleosomes can be seen by comparing their structures: The backbone of the archeal tetramer nearly superimposes over that of the (H3-H4)2 tetramer. When archaeal nucleosomes are reconstituted to form chromatin, the resulting fiber behaves similarly to “tetrasomes” of (H3-H4)2, and when mapped in vivo, they show phasing downstream from transcriptional start sites analogous to what is seen for eukaryotic nucleosomes (Ammar et al. 2012). Therefore, it is thought that eukaryotic nucleosomes evolved from an archaeal ancestor by doubling the number of subunits to allow for a second DNA wrap, and by acquisition of histone tails. In addition, DNA wraps into a right-handed superhelix around archaeal cores, but into a left-handed superhelix around eukaryotic cores.
Figure 2.
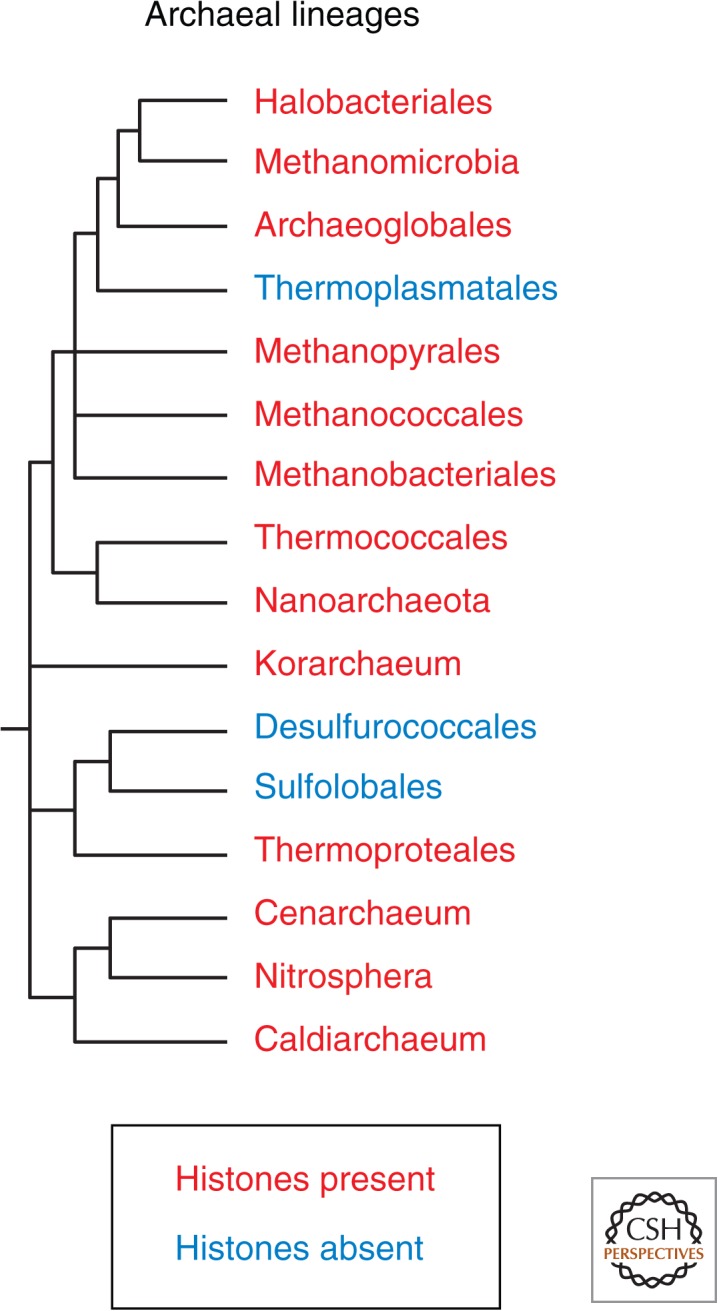
Archaeal cladogram indicating the presence of histones in all ancestral clades. Losses are attributable to horizontal transfer of HU proteins from bacteria. (Modified, with permission, from Brochier-Armanet et al. 2011, © Elsevier.)
Further insight into the origin of the eukaryotic nucleosomes comes from examination of the subunit structures of archaeal nucleosomes. Whereas most archaeal histones are undifferentiated monomers or are differentiated into structurally interchangeable variants that come together to form a tetramer, some are head-to-tail dimeric fusions that come together to form a dimer of fused dimers. When two of these fused dimers assemble into a nucleosome particle, each member of the fused pair is in a structurally distinguishable position. By occupying distinct positions in the particle, each member of the archaeal fused dimer will evolve independently, allowing it to adapt to a single position in the nucleosome particle. In contrast, monomers that occupy interchangeable positions are not free to adapt to particular positions. Indeed, the two members of archaeal dimers have diverged from one another in both independent lineages in which they are found. This process provides a possible scenario for the differentiation of an ancestral histone-fold domain protein into four distinct subunits that occupy distinct positions in the eukaryotic nucleosome. Like their presumed archaeal ancestors, eukaryotic histones form dimers, where H2A dimerizes with H2B, and H3 with H4 (which also stably tetramerizes in solution). The structural backbone of an archaeal histone dimer superimposes with those of H2A-H2B and H3-H4 at 2-Å resolution, with the first member of the dimeric repeat superimposing on H2A or H3 and the second member superimposing on H2B or H4. So, although all four eukaryotic histones lack significant sequence similarity to one another and to archaeal histones, the striking structural superposition of dimeric units suggests that eukaryotic histones evolved and differentiated from simpler archaeal ancestors.
The asymmetry of H2A-H2B and H3-H4 dimers, which appears to have originated from archaeal tandem dimers, could have led the way to subsequent diversification of eukaryotic histone variants. Both H2A and H3 correspond to the first member of archaeal tandem histone dimers and both have subsequently diversified multiple times in eukaryotic evolution. In contrast, H2B and H4 correspond to the second member and have shown little (H2B) or no (H4) functional diversification. Both H3 and H2A make homodimeric contacts in the octamer (Fig. 3), whereas H4 and H2B only contact other histones. As a result, changes in the residues involved in homodimerization of either H2A or H3 can potentially resist formation of mixed octamers, allowing nucleosomes containing an H2A or H3 variant to evolve independently of parental nucleosomes. In general, structural features that facilitated independent evolution of subunits may have been prerequisites for diversification of nucleosome particles.
Figure 3.

Location of histones H3 (blue) and H2A (brown) in the nucleosome core particle. Differences between variants are highlighted in yellow. (Reprinted, with permission, from Henikoff and Ahmad 2005.)
Although we can rationalize the descent of the eukaryotic core histones from archaeal tandem dimers, other basic questions remain. Where did histone tails come from? Did (H3-H4)2 tetrasomes evolve before acquiring flanking H2A-H2B dimers, or perhaps the four core histones evolved first as (H2A-H2B-H4-H3) “hemisomes” before doubling to form the octamer, or was there some other evolutionary progression from tetramer to octamer? Did these events occur before, during, or after the evolution of the eukaryotic nucleus? Did the emergence of octameric nucleosomes with two DNA wraps allow for the tight packaging of mitotic chromosomes, a eukaryotic-specific invention? Perhaps the sequences of more archaea or of primitive eukaryotes will reveal intermediate forms that can answer these questions.
3. BULK HISTONES ARE DEPOSITED AFTER DNA REPLICATION
The packaging of essentially all DNA in a eukaryotic cell into nucleosomes requires that chromatin is duplicated when DNA replicates. Thus, canonical histones are produced during the DNA synthesis (S) phase of the cell cycle. S-phase coupling of histone synthesis to DNA synthesis is under tight cell-cycle control (Marzluff and Duronio 2002). This is especially evident in animals, in which special processing of histone transcripts by the U7 small nuclear ribonuclear protein complex, and messenger RNA (mRNA) stabilization by the stem-loop-binding protein (SLBP) contributes to the tight coordination of histone synthesis with DNA replication. The need for rapid and massive production of histones during S phase is very likely responsible for the fact that replication-coupled (RC) histones in animals are encoded in clusters that comprise many histone genes. For example, there are 14 H4 genes in the human genome, most of which are found in two major clusters, where these H4 genes are interspersed with other RC histone genes (Marzluff et al. 2002). In animals, RC histones are recognizable by the presence of a 26-bp 3′ sequence that forms a stem-loop for recognition by SLBP when transcribed into histone mRNA. Canonical plant histones are also encoded by multiple genes and are deposited during S phase, although plant histone transcripts are polyadenylated and there does not appear to be a counterpart to SLBP.
To the extent that epigenetic inheritance results from inheritance of a chromatin “state,” the process of RC nucleosome assembly has been of intense interest. The biochemistry of the process was elucidated with the development of in vitro systems that could assemble nucleosomes onto replicating DNA. These studies revealed that a three-subunit complex, CAF-1 (chromatin assembly factor 1), acts as a histone chaperone that facilitates the incorporation of H3-H4 as a first step in nucleosome assembly (reviewed in Loyola and Almouzni 2004). CAF-1 was shown to interact with the replication processivity clamp, PCNA, which implies that DNA replication and RC assembly occur in close proximity (Fig. 4). Work in budding yeast revealed that none of the subunits of complexes involved in RC assembly in vitro is essential for growth, suggesting that in vivo, there are redundant mechanisms for RC assembly. The fact that much of yeast chromatin is assembled in a replication-independent (RI) manner (Altheim and Schultz 1999) provides a rationale for this evident redundancy. As we shall see, histone variants are typically deposited by RI nucleosome assembly.
Figure 4.

Distribution of old and new nucleosomes at a replication fork. Old nucleosomes (gray disks) are randomly distributed behind the replication fork and new nucleosomes (cyan disks) are deposited in the gaps. CAF-1-mediated nucleosome assembly is depicted on the leading and lagging strand in magnification. DNA polymerase (green); replication processivity clamp, PCNA (gray ring); histone H3-H4 tetramers (cyan); newly synthesized DNA (red lines).
RC assembly is not completely redundant in budding yeast. An intriguing finding is that absence of the large CAF-1 subunit leads to loss of epigenetic silencing at telomeres (Loyola and Almouzni 2004). Moreover, in human cells, depletion of CAF-1 results in the deposition of H3.3 at sites of DNA replication (Ray-Gallet et al. 2011). The connection between RC assembly and epigenetic silencing has been extended to Arabidopsis, in which loss of CAF-1 subunits results in a variety of defects attributable to loss of epigenetic memory (Kaya et al. 2001). Although the mechanistic basis for these observations is unknown, it seems clear that the proper deposition of new nucleosomes behind the replication fork is important for maintaining an epigenetically silenced state. An example of the importance of replication-coupled assembly for maintaining a developmental state is the observation that the identity of one of two sister neurons in Caenorhabditis elegans is disrupted by mutating either the H3-H3 dimerization interface or CAF-1 (Nakano et al. 2011).
A prerequisite for epigenetic inheritance of a nucleosome state is that preexisting nucleosomes must be distributed to daughter chromatids following replication. Indeed this is the case; classical studies have shown that old nucleosomes are inherited intact and evidently at random to daughter chromatids (Annunziato 2005). However, more recent studies of particular cell types and particular loci have challenged this dogma. Germline stem cells in Drosophila male early embryos show asymmetric inheritance of RC H3, but not RI H3.3, whereby old nucleosomes remain in the stem cell and new nucleosomes segregate to the differentiating daughter cell (Tran et al. 2012). Moreover, although RC H3 was not found to split at replication (Xu et al. 2010), the small amount of RI H3.3 that splits was shown to be enriched at active human genes and cell-type-specific enhancers (Huang et al. 2013). Taken together, these two findings suggest that RC nucleosome assembly is profoundly involved in initiating developmental decisions and RI assembly in epigenetic inheritance, resuscitating ideas proposed long before these pathways were elucidated (Weintraub et al. 1976). More work is needed to test these exciting possibilities and to explore the importance of nucleosome deposition pathways in establishing and maintaining epigenetic states (Jenuwein 2001; Henikoff and Ahmad 2005).
4. VARIANT HISTONES ARE DEPOSITED THROUGHOUT THE CELL CYCLE
As we have seen, core histones can be classified based on their ancestral sequence and position in the nucleosome. Linker histones are characterized by a winged-helix domain, rather than a histone-fold domain, and bind to the linker DNA that separates nucleosomes (Wolffe 1992). Although minor variants of these canonical histones exist, they appear to be interchangeable with the major form. For example, mammalian H3.1 and H3.2 differ by a single amino acid that is not known to impart different functional properties to the two isoforms. The existence of multiple genes that produce large amounts of canonical histones for S-phase deposition is typical of eukaryotic genomes. The near ubiquity and overwhelming abundance of canonical S-phase histones has resulted in relatively little attention being paid to histone variants until recently.
The renaissance of interest in histone variants came in part from the realization that they differ from canonical S-phase histones in ways that can lead to profound differentiation of chromatin. One way that they differ is in their mode of incorporation into chromatin. RC assembly incorporates new nucleosomes into gaps between old nucleosomes genome-wide, whereas RI assembly involves local replacement of an existing nucleosome or subunit (Marzluff et al. 2002). RI assembly therefore has the potential of switching a chromatin state by replacing a canonical histone with a variant. Replacing one histone with another also could erase or alter the pattern of posttranslational modifications. Therefore, RI assembly can potentially reset epigenetic states that are thought to be mediated by histones and their modifications. Recent progress in studying histone variants and the processes by which they are deposited has led to new insights into the basis for epigenetic inheritance and remodeling. Below we discuss features of particular histone variants that contribute to chromatin differentiation and might be involved in propagating epigenetic information.
5. CENTROMERES ARE IDENTIFIED BY A SPECIAL H3 VARIANT
A defining feature of the eukaryotic chromosome is the centromere, which is the site of attachment of spindle microtubules at mitosis. The first centromeres to be described in molecular detail were those of budding yeast (Saccharomyces cerevisiae), in which a 125-bp sequence is necessary and sufficient for centromere formation (Amor et al. 2004b). However, centromeres of plants and animals are very different, typically consisting of megabase arrays of short tandem repeats. Unlike the situation for budding yeast, the role of DNA sequence at these complex centromeres is uncertain because fully functional human neocentromeres are known to form spontaneously at ectopic sites that entirely lack sequences resembling centromeric repeats (Fig. 5A). These and other observations argue against a direct role of DNA sequence in determining the location of centromeres (see Allshire and Ekwall 2014).
Figure 5.

cenH3s at centromeres of eukaryotes. (A) Human neocentromeres (indicated by an arrow) lack centromeric α-satellite DNA, but have CENP-A and heterochromatin. Anti-CENP-A staining in green and Anti-CENP-B staining in red (which marks α-satellite DNA) identifies a Chromosome 4 neocentromere that lacks α-satellite (main panel). This Chromosome 4 is otherwise normal, having been transmitted for at least three meiotic generations in normal individuals. Inset shows anti-HP1 staining, which indicates that despite the lack of satellite DNA, heterochromatin forms around active neocentromeres (indicated by arrow). (Reprinted, with permission, from Amor et al. 2004a, © National Academy of Sciences.) (B) Drosophila melanogaster anti-cenH3 antibody (red) stains centromeres in metaphase chromosomes and throughout interphase. (Image courtesy of Suso Platero.) (C) C. elegans anti-cenH3 antibody (green) stains the end-to-end holocentromeres of prophase chromosomes (red). (Image courtesy of Landon Moore.)
A key insight into the basis for centromere identity and inheritance came from the identification of a histone H3 variant, CENP-A which was found to localize specifically to centromeres and be incorporated into nucleosomal particles in place of H3 itself (Palmer et al. 1991). Remarkably, CENP-A remains associated with centromeres during the transition from histones to protamines during spermatogenesis, when essentially all other histones are lost (Palmer et al. 1990). This early observation in the study of CENP-A suggested that CENP-A contributes to centromere identity of the male genome. The generality of this insight was not fully appreciated until it was realized that CENP-A is a much better marker for centromeres than DNA sequence (Fig. 5) (Amor et al. 2004b), and that counterparts of CENP-A (cenH3s) can be found in the genomes of all eukaryotes (Talbert and Henikoff 2010). So, although budding yeast centromeres are determined by a 125-bp consensus sequence, this is also the site of a centromeric nucleosome that contains the cenH3 variant. In fission yeast (Schizosaccharomyces pombe), an array of cenH3-containing nucleosomes occupies the central core region of the centromere flanked by H3-containing nucleosomes that display heterochromatic features (see Fig. 3 of Allshire and Ekwall 2014; Amor et al. 2004b). In flies and vertebrates, cenH3s are present in arrays that alternate with H3-containing arrays that display a unique pattern of histone modifications (Sullivan and Karpen 2004). Alternation can account for the fact that centromeres occupy only the outside edge of the centromeric constriction of metaphase chromosomes. This is consistent with the observation that in worm “holokinetic” chromosomes, microtubules attach throughout the length of each anaphase chromosome, and cenH3 occupies the leading edge all along its length (Fig. 5C) (Malik and Henikoff 2003). Indeed a unique cenH3 variant is found to precisely mark the centromere in nearly all eukaryotes (Fig. 6A). This apparent ubiquity, and the presence of centromeres to perform mitosis in all eukaryotes, raises the possibility that the first canonical H3 evolved from a cenH3.
Figure 6.

Histone variant phylogenies. Histone sequences from selected species were multiply aligned and neighbor-joining trees were produced using the EBI server (http://www.ebi.ac.uk/Tools/phylogeny). (A) Histone H3s. (B) Histone H2As. Note that there are no clear phylogenetic distinctions between RC H3 and RI H3.3, and between RC H2A and RI H2A.X. (C) H1 variants from diverse eukaryotes show a “star” phylogeny, which suggests that they are functionally interchangeable. (C, Modified from Talbert et al. 2012.)
Genetic experiments in a variety of eukaryotes have confirmed the essentiality of cenH3 for formation of the kinetochore and for chromosome segregation (Amor et al. 2004b). Because they remain in place throughout the cell cycle, cenH3-containing nucleosomes form the foundation for assembly of other kinetochore proteins during mitosis and meiosis (see Allshire and Ekwall 2014). An outstanding question in chromosome research is just how these proteins interact to provide a linkage between the centromere and spindle microtubules that can hold up to the strong pulling forces exerted on kinetochores at anaphase. Several dozen kinetochore-specific proteins have been identified in yeast (for more detail, see Allshire and Ekwall 2014), although exactly how they interact with cenH3-containing nucleosomes and other foundation proteins, such as CENP-C, remains unclear.
The evolution of cenH3s is unlike that of any other histone class. Whereas histone H3 is almost invariant in sequence, which reflects extraordinarily strong purifying selection on every residue, cenH3s are evolving rapidly, especially in plant and animal lineages (Talbert and Henikoff 2010). This is most evident from the amino-terminal tails, which differ in length and sequence to such an extent that they cannot be aligned between the cenH3s of different taxonomic groups. Even the histone-fold domain of cenH3 is evolving orders of magnitude faster than that of H3. What is the reason for this striking evolutionary difference between an H3 that functions at centromeres and an H3 that functions everywhere else?
Rapidly evolving regions of Drosophila and Arabidopsis cenH3 genes display an excess of replacement nucleotide substitutions over what would be expected from the rate of synonymous substitutions (Malik and Henikoff 2009). This excess is a hallmark of adaptive evolution. Adaptive evolution in plants and animals is also seen for another major centromere foundation protein, CENP-C (Malik and Henikoff 2009). Although adaptive evolution is well documented for genes involved in genetic conflicts such as arms races between hosts and parasites, these are the only known essential single-copy genes that are adaptively evolving in any organism. In the case of cenH3 and CENP-C, the regions of adaptive evolution correspond to regions of DNA binding and targeting. This suggests that the major centromere-binding proteins are adapting to the evolving centromeric DNA, thus allowing centromeric chromatin to interact with the conserved kinetochore machinery that connects the centromere to spindle microtubules. It has been proposed that centromeres compete during female meiosis to be included in the egg nucleus rather than being lost as polar bodies (Malik and Henikoff 2009). An arms race would develop leading to expansion of centromeres, probably by unequal crossing-over between sister chromatids. Host suppression of this meiotic drive process by cenH3 and CENP-C would lead to an excess of replacement changes in regions that interact with DNA. Organisms in which there is no opportunity for centromeres to compete, such as budding yeast, would not undergo centromere drive, and this might account for the fact that they have small centromeres, and their cenH3 and CENP-C proteins are under strong purifying selection.
Thus, we see that a special region of the genome, the centromere, is distinguished by a single histone variant class, whose sequences reveal remnants of an arms race that may have led to the extraordinary complexity of centromeres. The RI assembly process that targets new cenH3-containing nucleosomes to centromeres every cell cycle has been elucidated by the discovery and detailed characterization of the related Scm3 (yeast) and HJURP (mammalian) cenH3-specific chaperones (Stoler et al. 2007; Dunleavy et al. 2009; Foltz et al. 2009). Detailed biochemical and structural characterization of Scm3/HJURP complexes (Shuaib et al. 2010; Cho and Harrison 2011; Hu et al. 2011) indicates a role in CenH3 nucleosome assembly that parallels that of other nucleosome assembly complexes, discussed further in Almouzni and Cedar (2014). Centromeric nucleosomes show a remarkable lack of sequence specificity in that they not only can faithfully localize to neocentromeres that are completely unlike native centromeres (Fig. 5A), but also the yeast homolog Cse4 can functionally replace human CENP-A (Wieland et al. 2004). It is extraordinary that our centromeres have remained in the same positions for tens of millions of years without any evident sequence determinants involved in the process that maintains them. To the extent that epigenetics refers to inheritance that does not depend on DNA sequence, the inheritance of centromeres on a geological timescale is the most extreme form imaginable. Yet, we are still seeking a mechanism to explain how they have maintained themselves for even a single cell cycle.
There is now general agreement in the centromere field that the cenH3 nucleosome is the key to understanding the epigenetic inheritance of centromeres (Black and Cleveland 2011; Henikoff and Furuyama 2012). It is not only necessary for recruitment of the other structural components of the centromere, in some experimental systems it is also sufficient (Guse et al. 2011; Mendiburo et al. 2011). However, its molecular structure has been the subject of controversy for several years. In vivo evidence from flies, humans, and yeast is most consistent with a right-handed hemisome (Henikoff and Furuyama 2012), whereas several groups have shown that reconstitution of cenH3-containing particles generally results in the formation of a partially unwrapped left-handed octameric nucleosome (Black and Cleveland 2011). Indeed, since 2007, the authors of this article coauthored the studies that provided the first evidence for nonoctameric cenH3 particles (Dalal et al. 2007; Mizuguchi et al. 2007), but the composition and structure of these proposed particles were completely different! In light of the continuing controversy, we leave a final resolution of this important issue to the future.
6. THE REPLACEMENT HISTONE VARIANT H3.3 IS FOUND AT ACTIVE CHROMATIN
Like centromeres, transcriptionally active chromatin is thought to be maintained epigenetically and is enriched in an H3 variant, H3.3, which is the substrate for RI deposition (Filipescu et al. 2013). H3.3 is very similar in sequence to the canonical forms of H3, differing by only four amino acids. With so few differences, it might have been assumed that these two forms are interchangeable. However, H3.3 is deposited exclusively by RI nucleosome assembly, whereas H3 is deposited only at replication foci in an RC manner. This difference between the two variants is encoded in the protein itself, with three of the four differences between H3 and H3.3 evidently involved in preventing H3 from being deposited by an RI pathway (illustrated in the α-helix 2 of Fig. 3). Purification of soluble nucleosome assembly complexes confirmed that these two forms participate in distinct assembly processes: H3.1 copurifies with CAF-1 for RC assembly, and H3.3 copurifies with other components, including the HirA and Daxx histone chaperones, and participates in RI assembly.
Although the four-amino-acid difference might seem practically insignificant, when one considers that humans, flies, and clams have precisely the same H3.3 sequence, these differences from H3 stand out. Phylogenetic analysis reveals that the H3/H3.3 pair evolved at least four separate times during eukaryotic evolution, in plants, animals/fungi, ciliates, and apicomplexans (Fig. 6A) (Talbert and Henikoff 2010). Despite having a separate origin from animals and fungi, the animal H3/H3.3 pair and the pair from plants (called H3.1 [RC] and H3.2 [RI]—to avoid confusion, we will refer to all RC isoforms as H3 and all RI isoforms as H3.3) are strikingly similar. The same cluster of amino acids (positions 87–90) that prevents RI deposition of H3 in Drosophila are found to differ in plants, and the remaining difference in animals (position 31 is Ala for H3 and either Ser or Thr for H3.3) is also found in plants. Fungi are especially interesting. Ancestrally, they have both H3 and H3.3; however, ascomycetes, which include yeasts and molds, have lost the H3 form. Thus, the obligate RC form of histone 3 that has received the most attention in animals is not even present in yeast.
Studies of H3.3 in bulk chromatin showed that it is enriched in transcriptionally active chromatin fractions (Filipescu et al. 2013). However, various factors contributed to the obscurity of this potential “mark” of active chromatin during a time of great excitement in the chromatin field when it was realized that histone modifications can distinguish active from silent chromatin. For one thing, no antibodies were available that could effectively distinguish H3 from H3.3 in chromatin (positions 87–90 are blocked by the DNA gyres in the nucleosome), whereas excellent antibodies against many different posttranslational modifications were readily available. Also, the seemingly slight sequence differences between H3 and H3.3 did not suggest any fundamental distinctions in chromatin, whereas histone modifications were mostly on tail lysines that were known to affect chromatin interactions or to bind chromatin-associated proteins. This perception that the two histone 3 forms should be interchangeable was confirmed by the finding in Tetrahymena and Drosophila that the S-phase form can in general substitute for its replacement counterpart. Finally, the influential “histone code” hypothesis envisioned nucleosomes as fixed targets of modification enzymes during chromatin differentiation (Jenuwein and Allis 2001). However, it has become increasingly evident that chromatin is highly dynamic, and even heterochromatin-associated proteins bind with residence times of a minute or less (Phair et al. 2004). It appears that the chromatin of actively transcribed genes is in constant flux, characterized by continual histone replacement (Dion et al. 2007). The three core amino acid differences that distinguish H3 and H3.3 make H3.3-H4 dimers the substrate for RI assembly, and RI assembly itself profoundly changes chromatin. As a result of this process, actively transcribed regions become marked by H3.3 (Fig. 7), and evidence for this process comes from the observation of RI replacement of H3 methylated on lysine-9 (H3K9me) with tagged H3.3 at RNA polymerase I and II (RNA Pol I and II) transcribed loci (Schwartz and Ahmad 2005).
Figure 7.

H3.3 preferentially localizes to actively transcribed regions of Drosophila polytene chromosomes. DAPI staining (red) shows the DNA banding pattern (left), and H3.3-GFP (green) localizes to interbands (middle), which are sites of RNA Pol II localization. The merge (Schwartz and Ahmad 2005) is shown on the right. In each image, the shorter arrow points to a decondensed interband that is enriched in H3.3, and the longer arrow points to a condensed band that lacks H3.3.
The dynamic nature of chromatin at active loci results in the erasure of preexisting histone modifications, and yet histone modification states persist through multiple rounds of cell division. Therefore, the enzymes responsible for modifying histones must be targeted to their sites of action. For histone modifications that are typically associated with transcriptionally active chromatin, this is achieved by association with the carboxy-terminal domain (CTD) of RNA Pol II, a tandem array of YSPTSPS heptamers. For example, the Set1 H3K4 methyltransferase associates with the CTD when it is heavily phosphorylated on Serine-5 during transcriptional initiation, and so encounters its substrate primarily near initiation sites. Similarly, the Set2 H3K36 methyltransferase associates with the CTD when it becomes heavily phosphorylated on Serine-2 during transcriptional elongation and encounters its substrate within gene bodies. When a nucleosome is evicted and replaced with unmodified histones during transcriptional elongation, the newly deposited H3.3 is therefore modified appropriately.
It appears that an analogous process maintains histone modifications that are typically associated with silent chromatin. Nucleosomes that are lost at sites of short-period tandem repeats, such as occur at mammalian telomeres and pericentric regions, are replaced by the Daxx H3.3-specific histone chaperone complex and the ATRX ATP-dependent nucleosome remodeling protein (Fig. 8) (Drane et al. 2010; Goldberg et al. 2010). ATRX has a bifunctional histone tail recognition domain with high affinity for an H3 tail that is both unmethylated at K4 and trimethylated at K9 (Eustermann et al. 2011), and so likely is recruited to telomeric sites that are enriched for H3K9me and lack H3K4me. Telomeres are also enriched for heterochromatin-associated protein 1 (HP1), which recruits the Su(var)3-9 H3K9 methyltransferase, and binds its H3K9 methylated product (Hines et al. 2009), and so an enzyme that methylates the tails of replacement H3.3 at telomeres is present at a high local concentration in which new H3.3 is incorporated. This implies that all of the components necessary for maintaining H3K9 methylation are present at telomeres: the enzyme that performs the modification, the modification-specific-binding module on the machine that uses ATP to provide energy for the replacement process, and the fresh unmodified H3.3 substrate that becomes incorporated into the new nucleosome (Fig. 8). It seems likely that a similar process occurs at other sites of short-period tandem repeats in DNA where nucleosomes frequently turn over, insofar as ATRX is also abundant in mammals at C + G-rich sites (Law et al. 2010), which are found at most promoters, and the Drosophila ortholog of ATRX, XNP, is abundantly present at a single site of a (GATA)n repeat where H3.3 is actively incorporated (Schneiderman et al. 2009). The incorporation of H3.3 nucleosomes at sites of telomeric heterochromatin belies the common notion that H3.3 is a “mark” of active chromatin. As the general substrate for replacement of nucleosomes wherever they are lost, the finding that H3.3 is mostly incorporated at active genes rather implies that these are sites where nucleosome turnover is most intense. This generic replacement function of H3.3 has important relevance to human disease as described in Section 15.
Figure 8.

Model for maintenance of histone modifications by the concerted action of multiple chromatin regulators via RI replacement with H3.3. We address the question of how a histone modification can be inherited when a nucleosome is lost and replaced. (A) The Suv39h H3K9 methyltransferase (an ortholog of fly Su(var)3-9) is recruited by HP-1 protein, which binds specifically to methylated H3K9. To perpetuate this mark when the nucleosome turns over, we speculate that the ATRX ATPase is recruited to the site via its ATRX-DNMT3-DNMT3L (ADD) domain, which binds with high specificity to methylated H3K9 on tails that entirely lack H3K4 methylation (because there are no H3K4 methyltransferases in this region of the genome). (B) ATRX provides the energy of ATP and works together with the H3.3-specific DAXX histone chaperone complex to incorporate the new nucleosome (Goldberg et al. 2010), or half-nucleosome in the case of partial eviction (Xu et al. 2010). The high local concentration of Suv39h results in a new nucleosome with the same H3K9 methylation as the nucleosome that was lost.
7. H3.3 FUNCTIONS IN THE GERMLINE
When cells exit the cell cycle and differentiate, they no longer produce or incorporate S-phase histones, and H3.3 accumulates as a result. For example, H3.3 accumulates in rat brains to a level of 87% of the histone 3 by the time that rats are 400 days old (Pina and Suau 1987). This classical observation suggested that replacement by H3.3 has no functional significance except to prevent holes in the nucleosomal landscape. Consistent with this view, H3.3 has been found to be nonessential for Drosophila development, as flies lacking both H3.3 genes develop normally to the pupal stage, with occasional adult escapers that die shortly after eclosion but show no specific morphological defects (Hodl and Basler 2009; Sakai et al. 2009). Moreover, H3.3 can functionally substitute for H3 and allow at least some developmental decisions to be made in Drosophila embryos, further suggesting that RC and RI substrates are largely interchangeable (Hodl and Basler 2012). It appears that histone replacement in H3.3 null proliferating cells can be accomplished by incorporating H3 nucleosomes using the RI pathway. However, there is no germline development in flies lacking key RI pathway components, such as HirA (e.g., Fig. 9). Females that lack the ChD1 ATP-dependent nucleosome remodeler protein are also sterile, evidently because ChD1 is required in the zygote for sperm nuclei decondensation and the replacement of protamines by maternally encoded RI histones (Orsi et al. 2009). This essential germline function of the RI pathway is conserved in mammals, in which H3.3 is required for remodeling of both maternal and paternal gametes (Santenard et al. 2010; Akiyama et al. 2011). Similar RI processes have been documented in both C. elegans and Arabidopsis, in which maternal H3.3 is incorporated into the paternal genome of the zygote (Ooi et al. 2006; Ingouff et al. 2007). Therefore, germline remodeling via RI assembly of H3.3 nucleosomes is a universal process that has evolved in both animals and plants, most likely to “reset” the chromatin landscape to a totipotent state.
Figure 9.

Models for RI replacement or exchange. A large molecular machine (either the SWR1 complex or RNA polymerase) partially or completely unravels a nucleosome during transit. The result is either retention of heterodimeric subunits, such as the FACT-facilitated transfer of H2A-H2B from in front of RNA polymerase to behind (Formosa et al. 2002; Belotserkovskaya et al. 2003) or loss of a heterodimer. In the latter case, chromatin repair replaces the lost heterodimer with either H3.3-H4 (left) or H2A.Z-H2B (right).
Remodeling events in the zygote, where RI assembly of H3.3 nucleosomes plays a key role, is analogous to nuclear reprogramming, which can be artificially induced in Xenopus eggs, mouse embryonic stem cells, and induced pleuripotent cells. In Xenopus, transfer of an embryonic nucleus into an enucleated egg can result in the production of mostly normal embryos that nevertheless sometimes (mis)express genes that were active in the differentiated donor nucleus (Ng and Gurdon 2008). The lack of observable gene expression during the intervening 12 embryonic divisions implies that the persistence of an epigenetic mark maintains the memory of prior gene activity. Overexpression of H3.3 in developing embryos improved epigenetic memory and mutation of H3.3K4 to glutamine erased memory of the active state, whereas no effect was seen using a general DNA methyltransferase inhibitor. Further evidence for the importance of H3.3 in nuclear reprogramming comes from the finding that the shift from somatic to oocyte transcription does not require replication, but does require transcription and the H3.3-specific chaperone, HirA (Jullien et al. 2012). It is attractive to think of H3.3 and its histone partners as general mediators of totipotency both in the zygote and during nuclear reprogramming, insofar as histones are likely to be most accessible to the enzymes that posttranslationally modify their tails during nucleosome assembly (e.g., Fig. 8).
8. PHOSPHORYLATION OF H2A.X FUNCTIONS IN DNA DOUBLE-STRAND BREAK REPAIR
The H2A histones also comprise a family of distinct variants found throughout eukaryotes (Fig. 6B). The H2A.X variant is defined by the presence of a carboxy-terminal amino acid sequence motif, SQ(E or D)Ø, in which Ø indicates a hydrophobic amino acid. The serine in this sequence motif is the site of phosphorylation, producing a modified protein designated “γH2A.X.” The dynamic nature of chromatin, and H2A.X phosphorylation, is especially evident when double-strand breaks (DSBs) occur in DNA (Morrison and Shen 2005). The lethality of even a single double-stranded (ds) break requires immediate action to repair the lesion and restore the continuity of the double helix. The detection of a ds break normally occurs within a minute or so of its formation and this, in turn, triggers the rapid phosphorylation of H2A.X in the immediate vicinity of a break site. This phosphorylation is performed by members of the phosphoinositol 3-kinase-like kinase family. Following this initial event, H2A.X phosphorylation then spreads quickly along the chromosome marking a relatively large chromatin domain surrounding the break. Finally, the ds break is eventually repaired by either homologous recombination or nonhomologous end-joining and the phosphorylation mark is removed.
Phosphorylation of H2A.X is not essential for detection or repair of DSBs because deletion of the gene or mutation of the target serine residue does not abolish repair. However, H2A.X is not just a marker of damage because such mutants have reduced efficiency of repair and are hypersensitive to radiation damage and genotoxic agents. Currently, H2A.X is thought to function in ds break repair in at least two ways. First, it may help recruit or retain proteins required for repair at the site of the break (Morrison and Shen 2005). Second, it may stabilize the chromosome surrounding the broken ends through the recruitment of cohesin, the protein complex responsible for keeping sister chromatids together (Lowndes and Toh 2005).
The evolution of H2A.X is unlike that of other histone variants. Although a gene for H2A.X is found in nearly all eukaryotes, it has had multiple relatively recent divergences from H2A (Fig. 6B) (Malik and Henikoff 2003; Talbert and Henikoff 2010). For example, the version of H2A.X found in Drosophila is different from that found in another dipteran insect, Anopheles. Some organisms, such as yeasts, have an H2A.X but lack an H2A, consistent with the possibility that most present-day canonical H2As have evolved from ancestral H2A.Xs (Talbert and Henikoff 2010). Presumably, the ability to evolve either a canonical H2A from an H2A.X or vice versa is a consequence of the simplicity of the SQE motif. Depending on selective constraints, the loss or gain of such a simple motif at the carboxyl terminus of a protein might occur repeatedly over evolutionary time. Occasional loss of an existing H2A.X with a newly minted version might be fueled by the need for H2A.X to be very uniformly distributed because DSBs can occur anywhere in the genome. If mutations occur in an existing H2A.X gene that reduce its similarity to the canonical H2A in such a way that its assembly becomes less efficient or uniform, then there will be strong selection to replace it with a version that is more similar to canonical H2A. This rationale could help account for the exceptional case of Drosophila H2A.X, which unlike other eukaryotes, is not derived from its canonical H2A, but rather from the distant H2A.Z variant lineage (Baldi and Becker 2013). If all that is necessary to be an H2A.X is to be in the H2A position in a nucleosome and to have the carboxy-terminal motif for phosphorylation, then an H2A.Z can evolve this capability.
DSB repair is clearly the universal function of H2A.X phosphorylation, and there would seem to be no stable epigenetic aspect to this process. However, H2A.X null mice are sterile, and cytological examination of mammalian spermatogenesis has revealed a striking epigenetic feature in which H2A.X is specifically phosphorylated on the XY bivalent (Fig. 10) (Fernandez-Capetillo et al. 2003). This chromosome pair occupies a distinct “sex body” during meiotic prophase, which has been implicated in silencing of sex-linked genes during male meiosis. H2A.X phosphorylation is essential for normal sex-body formation, and H2A.X-deficient spermatocytes fail to pair or condense and fail to inactivate X and Y genes during meiosis. H2A.X phosphorylation of the XY bivalent is distinct from the process that occurs at DSBs. XY phosphorylation in the sex body does not require breaks, but rather occurs most conspicuously at unpaired regions of the chromosomes. The mechanisms whereby H2A.X phosphorylation is targeted to unpaired chromosomes and how this event leads to condensation, pairing, and silencing are currently unknown. However, it is interesting to speculate that this role may be related to its ability to interact with and recruit cohesin.
Figure 10.

Pachytene stage of spermatogenesis showing the dependence of sex-body formation on H2A.X. In normal mammalian spermatocytes, a nuclear structure, the sex body (arrow, green, in right panels), is seen to encompass the unpaired XY bivalent (labeled in left panels). The synaptonemal complex, which aligns paired chromosomes, is stained red. H2A.X is normally enriched in the sex body (H2A.X+/+). In H2A.X−/− spermatocytes, the sex body does not form and a sex-body epitope becomes dispersed (lower right). Scale bar, 10 µm. (Images courtesy of Shantha Mahadevaiah and Paul Burgoyne; Fernandez-Capetillo et al. 2003.)
9. H2A.Z PLAYS DIVERSE ROLES IN CHROMATIN REGULATION
Histone variant H2A.Z is found throughout most eukaryotic lineages and it has been intensely studied for its structure and function in chromatin biology (Zlatanova and Thakar 2008; Draker and Cheung 2009; Marques et al. 2010; Talbert et al. 2012). H2A.Z diverged from an ancestral H2A early in evolution and shares only ∼60% similarity with its major histone H2A counterpart. Consistent with this separate lineage, genetic experiments in yeast, plants, flies, and mammals have shown that histones H2A and H2A.Z have evolved to play separate nonoverlapping functions. H2A.Z is an essential histone in most organisms, from ciliated protozoans to mammals. However, in budding and fission yeasts, cells that carry a deletion of the single-copy H2A.Z gene are viable, although the null mutants show a variety of conditional lethal phenotypes. Plants, as exemplified by Arabidopsis thaliana, have three closely related H2A.Z genes, HTA8, HTA9, and HTA11, with roughly 90% identity and a more distantly related HTA4 gene (Talbert and Henikoff 2010). There appears to be functional redundancy among these genes as organisms with single deletions of either HTA8, HTA9, or HTA11 are normal, whereas the hta9 hta11 double mutant shows developmental defects. Interestingly, vertebrates have evolved two closely related H2A.Z variants, H2A.Z.1 and H2A.Z.2, which differ at only three amino-acid residues (Dryhurst et al. 2009; Matsuda et al. 2010; Mehta et al. 2010). These two variants show different patterns of chromatin distribution (Dryhurst et al. 2009) and are apparently nonredundant as the single deletion of H2A.Z.1 in the mouse is lethal (Faast et al. 2001).
Evidence for functional diversification of H2A.Z comes from the discovery of an alternatively spliced form of human H2A.Z.2 mRNA that results in a protein with a reduced CTD. This shorter H2A.Z isoform destabilizes nucleosomes and is most enriched in brain (Bonisch et al. 2012), in which H2A.Z, like H3.3, is known to be especially abundant (Pina and Suau 1987). Although this particular isoform appears to be limited to primates, evidence for a longer alternatively spliced isoform with potential for nucleosome destabilization was reported in carp brain (Simonet et al. 2013). With the increasing popularity of RNA-seq for identification of alternatively processed variants, we expect that other examples of potential H2A.Z functional diversification will be discovered.
The high-resolution structure of an H2A.Z-containing nucleosome reveals several unique properties of the variant (Suto et al. 2000). Compared with H2A nucleosomes, H2A.Z presents an extended acidic patch domain on the surface of the nucleosome and mutational studies have shown this to have functional significance. The acidic patch is then part of a larger “docking domain,” an essential part of the protein necessary for interaction with H3 in the nucleosome. Like other histones, H2A.Z is subject to a variety of posttranslational modifications, including acetylation, ubiquitylation, and sumoylation. There is strong genetic and biochemical evidence that these modifications affect the localization, dynamics, and function of H2A.Z nucleosomes (Talbert and Henikoff 2010).
H2A.Z has been linked to a wide variety of different and sometimes contradictory nuclear functions, including transcriptional activation, transcriptional repression, RNA Pol II elongation, heterochromatin, antisilencing, cell-cycle control, DNA replication, DNA damage repair, chromosome segregation, and genome integrity (Zlatanova and Thakar 2008; Altaf et al. 2009; Marques et al. 2010; Talbert and Henikoff 2010; Xu et al. 2012; Adkins et al. 2013). It likely has direct mechanistic roles in transcription initiation, elongation, antisilencing, and DNA damage repair. In some other cases, evidence suggests that H2A.Z function is manifest indirectly through its involvement in transcription. For example, delay in the G1-S transition observed in budding yeast deleted for H2A.Z is likely due to the misregulation of cyclin gene expression and not defects in DNA replication initiation (Dhillon et al. 2006). In S. pombe, and perhaps other organisms, H2A.Z cooperates with heterochromatin-silencing factors (Clr4/SUV39H) to enforce RNA-processing fidelity and prevent deleterious antisense transcription (Zofall et al. 2009). Loss of this enforcement may account for the genome instability and chromosome segregation defects observed in H2A.Z deletion mutants. Indeed, at least part of the chromosome segregation defects observed in H2A.Z mutants of S. pombe might be caused by decreased transcriptional expression of the centromere protein, CENP-C (Hou et al. 2010). Nevertheless, such examples of indirect function are relatively rare. Indeed, ruling out indirect activities for any chromatin regulator is a challenging problem, particularly for H2A.Z in organisms for which it is essential.
10. H3.3 AND H2A.Z OCCUPY DISCRETE CHROMATIN LOCATIONS
Much of our understanding of histone variant function is inferred from its patterns of genomic chromatin occupancy. H3.3 makes up ∼15%–25% of total H3 protein and H2A.Z makes up ∼5%–10% of the total H2A protein in most organisms examined to date. Abundances increase when cells exit from the cell cycle and no longer replicate their DNA, such as during development (Pina and Suau 1987). These variants are widely, but not uniformly, distributed throughout the genome. High-resolution chromatin immunoprecipitation experiments in a number of model organisms have revealed that both H3.3 and H2A.Z preferentially occupy nucleosomes that flank gene promoters and both are particularly enriched at the +1 nucleosome bordering transcriptional start sites (TSSs). They are often enriched at the −1 or −2 nucleosome as well and thus flank a nucleosome-depleted region at the TSS (Talbert and Henikoff 2010). In animals, the H3.3 over gene bodies correlates with transcriptional levels, suggesting that it replaces nucleosomes that are occasionally lost during transcription (illustrated in the left panel of Fig. 9). Direct evidence for this interpretation comes from measuring nucleosome turnover by metabolic labeling, which showed nucleosome turnover patterns closely matching H3.3 patterns genome-wide (Deal et al. 2010).
H2A.Z has been mapped genome-wide in a variety of eukaryotes. In budding yeast, nematodes, and plants, H2A.Z occupancy around the promoter is correlated with nontranscribing genes “poised” for activation (Zhang et al. 2005; Mavrich et al. 2008; Whittle et al. 2008; Kumar and Wigge 2010). However, in flies and mammals, promoter H2A.Z occupancy appears to correlate more with actively transcribing genes (Barski et al. 2007; Mavrich et al. 2008; Hardy et al. 2009; Hardy and Robert 2010; Kelly et al. 2010), similar to the situation for H3.3. Although preferentially found at promoters and regulatory sites, H2A.Z nucleosomes can also be found at lower frequency in gene bodies and elsewhere (Hardy et al. 2009; Weber et al. 2010; Santisteban et al. 2011). Enrichment of H2A.Z over gene bodies closely corresponds to that of chromatin that is extracted with low salt, suggesting that H2A.Z changes the physical properties of nucleosomes (Weber et al. 2010).
H2A.Z is also specifically deposited near or within heterochromatin. In budding yeast, H2A.Z is enriched near telomeres where it serves as an antisilencing factor. Deletion of the H2A.Z gene results in extended spreading of silent chromatin inward from the telomeres and this defect can be suppressed by the additional deletion of genes encoding the silencing factors themselves (see Grunstein and Gasser 2013 for more detail). Indeed, this function may act globally, in parallel with the Set1 histone H3 methyltransferase, to prevent large-scale aberrant distribution of silencing factors (Venkatasubrahmanyam et al. 2007). In metazoans, H2A.Z is also localized in facultative and constitutive heterochromatin, the inactive X chromosome, transposable elements, and pericentric heterochromatin (Greaves et al. 2007; Draker and Cheung 2009; Boyarchuk et al. 2011; Zhang and Pugh 2011).
In contrast to the chromosomal features that are correlated with H2A.Z, there is a remarkable anticorrelation between histone H2A.Z nucleosome occupancy and DNA methylation (Zilberman et al. 2008; Kobor and Lorincz 2009; March-Diaz and Reyes 2009; Conerly et al. 2010; Edwards et al. 2010; Zemach et al. 2010; see Li and Zhang 2014 for a discussion of DNA methylation). There is strong evidence that this mutual antagonism is causal and not simply correlative. Mutants in A. thaliana with decreased DNA methylation show an increase in H2A.Z occupancy at loci where it is normally not found, independent of transcriptional activity. Conversely, mutants defective in H2A.Z deposition show increased DNA methylation over gene bodies normally occupied by H2A.Z nucleosomes. While the precise molecular pathways that account for this mutual exclusion remain to be elucidated, this functional relationship has important implications for development and carcinogenesis. For example, the stochastic or environmentally effected loss of H2A.Z from the promoter of a tumor-suppressor gene could well contribute to locally increased DNA methylation and heritable epigenetic repression.
11. H2A.Z NUCLEOSOME OCCUPANCY IS DYNAMIC AND CHANGES THE PROPERTIES OF CHROMATIN
The dynamic exchange of H2A.Z nucleosomes in chromatin appears to be an important part of its function. (See Becker and Workman 2013 for an in-depth discussion of histone exchange.) Unlike the major core histones, H2A.Z expression is not restricted to S phase and it can be incorporated into chromatin independent of DNA replication. The deposition of H2A.Z into nucleosomes is performed by multisubunit protein complexes, which have been conserved throughout the eukaryotic kingdom (Lu et al. 2009; March-Diaz and Reyes 2009; Morrison and Shen 2009). First identified in budding yeast, the SWR1 complexes contain, as their catalytic subunits, homologs of the protein Swr1, a member of the SWI/SNF family of ATP-dependent chromatin remodelers. The substrate for SWR1 is an H2A.Z-H2B dimer, which is used to replace one of the existing H2A-H2B dimers in the nucleosome in an ATP-dependent exchange reaction (Fig. 9). This reaction is stepwise and unidirectional, in vitro, resulting in the complete replacement of H2A-H2B dimers with H2A.Z-H2B dimers (Luk et al. 2010). In vivo, unidirectional replacement of H2A with H2A.Z by SWR1 is enforced by acetylation of H3K56, which allows the reverse reaction to occur, resulting in local reduction in H2A.Z incorporation, thus modulating transcription (Watanabe et al. 2013). SWR1 is likely dedicated to the task of replacing H2A with H2A.Z because the effects of eliminating SWR1 function are similar to the effects of deleting the gene encoding H2A.Z itself. Indeed, the activity of the SWR1 complex is actually deleterious to the cell in the absence of its H2A.Z-H2B substrate (Halley et al. 2010; Morillo-Huesca et al. 2010).
The removal of H2A.Z from chromatin proceeds by at least two pathways. Nucleosome exchange and eviction occurs in many contexts such as the remodeling and eviction of complete nucleosomes that can happen at promoters. Any H2A.Z that is a part of those nucleosomes will be removed as well. However, there is also evidence that H2A.Z-H2B dimers may be specifically removed from nucleosomes by the INO80 complex, a close relative of SWR1 (Morrison and Shen 2009; Papamichos-Chronakis et al. 2011). In budding yeast, the elimination of INO80 results in the global mislocalization of H2A.Z and a decrease in its apparent exchange rate. Additional mutational results are consistent with this interpretation. In vitro, purified INO80 is reported to catalyze the replacement of a nucleosomal H2A.Z-H2B dimer with a canonical H2A-H2B dimer, that is, the reverse of the SWR1 reaction (Luk et al. 2010; Papamichos-Chronakis et al. 2011), and this reaction may also be regulated by acetylation of H3K56 (Watanabe et al. 2013).
The factors that determine where H2A.Z is deposited are incompletely understood. At present, there is little evidence that H2A.Z templates its own deposition epigenetically (Viens et al. 2006). In budding yeast, a DNA sequence related to the binding site of transcription factor Reb1 is able to target the enrichment of H2A.Z at ectopic sites, independent of Reb1 (Raisner et al. 2005). In other cases, transcription factors themselves are implicated in targeting H2A.Z deposition (Updike and Mango 2006; Zacharioudakis et al. 2007; Gevry et al. 2009). The unifying theme underlying the function of these various factors appears to be the creation of a nucleosome-depleted region necessary for H2A.Z deposition, although how this recruits H2A.Z is not currently known (Hartley and Madhani 2009). Interestingly, SWR1 complexes often contain homologs of yeast Brd1, a protein containing dual bromodomain motifs capable of binding acetylated lysines. Thus, SWR1 may be recruited to, or stabilized at, chromatin neighborhoods rich in acetylated histones. H2A.Z deposition may also be blocked at specific loci. The S. pombe SWR1 complex contains a regulatory subunit, Msc1, which is dispensable for the loading of H2A.Z at promoter nucleosomes, but which is required to prevent H2A.Z deposition within the chromatin of the inner centromere and subtelomeric regions (Buchanan et al. 2009; Zofall et al. 2009). It has also been suggested that an additional pathway directing localized occupancy by H2A.Z may involve its random deposition and then specific eviction, perhaps as a consequence of transcription (Hardy and Robert 2010).
In any case, the consequence of H2A.Z deposition and replacement can be complex. Because of the exchange reactions, nucleosomes in cellular chromatin may be “ZZ,” “ZA,” or “AA,” containing two, one, or zero H2A.Z-H2B dimers, respectively (Luk et al. 2010; Weber et al. 2010). In Drosophila, the pattern of ZZ and ZA nucleosome occupancy is different, and homotypic H2A.Z nucleosomes are enriched over the bodies of active genes, perhaps as a consequence of transcription elongation. In mouse trophoblast cells, there are distinct changes in the promoter content of ZZ and ZA nucleosomes during G1, S, and M phase, independent of actual transcription activity, suggesting a major remodeling pathway dependent on the cell-division cycle (Nekrasov et al. 2012). Differential posttranslational modifications further enrich the situation as H2A.Z acetylation has been linked with both SWR1 and INO80 functions (Millar et al. 2006; Papamichos-Chronakis et al. 2011).
A quantitative evaluation of the +1 nucleosome in yeast revealed that at steady state the relative abundance of ZZ, ZA, and AA nucleosomes is roughly 32%, 24%, and 44% respectively (Luk et al. 2010). How can H2A.Z drive chromatin function with this level of heterogeneity? One explanation is that H2A.Z nucleosomes are in dynamic exchange at these sites. In budding yeast, nucleosomes with rapid turnover were identified by the kinetic incorporation of newly synthesized histone H3. These “hot” nucleosomes preferentially map to promoter regions, including the nucleosomes at the TSS, which are enriched in H2A.Z histones (Dion et al. 2007). Remarkably, in budding yeast H2A.Z appears to increase the global turnover of nucleosomes and not simply those into which it is most abundantly incorporated (Dion et al. 2007; Santisteban et al. 2011). The mechanistic basis for this influence is currently unknown.
The stability of H2A.Z-containing nucleosomes in vitro has been examined in many studies with contrasting results (Zlatanova and Thakar 2008; Talbert and Henikoff 2010). In vivo it is clear that not all H2A.Z nucleosomes are created equal and that the lability of H2A.Z nucleosomes is affected by posttranslational modifications and the presence of other variant histones. Nucleosomes containing both H2A.Z and H3.3, especially H2A.Z/H2A nucleosomes with H3.3, are particularly unstable and sites occupied by these double-variant nucleosomes may be erroneously scored as entirely free of nucleosomes, depending on how the chromatin is isolated (Jin et al. 2009; Nekrasov et al. 2012). H2A.Z acetylation generally destabilizes nucleosomes in vitro and in vivo is associated with gene activation (Tanabe et al. 2008; Wan et al. 2009; Halley et al. 2010).
A particularly striking example of the differential properties of H2A.Z nucleosomes is the transcriptional response of cells to temperature. Plants possess a signaling pathway that senses ambient temperature and regulates gene expression (see Baulcombe and Dean 2014 for a discussion of plant responses to environmental factors). The developmental program for flowering, for example, is accelerated at higher temperature. To identify factors that regulate this response, A. thaliana was screened for mutants with a constitutive high temperature expression pattern and the mutations turned out to be in ARP6, which encodes one of the conserved subunits of the SWR1 complex (Kumar and Wigge 2010). Indeed, H2A.Z occupancy at the promoters of heat responsive genes was found to decrease with increasing temperature and this loss was independent of transcriptional activity per se. These findings have potentially broad implications for agriculture in a globally warming environment, insofar as depletion of H2A.Z in a model cereal phenocopies the ambient temperature response and impacts grain yield (Boden et al. 2013).
12. H2A.Z FUNCTIONS IN EPIGENETIC INHERITANCE
There is increasing evidence that H2A.Z participates in the heritable specification of chromatin function. Chromosomes undergo extensive remodeling each mitotic generation and the specification of silent and active chromatin domains across division cycles is essential for normal development. In budding yeast, H2A.Z is transiently lost from a subset of genes during telophase, as measured by chromatin immunoprecipitation and this displacement is required for the establishment of heterochromatin in an inducible model (Martins-Taylor et al. 2011). In mammalian cells most active transcription is repressed during mitosis and must be reactivated in the next cell cycle. The +1 nucleosome containing H2A.Z is a prime candidate for mediating this control and its translational positioning at TSSs has been examined in G0/G1-phase, S-phase, and M-phase cells (Kelly et al. 2010; Nekrasov et al. 2012). Indeed, the positions of these nucleosomes were found to be shifted during mitosis, altering the size of the nucleosome-depleted region and remodeling chromatin at the TSS. Together these features may help establish epigenetic memory during mitosis and rapid gene reactivation following cell division. The pathways regulating these cell-cycle behaviors of H2A.Z are currently unknown.
The location of a gene within the nucleus can be important for its expression. Positioning at the nuclear periphery is often associated with gene inactivation with the exception of localization at nuclear pores, which is a positive factor in expression (reviewed in Akhtar and Gasser 2007). Several genes in budding yeast, such as GAL1 and INO1, have been found to move to the nuclear periphery on activation and remain there for multiple mitotic cell cycles even after repression. It has been argued that this localization contributes to “transcriptional memory,” which is the observation that recently expressed genes are more rapidly reactivated than genes that have experienced long-term repression (Brickner 2009). The molecular mechanisms responsible for transcriptional memory are currently controversial (Zacharioudakis et al. 2007; Halley et al. 2010; Kundu and Peterson 2010). Nevertheless, H2A.Z appears to have a role in maintaining the localization of recently repressed genes at the nuclear periphery for multiple mitotic generations. In the case of INO1, this pathway requires a cis-acting DNA element in the INO1 promoter and interaction with the nuclear pore protein NUP100 to direct the loading of H2A.Z (Light et al. 2010). If the H2A.Z gene is deleted, recently repressed INO1 fails to remain at the nuclear periphery and becomes nucleoplasmic (Brickner 2009).
H2A.Z is also linked to the epigenetic specification of cell fates in mammalian stem cells (Creyghton et al. 2008; see Reik and Surani 2014 for more on stem cell epigenetics). Whole genome-mapping studies in mouse embryonic stem cells revealed that H2A.Z preferentially occupies the promoters of genes that are poised to direct development and differentiation when activated (Ku et al. 2012; Li et al. 2012; Hu et al. 2013). Indeed, the distribution of H2A.Z is coincident with that of Suz12, a component of the Polycomb group complexes, which have primary roles in development (see Grossniklaus and Paro 2014 for a detailed discussion of Polycomb group proteins). Inhibiting H2A.Z expression results in increased and more stable nucleosome occupancy at regulatory regions, decreased methylation of H3K4 and H3K27 at promoters and enhancers, the derepression of developmental target genes, and aberrant differentiation of embryoid bodies. These results are consistent with a model in which H2A.Z variant nucleosomes impart a dynamic instability to chromatin structure increasing access to chromatin-modification factors such as mixed lineage leukemia and Polycomb repressive complex 2 (PRC2), and to lineage-specific factors such as the transcription factor FoxA2 for endoderm/hepatic differentiation (Li et al. 2012) or RARα for neuronal development (Hu et al. 2013).
In theory, stem cells are at a higher risk of mutation because of their continued proliferation during the life of the organism. Cairns first proposed that stem cells might protect against this risk by ensuring the inheritance of the “oldest” DNA strands exclusively to the stem cell daughter as opposed to the differentiating daughter cell (Cairns 1975). There is experimental support for this “immortal-strand model” in a number of systems in which asymmetric self-renewal can be experimentally manipulated. Interestingly, histone H2A.Z expression is a biomarker for this immortal-strand inheritance (Huh and Sherley 2011). First, H2A.Z mRNA is down-regulated in the differentiating sister cell during asymmetric cell division. This observation is consistent with the fact that the H2A.Z gene is a target of Oct4 regulation and is down-regulated following differentiation of both mouse and human stem cell populations (Du et al. 2001; Shaw et al. 2009; Huh and Sherley 2011). More striking, however, is the apparent asymmetric distribution of H2A.Z entirely to the immortal DNA strands segregating to the stem cell sister, as detected by immunofluorescence (Huh and Sherley 2011). This turns out to be because of the “cloaking” of the H2A.Z histones on the mortal-strand chromatids inherited by the differentiating sister cell. That is, H2A.Z is actually present on both sets of chromosomes, but it is only accessible to detection by the anti-H2A.Z antibody on the set segregating to the stem cell sister. Interestingly, mild acid treatment before immunostaining removes whatever is blocking detection and reveals H2A.Z on both sets of chromosomes. The nature of the cloaking mechanism is currently unknown, but these observations place H2A.Z quite proximal to the mechanism of immortal-strand inheritance.
13. OTHER H2A VARIANTS DIFFERENTIATE CHROMATIN, BUT THEIR FUNCTIONS ARE AS YET UNKNOWN
Further diversification of H2A has occurred in vertebrates. In mammals, macroH2A and H2A.B represent unique lineages that appear to play roles in the epigenetic phenomenon of dosage compensation (discussed in detail in Brockdorff and Turner 2014). macroH2A is so-called because, in addition to the histone-fold domain and amino- and carboxy-terminal tails, it contains a more than 200-amino- acid carboxy-terminal globular domain (Ladurner 2003). macroH2A is enriched in discrete regions of the facultatively inactive X chromosome of human females that alternate with regions of constitutive heterochromatin (Fig. 11A) (Chadwick and Willard 2004). However, the role of macroH2A must be more diverse, insofar as macroH2A orthologs are present in many nonmammalian clades, and appear to be ancestral in the animal kingdom (Talbert and Henikoff 2010). In vitro, macroH2A reduces transcription factor access and excludes histone H1. The macro domain itself binds to the enzyme PolyADP ribose polymerase 1 and inhibits its activity in vitro (Nusinow et al. 2007), although exactly what is occurring in vivo to mediate gene repression is unclear.
Figure 11.
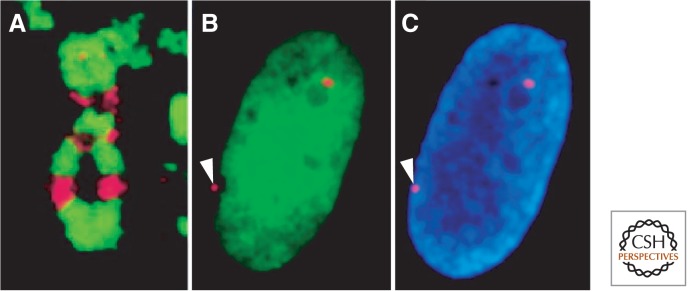
H2A variants and the inactive X chromosome of human females. (A) macroH2A (red) stains discrete regions of the inactive X chromosome that alternate with a marker for heterochromatin (histone H3K9me3). (B) H2A.B (green) is excluded from the inactive X chromosome (red dot with arrowhead pointing to it). (C) Same nucleus as in B, but stained with DAPI to show chromatin. (A, Reprinted, with permission, from Chadwick and Willard 2004, © National Academy of Sciences; B,C, reprinted, with permission, from Chadwick and Willard 2001, © 2001 The Rockefeller University Press. Originally published in Journal of Cell Biology 152: 375–384. doi: 10.1083/jcb.152.2.375.)
In contrast to macroH2A, H2A.B seems to be undetectable on the Barr body, but otherwise ubiquitous throughout the nucleus (Fig. 11B) (Chadwick and Willard 2001). The short wrap of DNA around H2A.B nucleosomes (Bao et al. 2004) and its enrichment at TSSs (Soboleva et al. 2012) is consistent with it playing a role in facilitating transcription. H2A.B and its closest relative, H2A.L (Govin et al. 2007), are rapidly evolving relative to other H2A variant classes, perhaps related to the fact that they are both testes-specific variants (Talbert and Henikoff 2010). The roles that H2A.B and H2A.L play in mammalian male germ cell development remain to be elucidated.
14. MANY HISTONES HAVE EVOLVED TO MORE TIGHTLY PACKAGE DNA
When it is no longer necessary to gain access to DNA for replication and transcription, chromatin typically becomes further condensed, and this often involves replacement of canonical histones. This is obviously the case for sperm, and in some lineages, histone paralogs have evolved specialized packaging roles. For example, sea urchin sperm contains H1 and H2B variants with repeated tail motifs that bind to the minor grooves of DNA (Talbert and Henikoff 2010), presumably an adaptation to tightly package chromosomes for inclusion into sperm heads. A similar adaptation is found in pollen-specific H2A variants in flowering plants. In vertebrates, a sperm-specific H2B specialized histone variant is found in mammalian testes, including an H2B paralog (subH2Bv) that localizes to the acrosome and a testes-specific H3 variant (Witt et al. 1996).
The replacement of histones during sperm maturation by protamines and other proteins provides a potential means of erasing epigenetic information in the male germline. However, evidence for transgenerational inheritance (Rakyan and Whitelaw 2003; discussed in more detail in Blewitt and Whitelaw 2013), especially in animals that lack DNA methylation, raises the possibility that a subset of nucleosomal histones survive this transition and transmit epigenetic information. As already pointed out, this is just what occurs for CENP-A at centromeres (Palmer et al. 1990), and it is possible that a small fraction of other variants, such as H3.3, remain with sperm for epigenetic inheritance of gene-expression information. Although our understanding of the process that replaces histones during sperm development is rudimentary, we expect that much more can be learned by understanding how CENP-A survives this transition.
Increased compaction also occurs in somatic cells that have finished dividing and undergo differentiation. In some cases, compaction involves quantitative and qualitative changes in linker histones. The stoichiometry of histone H1 relative to nucleosomes determines the average spacing within nucleosome arrays in vivo (Fan et al. 2003). In addition, the presence of H1 in chromatin promotes higher-order chromatin structure that generally inhibits transcription (Wolffe 1992). Linker histones are much more mobile than core histones in vivo. Residence times for H2A and H2B are hours in length, and cannot even be measured for H3 and H4, whereas the residence time of H1 is a few minutes (Phair et al. 2004). As a result, the incorporation of variant linker histones is unlikely to differentiate chromatin in a heritable manner. Rather, the role of H1 variants is thought to change the bulk properties of chromatin that can affect overall compaction (Wolffe 1992).
H1 variants share with core histones a distinction between RC and RI forms (Marzluff et al. 2002). RC variant forms of H1 appear to be interchangeable with one another, based on the fact that knockout mice lacking one or two of the five RC H1 variants are phenotypically normal (Fan et al. 2003). The functional interchangeability of histone variants is implicit in the “star” phylogeny of the H1 family, in which there is little if any evidence for evolutionary conservation of H1 branches in the tree (Fig. 6C). Rather, the greater divergence of H1 relative to core histone variants is likely to reflect weaker structural constraints, and the existence of multiple H1 genes in many lineages might represent an adaptation for regulating levels of linker histone. For example, in birds, the H1.0 linker histone variant (previously referred to as H5) is deposited during erythrocyte maturation, which accompanies extreme compaction of the nucleus. The mammalian H1.0 ortholog (previously referred to as H1°) is deposited at high levels in nondividing cells. Overexpression of H1.0 renders chromatin less accessible to nucleases than similar overexpression of a canonical form. The natural accumulation of H1.0 in nondividing cells might be a general mechanism for chromatin compaction as cells become quiescent.
15. HISTONE VARIANTS AND HUMAN DISEASE
The diverse roles of histone variants in basic epigenetic processes leads to the expectation that their loss or misexpression can result in disease, and indeed some examples of this have recently come to light. For example, ATRX, which is the DNA translocase component of the ATRX-Daxx-H3.3 pathway (Fig. 8), was originally identified as causing α-thalassemia mental retardation on the X (ATRX) syndrome. In this syndrome, loss of ATRX results in defects at the CpG island of the α-globin gene promoter, and can now be understood as resulting from frequent nucleosome loss at CpG islands (Law et al. 2010). Importantly, mutations causing the ATRX syndrome are most frequently found within the ADD histone tail-binding domain (Eustermann et al. 2011), strongly implicating H3/H3.3 tail interactions as being key causal factors in the syndrome. The study of ATRX syndrome has also led to the realization that another histone variant, macroH2A, is also depleted from specific sites, including the α-globin CpG island (Ratnakumar et al. 2012).
A remarkable relationship has recently emerged between the ATRX-Daxx-H3.3 nucleosome assembly process and cancer that provides important insights into both the process itself and oncogenesis. Genomic sequencing of human pancreatic neuroendocrine tumors (PanNETs) revealed that ∼40% harbored likely loss-of-function mutations in either Daxx or ATRX (Jiao et al. 2011). Notably, all such tumors displayed a distinct phenotype called “ALT” (alternative lengthening of telomeres) in which telomeres show massive lengthening without induction of telomerase (see Fig. 1 of Liu et al. 2014; Heaphy et al. 2011a). Based on examination of more than 6000 human tumors, 3.73% showed ALT occurring in cancers of all different types (Heaphy et al. 2011b). ATRX or Daxx are mutated in the vast majority of cases of ALT examined by sequencing. Although the precise molecular basis for telomere lengthening in ALT is not well understood, lengthening is likely initiated by recombinational strand invasion between telomeres, which suggests that failure to replace a nucleosome at a simple sequence array can result in DNA breakage and strand invasion. In the case of PanNETs, ALT predicts a more favorable outcome than telomerase induction, as if the ALT pathway is less efficient than the telomerase pathway in preventing the senescence that occurs when telomeres erode (Jiao et al. 2011).
Sequencing of tumors has also revealed the very surprising presence of specific mutations in H3.3 itself in a large fraction of pediatric glioblastomas (the topic of Liu et al. 2014; also Schwartzentruber et al. 2012; Wu et al. 2012). One of the two H3.3 genes in these tumors is found to encode either K27M or G34R/V, and is most likely to be a gain-of-function mutation that drives accelerated tumorigenesis (see Fig. 1 of Liu et al. 2014). G34R/V is associated with ATRX loss and probably enhances ALT, whereas K27M can occur on an H3 as well, so would appear to be independent of the ATRX-Daxx-H3.3 pathway. Rather, K27 is a key substrate for methylation and acetylation involved in Polycomb silencing, and the presence of a nonmodifiable residue at position 27 of the H3/H3.3 tail causes gain-of-function inhibition of methylation by the EZH2 subunit of PRC2 (Lewis et al. 2013). It would then appear that the resulting global reduction in H3K27 trimethylation contributes to this aggressive pediatric tumor.
Misregulation of histone variant expression has also been implicated in cancer. In malignant melanoma cells, macroH2A levels are sharply reduced and restoration of macroH2A levels suppresses metastasis (Kapoor et al. 2010). Reduced levels of macroH2A result in up-regulation of many genes, including the CDK8 oncogene. macroH2A is also reduced in more rapidly proliferating lung cancers, and is elevated in cells undergoing senescence, which suggests that reduced senescence of tumor cells is a mechanism whereby loss of macroH2A contributes to oncogenesis (Sporn et al. 2009). It is possible that other defects in the expression of histone variants and in pathways that deposit them can contribute to cancer and other human diseases, but there is as yet insufficient evidence of causality as opposed to mere correlation. For example, excessively high levels of H2A.Z are found to correlate with poor outcomes in estrogen-positive breast cancer (Hua et al. 2008). In addition, CENP-A is found to be overexpressed and ectopically localized in a variety of cancers, suggesting that transient or permanent neocentromere formation might result in some of the aneuploidies that are hallmarks of cancer cells (Dalal 2009). We anticipate that as genome-wide technologies improve, other possible roles of histone variants incorporation pathways in human diseases will be uncovered.
16. CONCLUSIONS AND FUTURE RESEARCH
Histone variants provide the most fundamental level of differentiation of chromatin, and alternative mechanisms for depositing different variants can potentially establish and maintain epigenetic states. Histones H2A, H2B, H3, and H4 occupy distinct positions in the core particle as a result of an evolutionary process that began before the last common ancestor of eukaryotes. Key evolutionary innovations remain uncertain, including the emergence of an octamer from an ancestral tetramer, and we look forward to the sequencing of more archaeal and primitive eukaryotic genomes that might provide missing links. Subsequent elaborations of the four core histones into distinct variants have provided the basis for epigenetic processes, including development and chromosome segregation. For a full understanding of epigenetic inheritance, we need a better understanding of the processes that incorporate variants by replacing canonical histones. An important recent development is the initial characterization of RI assembly pathways dedicated to particular variants.
Centromeres are the most conspicuous examples of profoundly different chromatin that is attributable to special properties of a histone variant. Although it is clear that cenH3-containing nucleosomes form the foundation of the centromere, just how they are deposited in the same place every cell generation without any hint of sequence specificity is a major challenge for future research.
It is becoming evident that histone variants are also involved in epigenetic properties of active genes. Both H3.3 and H2A.Z are enriched at transcriptionally active loci, and understanding the assembly processes that are responsible for their enrichment is an exciting area of current research. The dynamic behavior of chromatin leads to the realization that transcription, chromatin remodeling, and histone modification might be coupled to nucleosome assembly and disassembly. The study of dynamic processes coupled to histone turnover is only at an early stage, and we look forward to technological advances in molecular biology, cytogenetics, biochemistry, and structural biology that can be harnessed to better understand the dynamic nature of chromatin.
In addition to these universal processes, histone variants are also involved in particular epigenetic phenomena. In the case of the mammalian X chromosome, three different H2A variants, phospho-H2A.X, macroH2A, and H2A.B have been recruited to participate in silencing or activation of genes for purposes of germline inactivation or dosage compensation. Understanding the function of these variants in epigenetic processes remains a major challenge for the future.
The availability of the first high-resolution structure of the nucleosome core particle (Luger et al. 1997) was a seminal advance in elucidating the properties of chromatin. By elaborating this basic structure in a way that has biological consequences, histone variants provide an opportunity to deepen our understanding of how these fascinating architectural proteins have evolved to play diverse roles in epigenetic processes.
Footnotes
Editors: C. David Allis, Marie-Laure Caparros, Thomas Jenuwein, and Danny Reinberg
Additional Perspectives on Epigenetics available at www.cshperspectives.org
REFERENCES
*Reference is also in this collection.
- Adkins NL, Niu H, Sung P, Peterson CL 2013. Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol 20: 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar A, Gasser SM 2007. The nuclear envelope and transcriptional control. Nat Rev Genet 8: 507–517. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Suzuki O, Matsuda J, Aoki F 2011. Dynamic replacement of histone h3 variants reprograms epigenetic marks in early mouse embryos. PLoS Genet 7: e1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Allshire RC, Ekwall K 2014. Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb Perspec Biol 10.1101/cshperspect.a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Almouzni G, Cedar H 2014. Maintenance of epigenetic information. Cold Spring Harb Perspec Biol 10.1101/cshperspect.a019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaf M, Auger A, Covic M, Cote J 2009. Connection between histone H2A variants and chromatin remodeling complexes. Biochem Cell Biol 87: 35–50. [DOI] [PubMed] [Google Scholar]
- Altheim BA, Schultz MC 1999. Histone modification governs the cell cycle regulation of a replication-independent chromatin assembly pathway in Saccharomyces cerevisiae. Proc Natl Acad Sci 96: 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammar R, Torti D, Tsui K, Gebbia M, Durbic T, Bader GD, Giaever G, Nislow C 2012. Chromatin is an ancient innovation conserved between Archaea and Eukarya. eLife 1: e00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor DJ, Bentley K, Ryan J, Perry J, Wong L, Slater H, Choo KH 2004a. Human centromere repositioning ‘in progress’. Proc Natl Acad Sci 101: 6542–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor DJ, Kalitsis P, Sumer H, Choo KH 2004b. Building the centromere: From foundation proteins to 3D organization. Trends Cell Biol 14: 359–368. [DOI] [PubMed] [Google Scholar]
- Annunziato AT 2005. Split decision: What happens to nucleosomes during DNA replication? J Biol Chem 280: 12065–12068. [DOI] [PubMed] [Google Scholar]
- Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN 1991. The nucleosomal core histone octamer at 3.1 A resolution: A tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci 88: 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi S, Becker PB 2013. The variant histone H2A.V of Drosophila—Three roles, two guises. Chromosoma 122: 245–258. [DOI] [PubMed] [Google Scholar]
- Bao Y, Konesky K, Park YJ, Rosu S, Dyer PN, Rangasamy D, Tremethick DJ, Laybourn PJ, Luger K 2004. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J 23: 3314–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837. [DOI] [PubMed] [Google Scholar]
- *.Baulcombe D, Dean C 2014. Epigenetic regulation in plant responses to the environment. Cold Spring Harb Perspec Biol 6: a019471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Becker PB, Workman JL 2013. Nucleosome remodeling and epigenetics. Cold Spring Harb Perspec Biol 5: 017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Black BE, Cleveland DW 2011. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Blewitt M, Whitelaw E 2013. The use of mouse models to study epigenetics. Cold Spring Harb Perspec Biol 5: 017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden SA, Kavanova M, Finnegan J, Wigge PA 2013. Thermal stress effects on grain yield in Brachypodium distachyon occur via H2A.Z-nucleosomes. Genome Biol 14: R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonisch C, Schneider K, Punzeler S, Wiedemann SM, Bielmeier C, Bocola M, Eberl HC, Kuegel W, Neumann J, Kremmer E, et al. 2012. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res 40: 5951–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyarchuk E, Montes de Oca R, Almouzni G 2011. Cell cycle dynamics of histone variants at the centromere, a model for chromosomal landmarks. Curr Opin Cell Biol 23: 266–276. [DOI] [PubMed] [Google Scholar]
- Brickner JH 2009. Transcriptional memory at the nuclear periphery. Curr Opin Cell Biol 21: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier-Armanet C, Forterre P, Gribaldo S 2011. Phylogeny and evolution of the Archaea: One hundred genomes later. Curr Opin Microbiol 14: 274–281. [DOI] [PubMed] [Google Scholar]
- *.Brockdorff N, Turner B 2014. Dosage compensation in mammals. Cold Spring Harb Perspec Biol 10.1101/cshperspect.a019406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan L, Durand-Dubief M, Roguev A, Sakalar C, Wilhelm B, Strålfors A, Shevchenko A, Aasland R, Shevchenko A, Ekwall K, et al. 2009. The Schizosaccharomyces pombe JmjC-protein, Msc1, prevents H2A.Z localization in centromeric and subtelomeric chromatin domains. PLoS Genet 5: e1000726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J 1975. Mutation selection and the natural history of cancer. Nature 255: 197–200. [DOI] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF 2001. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J Cell Biol 152: 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF 2004. Multiple spatially distinct types of facultative heterochromatin on the human inactive X chromosome. Proc Natl Acad Sci 101: 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho US, Harrison SC 2011. Recognition of the centromere-specific histone Cse4 by the chaperone Scm3. Proc Natl Acad Sci 108: 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S 2010. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res 20: 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA 2008. H2AZ is enriched at Polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 135: 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal Y 2009. Epigenetic specification of centromeres. Biochem Cell Biol 87: 273–282. [DOI] [PubMed] [Google Scholar]
- Dalal Y, Wang H, Lindsay S, Henikoff S 2007. Tetrameric structure of centromeric nucleosomes in interphase Drosophila cells. PLoS Biol 5: e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S 2010. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon N, Oki M, Szyjka SJ, Aparicio OM, Kamakaka RT 2006. H2A.Z functions to regulate progression through the cell cycle. Mol Cell Biol 26: 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion M, Kaplan T, Friedman N, Rando OJ 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Draker R, Cheung P 2009. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochem Cell Biol 87: 19–25. [DOI] [PubMed] [Google Scholar]
- Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A 2010. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryhurst D, Ishibashi T, Rose KL, Eirin-Lopez JM, McDonald D, Silva-Moreno B, Veldhoen N, Helbing CC, Hendzel MJ, Shabanowitz J, et al. 2009. Characterization of the histone H2A.Z-1 and H2A.Z-2 isoforms in vertebrates. BMC Biol 7: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Cong H, Yao Z 2001. Identification of putative downstream genes of Oct-4 by suppression-subtractive hybridization. Biochem Biophys Res Commun 282: 701–706. [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G 2009. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137: 485–497. [DOI] [PubMed] [Google Scholar]
- Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. 2010. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 20: 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, Jelinska C, Garrick D, Clynes D, Gibbons RJ, Rhodes D, et al. 2011. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol 18: 777–782. [DOI] [PubMed] [Google Scholar]
- Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JR, Taylor H, Matthaei K, Rathjen PD, Tremethick DJ, Lyons I 2001. Histone variant H2A.Z is required for early mammalian development. Curr Biol 11: 1183–1187. [DOI] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, Morin-Kensicki EM, Zhao J, Magnuson TR, Woodcock CL, Skoultchi AI 2003. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. MolCell Biol 23: 4559–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A 2003. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell 4: 497–508. [DOI] [PubMed] [Google Scholar]
- Filipescu D, Szenker E, Almouzni G 2013. Developmental roles of histone H3 variants and their chaperones. Trends Genet 29: 630–640. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, Cleveland DW 2009. Centromere-specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell 137: 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Ruone S, Adams MD, Olsen AE, Eriksson P, Yu Y, Roades AR, Kaufman PD, Stillman DJ 2002. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: Polymerase passage may degrade chromatin structure. Genetics 162: 1557–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, Gaudreau L 2009. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev 23: 1522–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govin J, Escoffier E, Rousseaux S, Kuhn L, Ferro M, Thevenon J, Catena R, Davidson I, Garin J, Khochbin S, et al. 2007. Pericentric heterochromatin reprogramming by new histone variants during mouse spermiogenesis. J Cell Biol 176: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves IK, Rangasamy D, Ridgway P, Tremethick DJ 2007. H2A.Z contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci 104: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Grossniklaus U, Paro R 2014. Transcriptional silencing by Polycomb-group proteins. Cold Spring Harb Perspec Biol 6: a019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Grunstein M, Gasser SM 2013. Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb Perspec Biol 5: 017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF 2011. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halley JE, Kaplan T, Wang AY, Kobor MS, Rine J 2010. Roles for H2A.Z and its acetylation in GAL1 transcription and gene induction, but not GAL1-transcriptional memory. PLoS Biol 8: e1000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Robert F 2010. Random deposition of histone variants: A cellular mistake or a novel regulatory mechanism? Epigenetics 5: 368–372. [DOI] [PubMed] [Google Scholar]
- Hardy S, Jacques PE, Gevry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, Robert F 2009. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet 5: e1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. 2011a. Altered telomeres in tumors with ATRX and DAXX mutations. Science 333: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH, et al. 2011b. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179: 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K 2005. Assembly of variant histones into chromatin. Ann Rev Cell Dev Biol 21: 133–153. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Furuyama T 2012. The unconventional structure of centromeric nucleosomes. Chromosoma 121: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines KA, Cryderman DE, Flannery KM, Yang H, Vitalini MW, Hazelrigg T, Mizzen CA, Wallrath LL 2009. Domains of heterochromatin protein 1 required for Drosophila melanogaster heterochromatin spreading. Genetics 182: 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodl M, Basler K 2009. Transcription in the absence of histone H3.3. Curr Biol 19: 1221–1226. [DOI] [PubMed] [Google Scholar]
- Hodl M, Basler K 2012. Transcription in the absence of histone H3.2 and H3K4 methylation. Curr Biol 22: 2253–2257. [DOI] [PubMed] [Google Scholar]
- Hou H, Wang Y, Kallgren SP, Thompson J, Yates JR 3rd, Jia S 2010. Histone variant H2A.Z regulates centromere silencing and chromosome segregation in fission yeast. J Biol Chem 285: 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Liu Y, Wang M, Fang J, Huang H, Yang N, Li Y, Wang J, Yao X, Shi Y, et al. 2011. Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev 25: 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, Ge K, Levens D, Crane-Robinson C, Zhao K 2013. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell 12: 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Kallen CB, Dhar R, Baquero MT, Mason CE, Russell BA, Shah PK, Liu J, Khramtsov A, Tretiakova MS, et al. 2008. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Syst Biol 4: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhang Z, Xu M, Li Y, Li Z, Ma Y, Cai T, Zhu B 2013. H3.3-h4 tetramer splitting events feature cell-type specific enhancers. PLoS Genet 9: e1003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh YH, Sherley JL 2011. Molecular cloaking of H2A.Z on mortal DNA chromosomes during nonrandom segregation. Stem Cells 29: 1620–1627. [DOI] [PubMed] [Google Scholar]
- Ingouff M, Hamamura Y, Gourgues M, Higashiyama T, Berger F 2007. Distinct dynamics of HISTONE3 variants between the two fertilization products in plants. Curr Biol 17: 1032–1037. [DOI] [PubMed] [Google Scholar]
- Jenuwein T 2001. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol 11: 266–273. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD 2001. Translating the histone code. Science 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. 2011. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G 2009. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet 41: 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien J, Astrand C, Szenker E, Garrett N, Almouzni G, Gurdon JB 2012. HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics Chromatin 5: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. 2010. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468: 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya H, Shibahara KI, Taoka KI, Iwabuchi M, Stillman B, Araki T 2001. FASCIATA genes for chromatin assembly factor-1 in Arabidopsis maintain the cellular organization of apical meristems. Cell 104: 131–142. [DOI] [PubMed] [Google Scholar]
- Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA 2010. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell 39: 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobor MS, Lorincz MC 2009. H2A.Z and DNA methylation: Irreconcilable differences. Trends Biochem Sci 34: 158–161. [DOI] [PubMed] [Google Scholar]
- Ku M, Jaffe JD, Koche RP, Rheinbay E, Endoh M, Koseki H, Carr SA, Bernstein BE 2012. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol 13: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SV, Wigge PA 2010. H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140: 136–147. [DOI] [PubMed] [Google Scholar]
- Kundu S, Peterson CL 2010. Dominant role for signal transduction in the transcriptional memory of yeast GAL genes. Mol Cell Biol 30: 2330–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladurner AG 2003. Inactivating chromosomes: A macro domain that minimizes transcription. Mol Cell 12: 1–3. [DOI] [PubMed] [Google Scholar]
- Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, et al. 2010. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143: 367–378. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Li E, Zhang Y 2014. DNA methylation in mammals. Cold Spring Harb Perspec Biol 6: a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Gadue P, Chen K, Jiao Y, Tuteja G, Schug J, Li W, Kaestner KH 2012. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 151: 1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light WH, Brickner DG, Brand VR, Brickner JH 2010. Interaction of a DNA zip code with the nuclear pore complex promotes H2A.Z incorporation and INO1 transcriptional memory. Mol Cell 40: 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Liu X, McEachron T, Schwartzentruber J, Wu G 2014. Histone H3 mutations in pediatric brain tumors. Cold Spring Harb Perspec Biol 6: a018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes NF, Toh GW 2005. DNA repair: The importance of phosphorylating histone H2AX. Curr Biol 15: R99–R102. [DOI] [PubMed] [Google Scholar]
- Loyola A, Almouzni G 2004. Histone chaperones, a supporting role in the limelight. Biochim Biophys Acta 1677: 3–11. [DOI] [PubMed] [Google Scholar]
- Lu PY, Levesque N, Kobor MS 2009. NuA4 and SWR1-C: Two chromatin-modifying complexes with overlapping functions and components. Biochem Cell Biol 87: 799–815. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Luk E, Ranjan A, Fitzgerald PC, Mizuguchi G, Huang Y, Wei D, Wu C 2010. Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S 2003. Phylogenomics of the nucleosome. Nat Struct Biol 10: 882–891. [DOI] [PubMed] [Google Scholar]
- Malik HS, Henikoff S 2009. Major evolutionary transitions in centromere complexity. Cell 138: 1067–1082. [DOI] [PubMed] [Google Scholar]
- March-Diaz R, Reyes JC 2009. The beauty of being a variant: H2A.Z and the SWR1 complex in plants. Mol Plant 2: 565–577. [DOI] [PubMed] [Google Scholar]
- Marques M, Laflamme L, Gervais AL, Gaudreau L 2010. Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics 5: 267–272. [DOI] [PubMed] [Google Scholar]
- Martins-Taylor K, Sharma U, Rozario T, Holmes SG 2011. H2A.Z (Htz1) controls the cell-cycle-dependent establishment of transcriptional silencing at Saccharomyces cerevisiae telomeres. Genetics 187: 89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Duronio RJ 2002. Histone mRNA expression: Multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol 14: 692–699. [DOI] [PubMed] [Google Scholar]
- Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ 2002. The human and mouse replication-dependent histone genes. Genomics 80: 487–498. [PubMed] [Google Scholar]
- Matsuda R, Hori T, Kitamura H, Takeuchi K, Fukagawa T, Harata M 2010. Identification and characterization of the two isoforms of the vertebrate H2A.Z histone variant. Nucleic Acids Res 38: 4263–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, et al. 2008. Nucleosome organization in the Drosophila genome. Nature 453: 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta M, Braberg H, Wang S, Lozsa A, Shales M, Solache A, Krogan NJ, Keogh MC 2010. Individual lysine acetylations on the N terminus of Saccharomyces cerevisiae H2A.Z are highly but not differentially regulated. J Biol Chem 285: 39855–39865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P 2011. Drosophila CENH3 is sufficient for centromere formation. Science 334: 686–690. [DOI] [PubMed] [Google Scholar]
- Millar CB, Xu F, Zhang K, Grunstein M 2006. Acetylation of H2AZ Lys 14 is associated with genome-wide gene activity in yeast. Genes Dev 20: 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Xiao H, Wisniewski J, Smith MM, Wu C 2007. Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell 129: 1153–1164. [DOI] [PubMed] [Google Scholar]
- Morillo-Huesca M, Clemente-Ruiz M, Andujar E, Prado F 2010. The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS One 5: e12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Shen X 2005. DNA repair in the context of chromatin. Cell Cycle 4: 568–571. [PubMed] [Google Scholar]
- Morrison AJ, Shen X 2009. Chromatin remodelling beyond transcription: The INO80 and SWR1 complexes. Nat Rev Mol Cell Biol 10: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Stillman B, Horvitz HR 2011. Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell 147: 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov M, Amrichova J, Parker BJ, Soboleva TA, Jack C, Williams R, Huttley GA, Tremethick DJ 2012. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat Struct Mol Biol 19: 1076–1083. [DOI] [PubMed] [Google Scholar]
- Ng RK, Gurdon JB 2008. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol 10: 102–109. [DOI] [PubMed] [Google Scholar]
- Nusinow DA, Hernandez-Munoz I, Fazzio TG, Shah GM, Kraus WL, Panning B 2007. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem 282: 12851–12859. [DOI] [PubMed] [Google Scholar]
- Ooi S, Priess J, Henikoff S 2006. Histone H3.3 variant dynamics in the germline of Caenorhabditis elegans. PLoS Genet 2: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi GA, Couble P, Loppin B 2009. Epigenetic and replacement roles of histone variant H3.3 in reproduction and development. Int J Dev Biol 53: 231–243. [DOI] [PubMed] [Google Scholar]
- Palmer DK, O’Day K, Margolis RL 1990. The centromere specific histone CENP-A is selectively retained in discrete foci in mammalian sperm nuclei. Chromosoma 100: 32–36. [DOI] [PubMed] [Google Scholar]
- Palmer DK, O’Day K, Trong HL, Charbonneau H, Margolis RL 1991. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc Natl Acad Sci 88: 3734–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL 2011. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144: 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, Brown DT, Hager G, Bustin M, Misteli T 2004. Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol 24: 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina B, Suau P 1987. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev Biol 123: 51–58. [DOI] [PubMed] [Google Scholar]
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD 2005. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan V, Whitelaw E 2003. Transgenerational epigenetic inheritance. Curr Biol 13: R6. [DOI] [PubMed] [Google Scholar]
- Ratnakumar K, Duarte LF, LeRoy G, Hasson D, Smeets D, Vardabasso C, Bonisch C, Zeng T, Xiang B, Zhang DY, et al. 2012. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev 26: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LE, et al. 2011. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell 44: 928–941. [DOI] [PubMed] [Google Scholar]
- *.Reik W, Surani A 2014. Germline and pluripotent stem cells. Cold Spring Harb Perspec Biol 10.1101/cshperspect.a019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, Ahmad K 2009. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr Biol 19: 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla ME 2010. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol 12: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban MS, Hang M, Smith MM 2011. Histone variant H2A.Z and RNA polymerase II transcription elongation. Mol Cell Biol 31: 1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiderman JI, Sakai A, Goldstein S, Ahmad K 2009. The XNP remodeler targets dynamic chromatin in Drosophila. Proc Natl Acad Sci 106: 14472–14477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BE, Ahmad K 2005. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev 19: 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231. [DOI] [PubMed] [Google Scholar]
- Shaw ML, Williams EJ, Hawes S, Saffery R 2009. Characterisation of histone variant distribution in human embryonic stem cells by transfection of in vitro transcribed mRNA. Mol Reprod Dev 76: 1128–1142. [DOI] [PubMed] [Google Scholar]
- Shuaib M, Ouararhni K, Dimitrov S, Hamiche A 2010. HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc Natl Acad Sci 107: 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet NG, Reyes M, Nardocci G, Molina A, Alvarez M 2013. Epigenetic regulation of the ribosomal cistron seasonally modulates enrichment of H2A.Z and H2A.Zub in response to different environmental inputs in carp (Cyprinus carpio). Epigenetics Chromatin 6: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboleva TA, Nekrasov M, Pahwa A, Williams R, Huttley GA, Tremethick DJ 2012. A unique H2A histone variant occupies the transcriptional start site of active genes. Nat Struct Mol Biol 19: 25–30. [DOI] [PubMed] [Google Scholar]
- Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, Ladurner AG 2009. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 28: 3423–3428. [DOI] [PubMed] [Google Scholar]
- Stoler S, Rogers K, Weitze S, Morey L, Fitzgerald-Hayes M, Baker RE 2007. Scm3, an essential Saccharomyces cerevisiae centromere protein required for G2/M progression and Cse4 localization. Proc Natl Acad Sci 104: 10571–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA, Karpen GH 2004. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol 11: 1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto RK, Clarkson MJ, Tremethick DJ, Luger K 2000. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat Struct Biol 7: 1121–1124. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S 2010. Histone variants—Ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11: 264–275. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Ahmad K, Almouzni G, Ausio J, Berger F, Bhalla PL, Bonner WM, Cande WZ, Chadwick BP, Chan SW, et al. 2012. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe M, Kouzmenko AP, Ito S, Sawatsubashi S, Suzuki E, Fujiyama S, Yamagata K, Zhao Y, Kimura S, Ueda T, et al. 2008. Activation of facultatively silenced Drosophila loci associates with increased acetylation of histone H2AvD. Genes Cells 13: 1279–1288. [DOI] [PubMed] [Google Scholar]
- Tran V, Lim C, Xie J, Chen X 2012. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338: 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updike DL, Mango SE 2006. Temporal regulation of foregut development by HTZ-1/H2A.Z and PHA-4/FoxA. PLoS Genet 2: e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD 2007. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci 104: 16609–16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viens A, Mechold U, Brouillard F, Gilbert C, Leclerc P, Ogryzko V 2006. Analysis of human histone H2AZ deposition in vivo argues against its direct role in epigenetic templating mechanisms. Mol Cell Biol 26: 5325–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Saleem RA, Ratushny AV, Roda O, Smith JJ, Lin CH, Chiang JH, Aitchison JD 2009. Role of the histone variant H2A.Z/Htz1p in TBP recruitment, chromatin dynamics, and regulated expression of oleate-responsive genes. Mol Cell Biol 29: 2346–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL 2013. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 340: 195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, Henikoff JG, Henikoff S 2010. H2A.Z nucleosomes enriched over active genes are homotypic. Nat Struct Mol Biol 17: 1500–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Worcel A, Alberts B 1976. A model for chromatin based upon two symmetrically paired half-nucleosomes. Cell 9: 409–417. [DOI] [PubMed] [Google Scholar]
- Whittle CM, McClinic KN, Ercan S, Zhang X, Green RD, Kelly WG, Lieb JD 2008. The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genet 4: e1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland G, Orthaus S, Ohndorf S, Diekmann S, Hemmerich P 2004. Functional complementation of human centromere protein A (CENP-A) by Cse4p from Saccharomyces cerevisiae. Mol Cell Biol 24: 6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt O, Albig W, Doenecke D 1996. Testis-specific expression of a novel human H3 histone gene. Exp Cell Res 229: 301–306. [DOI] [PubMed] [Google Scholar]
- Wolffe AP 1992. Chromatin: Structure and function. Academic, San Diego. [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, Zhu B 2010. Partitioning of histone H3-H4 tetramers during DNA replication-dependent chromatin assembly. Science 328: 94–98. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, Price BD 2012. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell 48: 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharioudakis I, Gligoris T, Tzamarias D 2007. A yeast catabolic enzyme controls transcriptional memory. Curr Biol 17: 2041–2046. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Pugh BF 2011. Genomic organization of H2Av containing nucleosomes in Drosophila heterochromatin. PLoS One 6: e20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Roberts DN, Cairns BR 2005. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S 2008. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456: 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, Thakar A 2008. H2A.Z: View from the top. Structure 16: 166–179. [DOI] [PubMed] [Google Scholar]
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SI 2009. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461: 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
WWW RESOURCES
- http://www.ebi.ac.uk/Tools/phylogeny. EBI server.