

Abstract
Heavy metal toxicity has serious adverse effects on the environment. The metal sequestering characteristics of a novel metal binding peptide (Glu-Cys)11 Gly+linker+hexahistidine (EC11:His6) was investigated to determine if it can absorb Cu2+ or Zn2+ cations. Molecular dynamics simulations were carried out using a model of 6 Cu2+ or Zn2+ and other ions enclosed in a fully hydrated simulation box with the designed peptide. Totally, 240 nano second (ns) simulations were done in three phases. Results showed that the selected linker is able to separate two domains of this peptide and that the carboxyl oxygens of Glu residues of EC11 in the designed peptide can absorb these ions. Sequestration of Cu2+ or Zn2+ ions by the designed peptide does not change overall tertiary and secondary structures of peptide.
Keywords: Molecular dynamics, Metal binding peptide
INTRODUCTION
Zinc (Zn) is the second most abundant transition metal in living organisms after iron and is an essential element necessary for plants (1), animals (2), and microorganisms (3). It is found in nearly 100 specific enzymes (4) and represented to all enzyme classes (1). Two examples of zinc-dependent enzymes are carbonic anhydrase and carboxy peptidase, which are vital to the processes of carbon dioxide regulation and digestion of proteins, respectively (5). Zn ions are often coordinated to the amino acid side chains of aspartic acid, glutamic acid, cysteine and histidine residues of proteins (6). In the brain, zinc is stored in specific synaptic vesicle by glutamatergic neurons and is able to modulate brain excitability (7,8). As zinc also can be a neurotoxin, this suggests that zinc homeostasis plays an important role in normal functioning of the brain and central nervous system (9). Consumption of excess zinc can cause ataxia, lethargy and copper deficiency (10). Zinc is normally found in small amounts in nature, but it can be released into the environment unduly through mining, metal processing activities (galvanized steel production), as well as burning coal and certain wastes. People living near industries using zinc could be exposed to higher levels of zinc by drinking water, breathing air and touching soil that contains the metal.
Copper is another essential element that is required for plants and animals. Easy interconversion of Cu(I) and Cu(II) has caused copper proteins to have diverse roles in biological electron transport and oxygen transportation processes (11). Tyrosinase, cytochrome C oxidase, and many superoxide dismutases are some examples of copper-containing enzymes present in animals including humans and plants. Gram quantities of copper salts taken can produce acute copper toxicity in humans, possibly due to redox cycling and the generation of reactive oxygen species that damage DNA (12,13). From another point of view, copper seems to be important for us. Microorganisms take essential metal ions, such as copper, from their environment. Antimicrobial peptides (AMPs) which are secreted by fungi, parasites, animals and humans are essential components of the first line of their defense mechanisms. One of the proposed mechanisms of action of these molecules is their ability to bind and sequester metal ions, thus preventing their uptake by microorganisms. This has been demonstrated clearly for the human neutrophil proteins such as calprotectin and lactoferrin (14,15). Based on this, copper sequestering peptides can be regarded as potential antimicrobial agents. In addition, such peptides may be useful for the treatment of diseases such as Wilson's disease in which patients have high levels of copper in the body (16).
Cysteine-rich peptides such as glutathione (GSH), metallothioneins (MTs) and phyto-chelatins can bind metal ions (such as zinc, copper, lead, mercury and cadmium) and sequester them in biologically inactive forms (17). MT is a family of cysteine-rich, low molecular weight (MW ranging from 500 to 14000 Da) proteins. They are localized to the membrane of the Golgi apparatus. Phyto-chelatins (PCs) are a family of short cysteinerich metal-chelating peptides with a structure of (γGlu-Cys)nGly (where, n varies from 2-11 depending on the species and conditions of exposure to metal ions). The PCs are found in many plants and yeasts. PCs are enzymatically synthesized from glutathione by phytochelatin synthase. Mechanisms of phytochelatin biosynthesis are not understood in sufficient details till now. Therefore, genetic engineering of plants for phytoremediation is not an easy technology (18). (Glu-Cys)nGlyare synthetic phytochelatins (ECs) with the standard α-linkage in their structure. In spite of phytochelatins, ECs can be easily synthesized on the ribosomal machinery (18) or prepared by peptide synthesizer(19).
Peptides are excellent materials for nano-biotechnology because they are small, fold at very fast rates nanosecond (ns) and can be easily synthesized by chemical methods or biological cloning (20).
Protein folding of fusion proteins and ion exchange between protein surface and bulk can be studied using molecular dynamics (MD) simulations. MD provides an atomic detailed description of the ion-protein time evolution (21).
Understanding the mechanism of synthetic phytochelatins folding is an important step for the rational design of chelating peptides. Also, owing to the experimental difficulties in studying the dynamics of small ions mobility on the protein surfaces on the molecular scale, ligand exchange reactions can only be studied by MD simulations. When only a small number of ions are present in the solution, their dynamics can be studied directly by monitoring the distance between each ion and the protein (21). In this research, we decided to study the folding and metal binding properties of a novel synthetic phytochelatin by theoretical methods prior to expressing this peptide for in vitro evaluations. Thus, design with this formula (Glu-Cys)11Gly+linker+ hexahistidine(EC11:His6) was synthesized. By preparing EC11 conjugated to other moieties, Baeand and coworker were able to design Lpp-OmpA-EC11 to anchor the peptide to the cell surface of certain microorganisms (17).
Our rational for the design of EC11:His6 was to prepare a fusion peptide to sequester heavy metals such as zinc and copper which exist in high quantities in the soil. Its folding mechanism and also its ability to chelate these metals near the protein surface were investigated using theoretical methods. More specifically, a gene encoding of this peptide was also constructed.
MATERIALS AND METHODS
MODELLER 9v2 was used for homology modeling of EC11 sequence [(Glu-Cys)11Gly] three-dimensional structure (22). Model templates were EC11 sequence alignments with other sequence (using BLAST site, copper thionein (PDB code: 1RJU) [Max score; 23.5%, Total score; 60.9%, Query coverage; 100%, E value: 7.2%] and elapid snake venom metalloproteases (PDB code: 3K7L) [Max score; 22.7%, Total score; 43.7%, Query coverage; 100%, E value: 11%]. Of the 100 models generated with MODELLER, the one corresponding to the lowest value of the probability density function (pdf) and the fewest restraints violations was selected for further analysis. After making a model for EC11 sequence, its C-terminal was linked to the N-terminal of a linker sequence. Crystal structure of the linker (PDB code: 1ej6C_2) with sequence “RGRWMARLARM” was taken from linker library (www.ibi.vu.nl) (23). This linker with a rigid-helical structure belongs to the C chain of a viral protein containing zinc.
The C-terminal linker was then linked to a hexahistidine sequence. This combination was named EC11:His6, containing totally 40 residues. The (Glu-Cys)11 sequence is negatively charged, so its attracted to hexahistidine with positive charges. Thus EC sequence in the putative fusion peptide would be expected to have a medium size and separated by a long linker sequence from the hexahistidine moiety. EC11 sequence was designed such that it could absorb ions, and also the hexahistidine tail added to this synthetic phytochelatin to facilitate extraction of it from the solution. Since hexahistidine tail may interfere with EC11 sequence, a helical linker was inserted between EC11 sequence and hexahistidine tail. Helical linkers are thought to act as rigid spacers to prevent non-native interaction between domains (24).
Phase I
MD simulation and molecular mechanic (MM) minimization were performed using GROMACS 4.5.3 package under a Gromos force field (G43A1) (25,26,27,28). MD simulations were carried out at the NPT ensemble and periodic boundary conditions. Van der Waals forces were treated by using a cut-off of 12 Å. The electrostatic interactions were calculated by the Particle-Mesh Ewald method with a 14 Å cut-off (29).
EC11:His6 was solvated by a layer of water molecules with a thickness of at least 1.2 nm in all directions. Common value for this parameter is 0.5-1 nm. However, in order to have enough water molecules and an appropriate ionic strength a value of 1.2 nm was used.
The size of simulation box was 5.0 × 7.6 × 5.8 nm and the protein was solvated by 7031 spc216 water molecules. The frequency to update the neighbor list was 10. The default protonation state of Gromacs package was used except histidines with positively charged residues helping extract the molecules from other compounds in experimental processes. As all 11 Glu were deprotonated (negative) and there exist six positively charged histidines in EC11 sequence and four positively charged arginines in the linker, the total charge of EC11:His6 reached to “-1”, and the ionic strength for the simulation box was set at 140 mM. The reason for choosing this ionic strength for this study was that this ionic strength is closed to biological ionic strength (30,31). This makes folding study and ion absorption in a simulated biological system possible. The system was then neutralized by adding 18 Na+ and 17 Cl- ions to the simulation box.
MD simulation was accomplished in four steps. In the first step, the entire system was minimized using the steepest descent followed by conjugate gradients algorithms. In the second step, the solvent and Na+ and Cl- ions were allowed to evolve. This was done through minimization and molecular dynamics in the NVT ensemble for 500 ps and in the NPT ensemble for 1000 ps in which initial configuration of the structure of EC11:His6 was kept fixed. In the third step, in order to obtain equilibrium geometry at 300 K and 1 atm, the system was heated at a weak temperature coupling (τ = 0.1 ps) and pressure coupling (τ = 0.5 ps). The Berendsen algorithm was used for thermostat and barostat during the equilibration phase (32). The LINCS algorithm was applied to constrain the lengths of hydrogen-containing bonds (33). The temperature of the system was then increased from 0 K to 300 K and the velocities at each step were reassigned according to the Maxwell-Boltzmann distribution at that temperature and equilibrated for 300 ps. In the final step, 80 ns MD simulation was performed at 300 K with a 2 femto second (fs) time step and therefore a folded structure was obtained. The thermostat and barostat for production phase were Nosé-Hoover thermostat and Parrinello-Rahmanbarostat (32).
After running of 80-ns MD simulation, the overall stereochemical quality of the final developed model for native EC11:His6 model was assessed by the program PROCHECK (34).
G-factor of the developed model was calculated using PROCHECK. Environment profile of the model was further checked with Verify-3D (Structure Evaluation Server) (35).
The secondary structural elements of the protein were calculated by do_dssp, which utilizes the DSSP program utility during MD simulation (36).
Principal component analysis (PCA) was performed using g_covar and g_anaeig modules of the GROMACS 4.5.3 package.
Phase II
The native EC11:His6 obtained from phase I, was centered in a box of water. The dimension of the box was 6.6 × 5.4 × 5.8 nm. Almost 6900 spc216 water molecules, 6 Zn2+(in an aqueous environment, hydrated Zn, e.g [Zn(H2O)6]2+ and dehydrated Zn2+ ions may be present which cannot be ignored) and 11 Cl- ions were added randomly to the box. In this case, the ionic strength remained constant (140 mM). Then, eighty ns MD simulation was performed while keeping all other parameters fixed.
Phase III
In this phase the native EC11:His6 obtained from phase I was centered in a box of water with the size of 5.8 × 5.4 × 6.6 nm with almost 6900 spc216 water molecules. 6 Cu2+ and 11 Cl- were randomly put in the box. These ions keep the ionic strength constant (140 mM). Then again 80-ns MD simulation was run with all parameters kept the same as before.
RESULTS
There were two reasons for selecting this peptide; the medium size desired for the final fusion peptide and the experimental facilities which make the construction of EC11:His6 gene possible. In order to reach the native structure of EC11:His6, MD simulations were performed. This method can give a description of full protein flexibility as it interacts with ions. It also applies to the laws of classical mechanics to compute the motion of particles in a molecular system. In this study, MD simulation was performed in three different phases.
In the following section, we investigate folding and ion absorption ability of EC11:His6 peptide.
Phase I
In phase I, the native structure of EC11:His6 was obtained after 80 ns by MD simulation. The root mean square deviation (RMSD) was calculated for protein backbone atoms using starting structure as a reference (Fig. 1). Variation in total energy versus time for 80 ns MD simulations was 0.0003 and the kinetics and potential energy fluctuated in equal and opposite direction for MD simulation in this phase (Fig. 2).
Fig. 1.
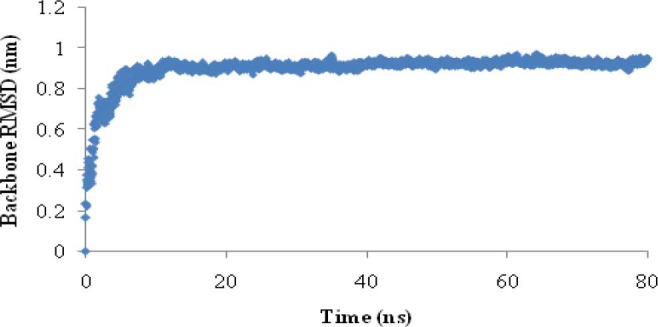
Root mean square deviation (RMSD) of protein backbone during 80 ns MD simulation in phase
Fig. 2.
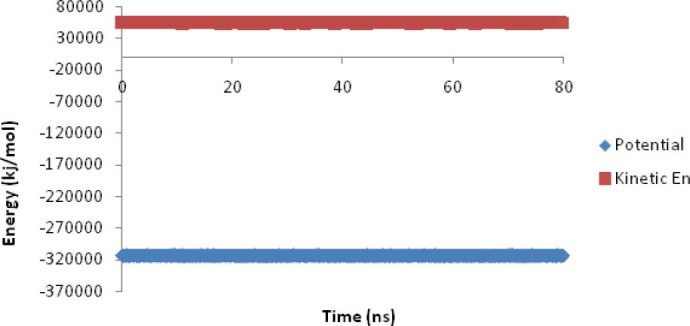
The kinetics and potential energies during 80 ns MD simulations in phase I.
Table 1 shows protein backbone RMSD, radius of gyration (Rg), temperature, potential energy and secondary structure elements at the last 60 ns MD simulations for EC11:His6 in the presence of Na+ and Cl- ions.
Table 1.
Results of the last 60 ns of 80 ns MD simulation for EC11:His6 in phases I, II and III.

The RMSDs of Na+ and Cl- at the last 60 ns of simulation in this phase were 3.46 ± 0.23 nm and 3.43 ± 0.25 nm, respectively. Minimum distance between Na+ and Cl- ions with peptide atoms at the last 60 ns in this phase was 0.49 ± 0.16 nm and 0.52 ± 0.23 nm, respectively.
In order to show whether the linker was able to separate two moieties of EC11:His6 from each other, the distance between the center of mass of EC11 sequence and the center of mass of hexahistidine sequence was calculated (Fig. 3). Results showed that this distance reached to 1.12 ± 0.03 nm after 12 ns and then remained stable throughout the simulation.
Fig. 3.
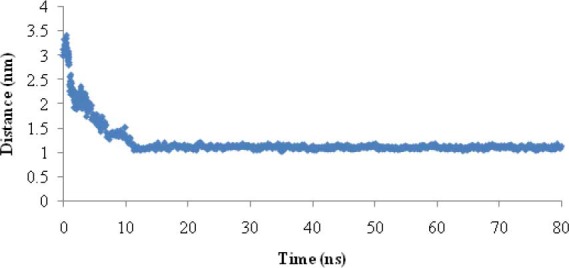
Distance between center of mass EC11 of sequence from center of mass hexahistidine sequence during 80 ns MD simulation.
Radial distribution function (RDF) or pair correlation function (r), describes how the atomic density varies as a function of the distance from one particular atom. The radial distribution function is a useful tool to describe the structure of a system, particularly of liquids. Fig. 4 shows the radial distribution functions of Na+ or Cl- to oxygen of Glus of EC11. This figure shows that at short distances (less than 0.4 nm) the RDF for Na+ reaches the maximum level.
Fig. 4.
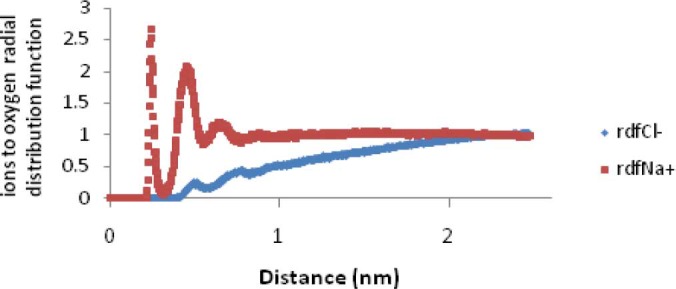
Na+ or Cl- to oxygen (of Glus of EC11) radial distribution function in phase I.
Table 2 shows the minimum distance between the carboxyl oxygen of glutamic acid residues and Na+ or Cl- in phase I at the last 60 (out of 80) ns MD simulation.
Table 2.
Minimum distances between carboxyl oxygen of glutamic acid residues of EC11 with ions and diffusion coefficients of ions in all phases during the last 60ns of MD simulation.

The calculated diffusion coefficients of the ions are reported in Table 2. These values are compatible with the experimental results (2.03 × 10-5 and 1.33 × 10-5 cm2/s for the Cl- and Na+ ions respectively) (40).
Ramachandran plot for native EC11:His6 model after 80 ns MD simulation was drawn and provided along with the plot statistics in Fig. 5. As it is seen, only two residues (Glu 5 and Glu 11) or about five percent of the residues are lying in the disallowed regions of the Ramachandran plot.
Fig. 5.

Ramachandran plot and plot statistics of native EC11:His6 model.
The G-factor value gives a measure of how far from the normal regions of the plot each residue lies. Low G-factors indicate residues in unlikely conformations. A typical G-factor is -1 and the overall G-factor in this phase was -1.11. Fig. 6 shows the G-factors of some properties of the final model. The first four eigenvalues in this phase were, respectively, 1.97, 0.59, 0.29 and 0.12. The other eigenvalues were trivial. Projections onto eigenvectors are overall coordinates that give information on how the system moves in the directions described by the eigenvectors (24). Projection of Cα motions of EC11:His6 during the first three principal components (PC) for 80 ns in this phase is shown in Fig. 7.
Fig. 6.
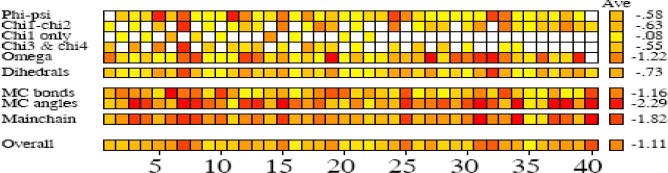
G-factors of some properties of final model in phase I.
Fig. 7.
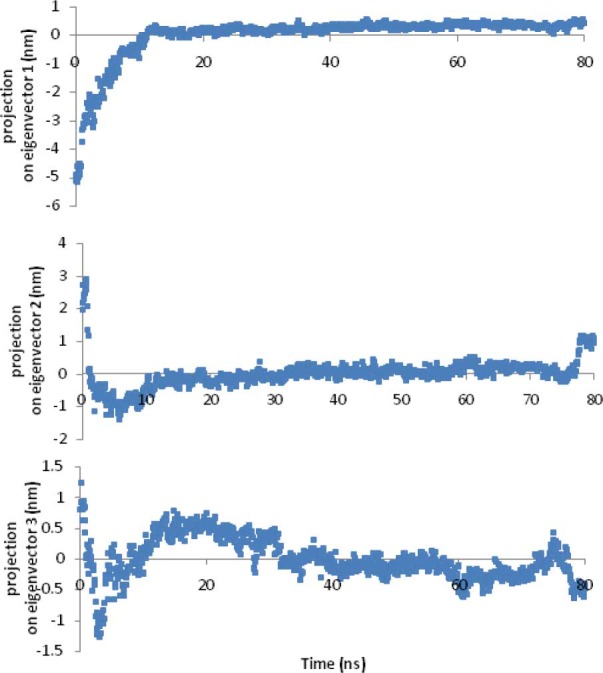
Projection of Cα motions of EC11:His6 during 80 ns MD simulation onto its first 3 principal components in phase I.
The cosine content was introduced as a measure of the closeness of the PC to a cosine shape, which appeared to be a good indicator for predicting whether or not a trajectory has sampled a free-energy landscape sufficiently for convergence (41). Cosine content of the first three PC at last 40 ns of this phase was, respectively, 0.15, 0.001, and 0.002 which is a good indicator of sampling.
Fig. 8 shows a two-dimensional plot of PC1-PC2 in phase I. Three states and a transition state (TS) during folding of EC11:His6 can be observed in this figure.
Fig. 8.
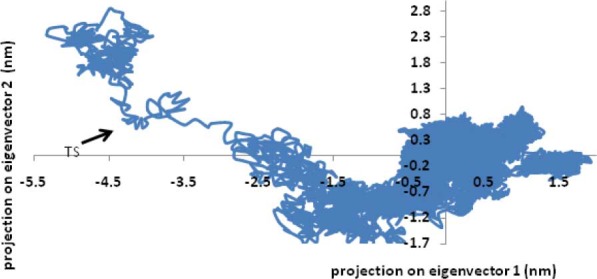
Projections of EC11:His6 structures found in 80 ns MD simulation on to the 1–2 eigenvector plane in Phase I.
Phase II
To investigate the interaction between Zn2+ and EC11:His6, the final structure obtained from phase I was used as the starting geometry for an 80 ns simulation in the presence of 6 Zn2+ and 11 Cl- ions. The variations in the total energy versus time for 80 ns MD simulations was about 0.00002 and the kinetics and potential energy fluctuated in equal and opposite direction for MD simulation in this phase. Table 1 shows the protein backbone RMSD, Rg, temperature, potential energy and secondary structure elements at the last 60 (out of 80) ns MD simulation in this phase.
Also the RMSDs of Zn2+ and Cl- at the last 60 ns of simulation in this phase were 2.44 ± 0.39 nm and 3.07±0.3 nm, respectively. The minimum distance between Zn2+ ions and the sulfur atoms of cysteine residues in phase II and at the last 60 ns of simulation was 0.65 ± 0.18 nm.
Fig. 9 shows the minimum distance between Zn2+ ions and the sulfur atoms of cysteine residues in phase II. Table 2 shows the minimum distance between the carboxyl oxygen of glutamic acid residues and Zn2+ and Cl- in phase II at the last 60 (out of 80) ns MD simulation.
Fig. 9.
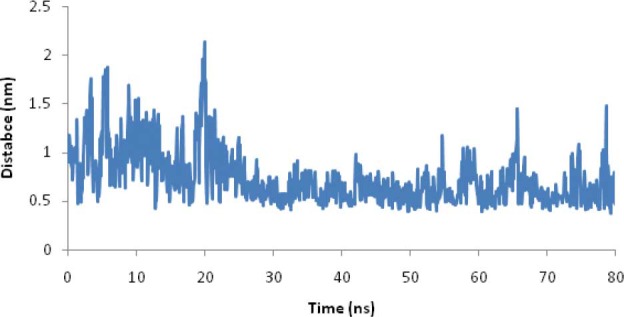
Minimum distance between Zn2+ ions and sulfur atoms of cysteines during 80 ns MD simulation in phase II.
In addition, the minimum distance between the EC11 sequence ([Glu-Cys]11Gly) and Zn2+ ions in the last 60 (out of 80) ns MD simulation was 0.42 ± 0.13 nm. Fig. 10 presents the simulation time versus the minimal distances between the ions and the carboxyl oxygens of glutamic acid residues of EC11 as a histogram.
Fig. 10.
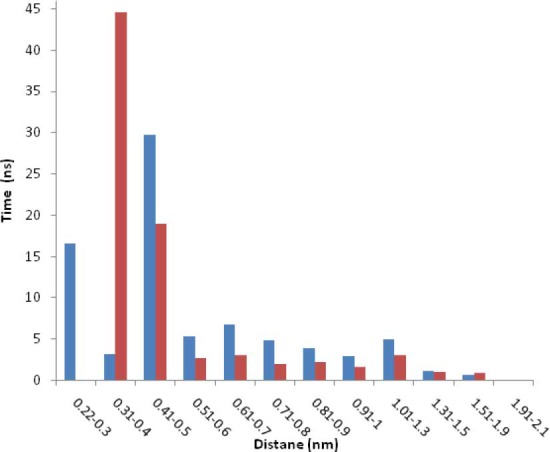
Detention time for Zn2+ ions and Cu2+ ions versus distance from carboxyl oxygen of glutamic acid residues of EC11.
Also, the minimum distance between the hexahistidine sequence and Zn2+ ions in the last 60 (out of 80) ns MD simulation was 1.17 ± 0.32 nm. Also minimum distance between the linker sequence (RGRWMARLARM) and Zn2+ ions in this phase was 0.92 ± 0.24 nm. Fig. 11 shows Zn2+ oxygen radial distribution function. The calculated diffusion coefficients of Zn2+ and Cl- ions are also reported in Table 2.
Fig. 11.
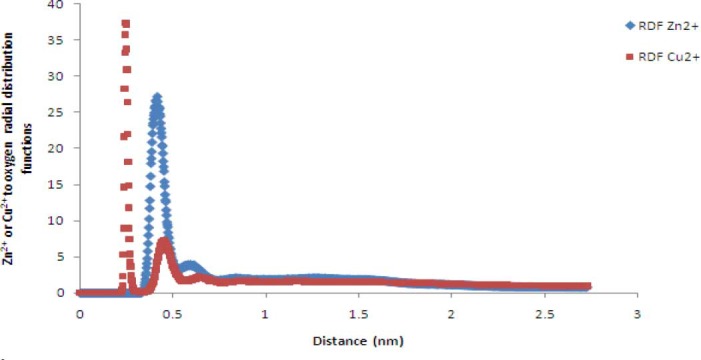
Zn2+ or Cu2+ to oxygen (of Glus of EC11) radial distribution functionsin phases II and III.
Phase III
In phase III, in order to investigate the interaction between Cu2+ and EC11:His6, the final structure obtained from the phase I was used for an 80 ns simulation in the presence of 6 Cu2+ and 11 Cl-.
The variations in total energy versus time for 80 ns of the MD simulations were 9 × 10-9 and the kinetics and potential energy fluctuated in equal and opposite direction for MD simulation of EC11:His6 with Cu2+. Table 1 shows the protein backbone RMSD, Rg, temperature, potential energy and helix elements at the last 60 (out of 80) ns MD simulation.
Also RMSDs of Cu2+ and Cl- at the last sixty ns of simulation in this phase were 2.72 ± 0.42 nm and 3.02 ± 0.29 nm, respectively.
Fig.12 shows the minimum distance between Cu2+ ions and the sulfur atoms of cysteine residues during 80 ns MD simulation in phase III. The average of this value at the last 60 ns of simulation was 0.83 ± 0.27 nm.
Table 2 shows the minimum distances between the carboxyl oxygen of glutamic acid residues and Cu2+ and Cl- ions in phase III at the last 60 (out of) 80 ns MD simulation. The calculated diffusion coefficients of the Cu2+ and Cl- in this phase are also reported in Table 2. Furthermore, the minimum distance between EC11 sequence and Cu2+ ions in the last 60 (out of 80) ns MD simulation was 0.5 ± 0.26 nm which is similar to the results obtained for Zn2+. Also, the minimum distance between the hexahistidine sequence and Cu2+ ions in the last 60 (out of 80) ns MD simulation was 1.12 ± 0.37 nm. Minimum distance between linker sequence and Cu2+ ions at the last 60 ns in this phase was 0.92 ± 0.26 nm.
Fig. 11 shows Cu2+ to oxygen radial distribution functions in phase III. This figure shows high probability of finding of Cu2+ at a distance about 0.4 nm from glutamic acid oxygens. The calculated diffusion coefficients of the Cu2+ and Cl- are reported in Table 2.
DISCUSSION
Phase I
The large backbone RMSD relative to starting structure (0.92 nm) shows large conformational changes of the peptide relative to starting structure. In addition, small changes (small standard deviation) of the backbone RMSD during the last 60 ns and also small changes in helix structure showed that the length of MD simulation was adequate for equilibration step and that simulation was stable under simulation condition (Fig. 1 and Table 1). Small variations in total energy versus time show that law of energy conservation is fulfilled in MD simulations (Fig. 2) (42). Small standard deviation of the RMSDs of Na+ and Cl- at the last 60 ns of simulation indicates that ions reached to stable positions during the last 60 ns MD simulation. Minimum distance between Na+ and Cl- ions with peptide atoms is consistent with the results of Friedman's study that showed in the case of S6 ribosomal protein, Na+ ion comes to 0.43 nm from protein surface (21). This proves that protein can attract Na+ and Cl- ions.
The distance between the center of mass of EC11 sequence and the center of mass of hexahistidine sequence demonstrates that the linker sequence is able to separate two moieties from each other (Fig. 3). The radial distribution functions (RDF) of Na+ or Cl- to oxygen of Glus of EC11 indicates that carboxyl oxygen of glutamic acid residues of EC11 can attract Na+ ions at 0.4 nm distance. When separation distance increases, the probability of finding Cl- ions and oxygen atoms at this distance increases. At long distances (e.g. 2.5 nm) RDF approaches to determine value, indicates that no long-rang order is present, so oxygen atoms cannot absorb Cl-. This is due to the strong repulsive forces between Cl- ions and oxygen atoms. (Fig. 4). The minimum distance between the carboxyl oxygen of glutamic acid residues and Na+ or Cl- in phase I also show that while carboxyl oxygens of EC11 are repelled Cl- ions will attract Na+ ions (Table 2). Ramachandran plot for native EC11:His6 model after 80 ns MD simulation shows that the generated model is correct and can be used in the next phases (Fig. 5). The value of G-factor means that the produced model is acceptable (Fig. 6).
The final step of testing of the model was the packing quality of each residue as evaluated by the Verify-3D method, which represents the profile achieved with respect to the residues. The compatibility score above zero in the Verify-3D graph in this phase (data not shown) is corresponded to acceptable side-chain environments. This suggests that the model has an overall self-consistency in terms of sequence-structure compatibility. The small values of all eigenvalues except the first two eigenvalues in this phase means that the first and the second PC are the most important indicators of the atomic fluctuations for this peptide. Cosine-shaped of the first three principal components in the first 20 ns can be interpreted as transition of the protein from one state to another during its folding. After a total of 20 ns, system reaches equilibrium and slower fluctuations accrue (Fig. 7). The small cosine content of the first three PC at last 40 ns of this phase was a good indicator of sampling.
Phase II
Small variations in total energy versus time show that the law of energy conservation is fulfilled in this phase. A comparison of helix structure between this phase and phase I show that in the presence of Zn2+ ions, no considerable changes in helix of the peptide occur. A small standard deviation of RMSDs of Zn2+ and Cl- at the last 60 ns of simulation in this phase indicates that Zn2+ and Cl- reached a stable state during the last 60 ns of MD simulation. The minimum distance between Zn2+ ions and the sulfur atoms of cysteine residues is about three times the length of Zn-S covalent bond in ZnS4 complex (0.27 nm) (43,44). However, since we have to protonate cysteines, Zn2+ ions are not able to come closer to sulfur atoms. Fig. 9 shows that the mercapto group of cysteine residues can absorb Zn2+ ions. In the detained state, the ions did not lose their freedom of motion; they were able to shuttle between nearby attractor sites, with a restriction that lowered their probability of moving far from the protein. This restriction is related to the electrostatic potential. The results of Table 2 also show that while carboxyl oxygens of EC11 are repelled Cl- ions will attract Zn2+ ions. They even attract Zn2+ more powerful than the mercapto group of cysteine residues. This is probably because carboxyl oxygen of glutamic acid residues has more negative charges than mercapto groups of cysteine residues. The minimum distance between the EC11 sequence and Zn2+ ions indicates that EC11 sequence can sequester Zn2+ ions.
Fig. 10 shows that Zn2+ rapidly shuttles between various carboxyl oxygens of EC11 attractor site and is confined to the vicinity of the ion attractors for a long duration (up to several hundreds of ps). This proves that the local electrostatic field strongly based on their Brownian motions. These values are consistent with the tendency of the ions to be retained near the protein, either at a Van Der Waals contact distance or separated by a single or multiple solvation layers (21). Then the protein was able to detain ions to its immediate vicinity, 0.6 nm or less from its surface, for time frames as long as hundreds of picoseconds. The minimum distance between the hexahistidine sequence and Zn2+ ions indicates that hexahistidine sequence cannot sequester Zn2+. Minimum distance between the linker sequence and Zn2+ ions shows that the linker cannot absorb Zn2+ ions. Radial distribution function of Zn2+ oxygen shows a high probability of finding Zn2+ at a distance about 0.4 nm of carboxyl oxygen of glutamic acid residues (Fig. 11).
Phase III
Small variations in total energy versus time and small standard deviation of RMSD in this phase indicated the simulation was correct. A comparison of helix structure between this phase and phase I show that in the presence of Cu2+ ions, helix structure of the peptide did not change (Table 1). Small standard deviation Cu2+ and Cl- (0.42 nm and 0.29 nm) indicates that ions reached a stable state after 20 ns MD simulation. The minimum distance between Cu2+ ions and the sulfur atoms of cysteine residues (0.83 ± 0.27 nm) is about four times the length of Cu-S covalent bond in CuS3 (0.219 nm) and CUS4 (0.231 nm) complexes (45). This distance is longer than Cu-S bond since this distance does not belong to a covalent bond and also sulfur atoms are protonated (Fig. 12).
Fig. 12.
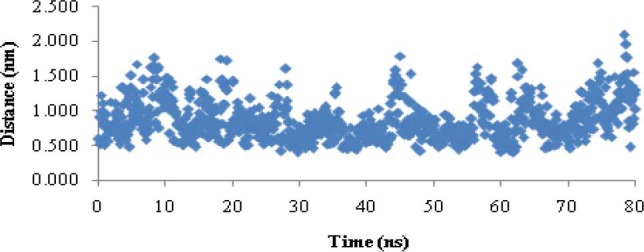
Minimum distances between Cu2+ ions and sulfur of cysteine residues during 80 ns MD simulation in phase III.
The minimum distances between the carboxyl oxygen of glutamic acid residues and Cu2+ and Cl- ions results reveal that while carboxyl oxygens of EC11 are repelled Cl- ions will attract Cu2+ ions (Table 2). Also carboxyl oxygens can almost attract Cu2+ ions more powerful than the mercapto group of cysteines. This is due to their more negative charges than those of the sulfur atom in mercapto groups of cysteine.
The minimum distance between EC11 sequence and Cu2+ ions proves that EC11 sequence can also sequester Cu2+ ions. Minimum distance between the hexahistidine sequence and Cu2+ ions (1.12 ± 0.37 nm) illustrate that hexahistidine sequence cannot sequester Cu2+ ions. This finding confirms the experimental works (16). Minimum distance between linker sequence and Cu2+ ions (0.92± 0.26 nm) shows that the linker sequence are not able to absorb Cu2+ ions. The results of Cu2+ to oxygen radial distribution function shows that carboxyl oxygens can absorb Cu2+ ions more powerful from Zn2+ ions (Fig. 11).
A comparison between the radius of gyration of phases II and III (Table 1) indicates that sequestration of Cu2+ or Zn2+ ions by the designed peptide does not change the radius of gyration or overall tertiary structure and secondary structure of the peptide. The experimental results confirm this finding. It has been shown that the addition of Cu2+ from 1 to 50 μM causes only slight changes in the circular dichroism spectrum of an AMP, microplusin. This indicates that ion binding does not significantly alter the protein conformation (16). Ion concentration in this study was more than 50 μM but during MD simulation, conformational change in protein might have occurred even though the final structures were the same.
The overall flexibility of EC11:His6 was calculated by the trace of the diagonalized covariance matrix of the Cα atoms positional fluctuations in three phases. It was 3.56, 1.47 and 1.11 in phase I, II and III, respectively. This indicates that EC11:His6 has a high degree of flexibility in phase I, but low degree flexibilities in phase II and III.
Baeand coworkers demonstrated that synthetic phytochelatins ranging from 8-20 cysteines (EC8, EC11 and EC20) have cadmium binding capability on the host cells (17). Experimental and molecular modeling studies have shown that the metal-sulfur connectivity and the overall structures of the metal binding sites are very similar for Cd-MT and Zn-MT (46,47). According to hard-soft metal ion concept, hard acids react preferentially with hard bases, and soft acids react preferentially with soft bases. Zn2+ is neither a hard nor soft but an intermediate metal ion. However, Cd2+ is a soft metal ion, and has a preference for soft donor atoms for example sulfur atoms of cysteine. This leads to the conclusion that EC11:His6 can also sequester Cd2+ (48,49).
The number of random coil, bend, turn and alpha helix structure of the designed peptide with and without Zn2+ and Cu2+ during 80 ns MD simulation were calculated (data not shown). However, generally no considerable changes occurred in the total secondary structure in the presence of Zn2+ or Cu2+ ions (Table 1).
CONCLUSION
Explicit solvent MD simulations were carried out to design a peptide under different environmental conditions. Its ability to fold and absorb Zn2+ and Cu2+ ions was then examined. These simulations revealed that the linker sequence “RGRWMARLARM” is able to separate two domains of the designed peptide so that there are no conflicts between them. Furthermore, there exists a fast exchange of the ions between the protein's surface and the bulk, indicating competition between two forces: the electrostatic attraction that favors the detainment and the entropic drive that prefers the free state of the ion. Throughout most of the simulation time, the ion diffuses in a Brownian motion in the bulk, but once an ion is trapped by the protein's Coulomb cage, it is drawn to the nearest attractor site. The obtained results show the dynamics of ions near the designed peptide in which the peptide can detain Zn2+ and Cu2+ to its immediate vicinity of EC11 residues. In spite of our expectations, the results confirm that Zn2+ and Cu2+ can be absorbed by the carboxyl oxygen of glutamic acids better than mercapto group of cysteine residues. The linker sequence and hexahistidine moieties are not capable to absorb Zn2+ and Cu2+ ions. Because of the similarity of Zn2+ and Cd2+, the absorption of Cd2+ by the designed peptide is also expected. When an ion is delayed near the protein for such a long period of time, its motion must be biased by the presence of the protein. Since EC11:His6 is able to absorb ions; it can be used as a nano bio filter attached to a polymer (or bed) to sequester Zn2+ or Cu2+ and other ions. Also, this fusion peptide as an antimicrobial agent appears to be a good candidate for further studies. It can also be used to treat heavy metal toxicity. The obtained results were consistent with other studies where residues that are up to 1.0–1.5 nm apart from each other could form proton attractive domains and share the proton among them at a very fast rate (21).
ACKNOWLEDGMENT
The authors thank the Biotechnology Institute of Shahrekord University. Financial support from the Research Council of the Shahrekord University is also acknowledged gratefully.
REFERENCES
- 1.Broadley MR, White PJ, Hammond JP. Zinc in plants. New Phytol. 2007;173:677–702. doi: 10.1111/j.1469-8137.2007.01996.x. [DOI] [PubMed] [Google Scholar]
- 2.Prasad AS. Zinc in human health: Effect of zinc on immune cells. Mol. Med. 2008;14:353–357. doi: 10.2119/2008-00033.Prasad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugarman B. Zinc and Infection. Rev Infect Dis. 1983;5:137–147. doi: 10.1093/clinids/5.1.137. [DOI] [PubMed] [Google Scholar]
- 4.Russell R, Beard JL, Cousins RJ. Washington DC: National Academies Press; 2001. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. [PubMed] [Google Scholar]
- 5.Greenwood NN, Earnshaw A, Earnshaw AN. 2nd ed. Oxford: Butterworth-Heinemann; 1997. Chemistry of the Elements; p. 1202. [Google Scholar]
- 6.Erik G, Brandt MH, Brinck T, Bergman T, Edholm O. Molecular dynamics study of zinc binding to cysteines in a peptide mimic of the alcohol dehydrogenase structural zinc site. PhysChemChem Phys. 2009;11:975–983. doi: 10.1039/b815482a. [DOI] [PubMed] [Google Scholar]
- 7.Bitanihirwe BKY, Cunningham MG. Zinc: the brain's dark horse. SYNAPSE. 2009;63:1029–1049. doi: 10.1002/syn.20683. [DOI] [PubMed] [Google Scholar]
- 8.Hambidge KM, Krebs NF. Zinc deficiency: a special challenge. Nutr J. 2007;137:1101–1105. doi: 10.1093/jn/137.4.1101. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima AS, Dyck RH. Zinc and cortical plasticity. Brain Res Rev. 2009;59:347–373. doi: 10.1016/j.brainresrev.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Fosmire GJ. Zinc toxicity. Am. J. Clin. Nutr. 1990;51:225–227. doi: 10.1093/ajcn/51.2.225. [DOI] [PubMed] [Google Scholar]
- 11.Lippard SJ, Berg JM. California: University Science Books, Mill Valley; 1994. Principles of bioinorganic chemistry. [Google Scholar]
- 12.Leszczyszyn OI, Evans CD, Keiper SE, Warren GZL, Blindauer CA. Differential reactivity of individual zinc ions in clusters from bacterial metallothioneins. InorgChimActa. 2007;360:3–13. [Google Scholar]
- 13.Li Y, Trush MA, Yager JD. DNA damage caused by reactive oxygen species originating from a copper-dependent oxidation of the 2-hydroxy catechol of estradiol. Carcinogenesis. 1994;15:1421–1427. doi: 10.1093/carcin/15.7.1421. [DOI] [PubMed] [Google Scholar]
- 14.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 15.Arnold RR, Russell JE, Champion WJ, Brewer M, Gauthier JJ. Bactericidal activity of human lactoferrin: differentiation from the stasis of iron deprivation. Infect Immun. 1982;35:792–799. doi: 10.1128/iai.35.3.792-799.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva FD, Rezende CA, Rossi DCP, Esteves E, Dyszy FH, Schreier S, et al. Structure and mode of action of microplusin, a copper II-chelating antimicrobial peptide from the cattle tick Rhipicephalus (Boophilus) microplus. J Biol Chem. 2009;284:34735–34746. doi: 10.1074/jbc.M109.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae W, Chen W, Mulchandani A, Mehra RK. Enhanced bioaccumulation of heavy metals by bacterial cells displaying synthetic phytochelatins. BiotechnolBioeng. 2000;70:518–524. doi: 10.1002/1097-0290(20001205)70:5<518::aid-bit6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Bae W, Mehra RK. Metal-binding characteristics of a phytochelatin analog (Glu-Cys)2Gly. J Inorg Biochem. 1997;68:201–210. [Google Scholar]
- 19.Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 20.Gnanakaran S, Nymeyer H, Portman J, Sanbonmatsu KY, Garcia AE. Peptide folding simulations. Cur OpinStructu.Bio. 2003;13:168–174. doi: 10.1016/s0959-440x(03)00040-x. [DOI] [PubMed] [Google Scholar]
- 21.Friedman R, Nachliel E, Gutman M. Molecular dynamics of a protein surface: Ion-residues interactions. Biophys J. 2005;89:768–781. doi: 10.1529/biophysj.105.058917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dauter Z, Wilson KS, Sieker LC, Moulis JM, Meyer J. ProcNatlAcad Sci. Vol. 93. USA: 1996. Zinc-and iron-rubredoxins from clostridium pasteurianum at atomic resolution: a high-precision model of a ZnS4 coordination unit in a protein; pp. 8836–8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grossfield A, Feller SE, Pitman MC. Proteins: structure, function, and bioinformatics. 2007;67:31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- 24.Van Aalten DMF, De Groot BL, Findlay JBC, Berendsen HJC, Amadei A. A comparison of techniques for calculating protein essential dynamics. J Comput Chem. 1997;18:169–181. [Google Scholar]
- 25.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 26.Xue F, Gu Z, Feng J. LINKER: a web server to generate peptide sequences with extended conformation. Nucleic acids Res. 2004;32:562–565. doi: 10.1093/nar/gkh422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George RA, Heringa J. An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 2002;15:871–879. doi: 10.1093/protein/15.11.871. [DOI] [PubMed] [Google Scholar]
- 28.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 29.Berendsen HJC, Van der Spoel D, Van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. ComputPhysCommun. 1995;91:43–56. [Google Scholar]
- 30.Lindahl E, Hess B, Van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 31.Hess B, Kutzner C, Van der Spoel D, Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 32.Darden T, York D, Pedersen L. Particle meshEwald: An N log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 33.Brenner B, Schoenberg M, Schalovich JM, Greene Le, Eisenberg E. Evidence for cross-bridge attachment in relaxed muscle at low ionic strength. ProcNatlAcadSci USA. 1982;79:7288–7291. doi: 10.1073/pnas.79.23.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold S, James PW, Minou B. Acidic polypeptides can assemble both histones and chromatin in vitro at physiological ionic strength. ProcNatlAcadSci USA. 1979;76:5000–5004. doi: 10.1073/pnas.76.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 36.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM.LINCS: a linear constraint solver for molecular simulations. J Comp Chem. 1997;18:1463–1472. [Google Scholar]
- 37.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J ApplCryst. 1993;26:283–291. [Google Scholar]
- 38.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 39.Kabsch WSC. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded geometrical features. Biopoly. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 40.Hille B. 2nd ed. Melbourne, Australia: Macmillan Education; 1984. Elementary properties of ions in solution in ion channels of excitable membranes. [Google Scholar]
- 41.Maisuradze GG, Liwo A, Scheraga HA. Principal component analysis for protein folding dynamics. J Mol Biol. 2009;385:312–329. doi: 10.1016/j.jmb.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollup SM, Salensminde G, Reuter N. WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics. 2005;6:52. doi: 10.1186/1471-2105-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kundu S, Roy D. Temperature-induced unfolding pathway of a type III antifreeze protein: Insight from molecular dynamics simulation. J Mol Graphics Modell. 2008;27:88–94. doi: 10.1016/j.jmgm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Ascone I, Nobili G, Benfatto M, Congiu-Castellano A. XAS characterization of the zn site of non-structural protein 3 (ns3) from hepatitis c virus. congiu-castellano, x-ray. Absorption Fine Structure-XAFS. 2007;13:319–321. [Google Scholar]
- 45.Ohmasa M, Suzuki M, Takeuchi Y. A refinement of the crystal structure of covellite, CuSMineralo J. 1977;8:311–319. [Google Scholar]
- 46.Szilágyi Z, Fenselau C. Molecular dynamics simulation of metallothionein-drug complexes. Drug MetabDispos. 2000;28:174–179. [PubMed] [Google Scholar]
- 47.Dudev T, Lim C. Metal binding and selectivity in zinc proteins. J Chin Chem Soc. 2003;50:1093–1102. [Google Scholar]
- 48.Jolly WL. New York. McGraw-Hill; Modern inorganic chemistry. ISBN 0-07-032760-2. [Google Scholar]
- 49.Pearson RG. Hard and soft acids and bases. J Am Chem Soc. 1963;85:3533–3539. [Google Scholar]