

Abstract
Shiga toxin (Stx), a major virulence factor of enterohemorrhagic Escherichia coli, binds to target cells through a multivalent interaction between its B-subunit pentamer and the cell surface receptor globotriaosylceramide, resulting in a remarkable increase in its binding affinity. This phenomenon is referred to as the “clustering effect.” Previously, we developed a multivalent peptide library that can exert the clustering effect and identified Stx neutralizers with tetravalent peptides by screening this library for high-affinity binding to the specific receptor-binding site of the B subunit. However, this technique yielded only a limited number of binding motifs, with some redundancy in amino acid selectivity. In this study, we established a novel technique to synthesize up to 384 divalent peptides whose structures were customized to exert the clustering effect on the B subunit on a single cellulose membrane. By targeting Stx1a, a major Stx subtype, the customized divalent peptides were screened to identify high-affinity binding motifs. The sequences of the peptides were designed based on information obtained from the multivalent peptide library technique. A total of 64 candidate motifs were successfully identified, and 11 of these were selected to synthesize tetravalent forms of the peptides. All of the synthesized tetravalent peptides bound to the B subunit with high affinities and effectively inhibited the cytotoxicity of Stx1a in Vero cells. Thus, the combination of the two techniques results in greatly improved efficiency in identifying biologically active neutralizers of Stx.
INTRODUCTION
Infection with enterohemorrhagic Escherichia coli (EHEC) causes bloody diarrhea and hemorrhagic colitis, sometimes followed by fatal systemic complications, such as acute encephalopathy and hemolytic-uremic syndrome (HUS) (1–6). In 2011, unprecedented outbreaks of E. coli O104:H4 occurred in the European Union (EU), particularly in Germany, with more than 4,000 cases of infection and 50 fatalities (7, 8). Since antibiotic use is controversial (9–11), novel therapeutic strategies against the infection are urgently required. EHEC produces Shiga toxin (Stx) as a major virulence factor; therefore, one of the most promising approaches is to develop an Stx neutralizer that effectively binds to and inhibits Stx.
Stx, a typical ribotoxin, is present in various forms that can be classified into two subgroups, Stx1 and Stx2, each of which has various closely related subtypes: Stx1a, -1c, and -1d and Stx2a, -2b, -2c, -2d, -2e, -2f, and -2g, respectively (12–14). Each Stx subtype consists of a catalytic A subunit and a B-subunit pentamer, which is responsible for high-affinity binding to the functional cell surface receptor Gb3 [Galα(1-4)-Galβ(1-4)-Glcβ-ceramide] (4, 15, 16), or Gb4 [GalNAcβ(1-3)-Galα(1-4)-Galβ(1-4)-Glcβ-ceramide], which is preferred by Stx2e (17). Each B subunit has three distinctive binding sites (sites 1, 2, and 3) for the trisaccharide moiety of Gb3 (18, 19), resulting in the formation of a multivalent interaction between the B-subunit pentamer and Gb3. This type of interaction is known to markedly increase the binding affinity a millionfold and is generally known as the “clustering effect.”
Previously, we developed a multivalent peptide library that can exert the clustering effect and identified Stx neutralizers with tetravalent peptides by screening this library based on high-affinity binding to specific receptor-binding sites (20–22). By targeting one of the receptor-binding sites (site 3) of subtype Stx2a which is most closely associated with high disease severity (23, 24), we identified four tetravalent peptides that bind to Stx2a with high affinity and specificity as novel peptide-based neutralizers (20). One of the neutralizers, PPP-tet, protected mice from a fatal dose of E. coli O157:H7 (20) and inhibited the lethal effect of intravenously administered Stx2a in a nonhuman primate model (25). Recently, by targeting receptor-binding site 1 of Stx1a, the most frequently observed subtype, we identified tetravalent peptide MMA-tet (22). Interestingly, MMA-tet strongly inhibited Stx1a and Stx2a with greater potency than that of PPP-tet as well as rescuing mice from the lethality caused by the infection by E. coli O157:H7, which produces both toxins. This multivalent peptide library technique, however, can yield only a limited number of binding motifs for the intended receptor-binding region of the B subunit, with redundancy of amino acid selectivity at some positions.
In this study, we established a novel technique to determine a wide range of binding motifs for the B subunit by directly screening hundreds of divalent peptides on a membrane whose structures were customized to exert the clustering effect. By targeting one of the receptor-binding sites (site 2) of the Stx1a B subunit, a site which plays a significant role in the receptor binding of Stx1a (18, 26), we successfully identified 11 peptide-based neutralizers of Stx1a using this novel technology combined with multivalent peptide library screening. Screening the multivalent peptide library alone could not identify a biologically active inhibitor of this site. Thus, the combination of the two techniques will provide a powerful strategy to develop customized neutralizers for a restricted area of the receptor-binding region of the B subunit, enabling the identification of tailored neutralizers for each Stx subtype with highly conserved structural similarity.
MATERIALS AND METHODS
Materials.
Recombinant Stx1a, histidine-tagged Stx1a B subunit (1BH), and 1BH with a single-amino-acid substitution (1BH-G62A) were prepared as described previously (27). The amino-PEG500-UC540 membrane (Intavis Bioanalytical Instruments AG, Germany) used for the spot synthesis of peptides was purchased from PerkinElmer, Tokyo, Japan. Porcine erythrocyte Gb3 and egg phosphatidylcholine (PC) were purchased from Wako Pure Industries, Osaka, Japan.
Peptides and peptide library screening.
Tetravalent peptides and tetravalent peptide libraries were synthesized using N-α-9-fluorenylmethoxy carbonyl (FMOC)-protected amino acids and standard BOP [benzotriazol-1-yloxytris(dimetylamino)phosphonium hexafluorophosphate]/HOB (1-hydroxybenzotriazole hydrate) coupling chemistry as described previously (20). A Met-Ala sequence was included at the amino terminus of each library peptide to verify the identity and origin of the peptides being sequenced and to qualify the peptides. Recombinant 1BH or 1BH-G62A (0.5 mg protein) bound to Ni2+ beads was incubated with 300 μg of a given library peptide in phosphate-buffered saline (PBS) overnight at 4°C. After extensive washing, the bound peptides were sequenced on an Applied Biosystems model 477A protein sequencer. To calculate the relative amino acid preference at each degenerate position, the corrected quantities of amino acids in the peptides recovered from the 1BH beads were compared with those recovered from the 1BH-G62A beads to calculate the abundance ratios of amino acids (20).
ELISA of the binding between 1BH and inhibitory peptides.
The indicated amounts of the tetravalent peptide dissolved in PBS were applied as a coating onto each well of a 96-well enzyme-linked immunosorbent assay (ELISA) plate and incubated for 24 h at 4°C. After blocking, the plate was incubated with 1BH or 1BH-G62A (1 μg/ml) for 1 h at room temperature. Bound 1BH was detected using rabbit anti-Stx1a antiserum as described previously (20).
Cytotoxicity assay.
Subconfluent Vero cells were cultured in a 96-well plate in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and treated with Stx1a (1 pg/ml) in the absence or presence of a given tetravalent peptide for 72 h at 37°C. The relative number of living cells was determined using cell counting kit 8 (Dojindo, Kumamoto, Japan) as described previously (22).
Spot synthesis of peptides on cellulose membrane.
Basic spot synthesis of peptides on a cellulose membrane was performed as described previously (28, 29) using the ResPep SL spot synthesizer (Intavis Bioanalytical Instruments AG, Cologne, Germany). The density of peptides synthesized on a membrane was controlled by using a mixture of FMOC-βAla-OH and Boc-βAla-OH (Watanabe Chemical Industries, Japan) at different ratios (100:0, 30:70, or 10:90, respectively) for the first cycle. t-Butyloxycarbonyl (Boc) is resistant to the deprotection procedure and inhibits the subsequent elongation reaction. The spacer length of the peptide was controlled by the number of amino hexanoic acids following the first βAla. After the addition of amino hexanoic acid(s), FMOC-Lys(FMOC)-OH (Watanabe Chemical Industries) was used for the next cycle to bifurcate the peptide chain for subsequent motif synthesis. This step was omitted for monovalent peptide synthesis. The successful synthesis of each peptide was confirmed by staining the membrane using bromophenol blue (1% in N,N′-dimethylformamide), which reacts to free amino residues. Free amino residues are produced only after the completion of all the reactions and before the deprotection of the side chain residues. After destaining with N,N′-dimethylformamide, the membrane was used in the binding assay. As a positive control for Stx B-subunit binding, Gb3 (0.1 μg) mixed with PC (1 μg) was directly spotted on the membrane.
Binding assay of 1BH or 1BH-G62A to divalent peptides synthesized on the membrane.
After blocking with 5% skim milk in PBS, the membrane (prepared as described above) was blotted with the indicated concentration of 125I-1BH or 125I-1BH-G62A (1 × 106 to 2 × 106 cpm/μg protein) for 1 h at room temperature. After extensive washing, the radioactivity bound to each peptide spot was quantitated as a pixel value using a BAS 2500 bioimaging analyzer system (GE Healthcare, Amersham, United Kingdom). In another detection system, the membrane was blotted with 1.0 μg/ml 1BH for 1 h at room temperature. After extensive washing, bound 1BH was detected using rabbit anti-Stx1a antiserum as described previously (22) and quantitated as a pixel value using ImageQuant LAS 500 (GE Healthcare).
Kinetic analysis of the binding between inhibitory peptides and immobilized 1BH.
The binding of tetravalent peptides to immobilized 1BH was quantified using a Biacore T100 system instrument (GE Healthcare Sciences, USA) as described previously (20). The resonance unit is an arbitrary unit (AU) used by the Biacore system. Binding kinetics were analyzed using Biaevaluation software, v1.1.1 (GE Healthcare).
RESULTS
Tetravalent peptide library screening identified a peptide motif that specifically binds 1BH through site 2.
A tetravalent peptide library is comprised of peptides containing a polylysine core that bifurcates at both ends with four randomized peptides (20). The library was screened for the ability to bind to wild-type 1BH but not to 1BH-G62A, which contains a mutation in one of the receptor-binding sites (site 2). Site 2 has been shown to play an essential role in the receptor binding of Stx1a (26). A tetravalent peptide library with a fixed Arg at position 4 (the XRX library) was used for the first round of selection, based on the previous observation that Stx1a prefers clustered Args in its binding motifs (22). As shown in Fig. 1A, Arg was strongly selected at positions 1, 5, and 7, and His was selected at position 5. Based on this result, a second tetravalent peptide library with clustered Args (the XRR library) was screened to further refine peptide selection. Lys was strongly selected at position 1, and both Arg and His were selected at position 3. Based on these results, we identified KRRRRRR as a candidate motif. A tetravalent form of this peptide with the same core structure was synthesized and referred to as KRR-tet. As shown in Fig. 1B, KRR-tet bound efficiently to 1BH, whereas the binding to 1BH-G62A was less efficient. Under the same conditions, MA-tet, which has the same core structure but lacks the binding motif, did not bind to either of the B subunits (data not shown). These results indicate that KRR-tet specifically binds to the B subunit through site 2. KRR-tet, however, was found to be cytotoxic due to its highly basic nature (Fig. 1C) and thus did not inhibit the cytotoxicity of Stx1a in Vero cells, in contrast to MMA-tet (Fig. 1D).
FIG 1.

Identification of a peptide motif that specifically binds to Stx1a B subunit through site 2 by using tetravalent peptide library screening. (A) The tetravalent peptide library was comprised of tetravalent peptides with a polylysine core bifurcating at both ends with four randomized peptides. The peptide library for the first screening had the sequence Met-Ala-X-X-X-R-X-X-X-Ala-U (U, amino hexanoic acid), where X indicates all amino acids except Cys. Screening of the library was performed to identify tetravalent peptides that bound to 1BH but not to 1BH-G62A. For the second screening, a peptide library with fixed Args at positions 4 to 7 (XRR library) was used. Values in parentheses indicate relative selectivities for the amino acids. Bold letters indicate amino acids that were strongly selected. Each screening was performed twice; representative values are shown. (B) The binding of 1BH or 1BH-G62A (1 μg/ml) to KRR-tet at the indicated concentrations was examined using ELISA (mean ± standard error, n = 3). (C) The effect of KRR-tet on the cell viability in Vero cells was examined by the cytotoxicity assay. Data are presented as a percentage of the control value (mean ± standard error, n = 4). (D) The effect of KRR-tet or MMA-tet on the cytotoxic activity of Stx1a (1 pg/ml) in Vero cells was examined by the cytotoxicity assay (mean ± standard error, n = 4).
Establishment of a technique to synthesize peptides on a membrane that can exert the clustering effect on the Stx1a B subunit.
KRR-tet has an Arg cluster at positions 4 to 7; this cluster is also observed in MMA-tet, indicating that the motif is commonly required for the efficient binding to the Stx1a B subunit. Based on this motif, we tried to identify a series of site 2-targeted binding motifs by establishing a novel technique in which hundreds of peptides with the Arg cluster were synthesized in a divalent form on a cellulose membrane and screened for high-affinity binding to 1BH but not to 1BH-G62A. We also optimized the structure of the peptide synthesized on the membrane (Fig. 2A). A divalent form of the peptide, KRRRRRR, was found to exert the clustering effect and exhibited markedly increased binding to 1BH compared to the monovalent form (Fig. 2B). Under the same conditions, Gb3 blotted on the membrane was specifically detected by 1BH. Using the divalent peptide, we found that a spacer of one amino hexanoic acid functioned most efficiently and that the higher density yielded higher binding efficacy (Fig. 2B). We further examined the binding of other divalent peptides. The divalent form of MMARRRR, a binding motif present in MMA-tet, also efficiently bound to 1BH in a dose-dependent manner when one amino hexanoic acid was used as a spacer (Fig. 2C). Similar density dependency was observed for the divalent peptide (Fig. 2B). The divalent form of AAARRRR, another binding motif previously determined for 1BH (22), could still efficiently bind to 1BH. In contrast, the divalent form of AAADDDD, in which all Args of AAARRRR were replaced with Asps, completely lost binding activity (Fig. 2C), confirming the requirement of the Arg cluster in the binding motif.
FIG 2.

Optimization of the structure of the peptides synthesized on a membrane to exert the clustering effect. (A) The structure of a monovalent or a divalent peptide synthesized on a cellulose membrane is shown as described in Materials and Methods (density: y = 10, 30, or 100%; spacer length: n = 1, 2, or 3; U indicates amino hexanoic acid; R = Met-Ala-[indicated motif]-Ala). (B) The monovalent or the divalent form of the KKKRRRR motif was synthesized with different spacer lengths in triplicate (y = 100%, upper panel). The divalent form of the KKKRRRR or MMARRRR motif was synthesized with different densities (n = 1, lower panel). The membrane was blotted with 1.0 μg/ml of 1BH. Bound 1BH was detected using rabbit anti-Stx1a antiserum and quantitated as a pixel value. Each value is shown after subtraction of the control value obtained without 1BH. (C) Divalent peptides with the MMARRRR, AAADDDD, or AAARRRR motif were synthesized on a membrane with different spacer lengths (y = 100%). The membrane was blotted with the indicated concentration of 125I-1BH, and the radioactivity bound to each peptide was detected (upper panel) and quantitated. Data obtained with 1.0 μg/ml of 125I-1BH are shown (lower panel).
Screening of divalent peptides synthesized on a membrane successfully identified a wide range of peptide motifs that specifically bind to the Stx1a B subunit through site 2.
Using the optimized conditions identified above, 380 divalent peptides containing the Arg cluster and shuffled amino acids at motif positions 1 to 3 were synthesized on a cellulose membrane. One group had the XXA-RRRR motif, and the other group had the AAX-RRRR motif, respectively, where X indicates a fixed amino acid as indicated (Fig. 3A). The divalent peptide with the original motif KRR-RRRR was also synthesized. The membrane was blotted with 125I-1BH or 125I-1BH-G62A (Fig. 3B), and the radioactivity bound to each peptide spot was quantitated and analyzed. Divalent peptides with a normalized value of 125I-1BH binding (1BH-binding value) greater than 1.14 were selected and then resorted in descending order of the product (1BH × ratio) of the 1BH-binding value and the normalized ratio (1BH/G62A ratio), as shown in Table S1 in the supplemental material. A total of 64 peptides with 1BH × ratio values greater than 1.3 were identified as candidate motifs. These 64 divalent peptides were synthesized on a membrane (Fig. 4A) and blotted with different concentrations of 125I-1BH or 125I-1BH-G62A (Fig. 4B). The radioactivity bound to each peptide was quantitated and analyzed (see Fig. S1 in the supplemental material). Among these divalent peptides, 11 motifs were selected based on the binding intensity and specificity of 1BH, excluding peptide motifs that had tandem basic amino acids in the first three amino acids (e.g., KHA or KKA). The exclusion was implemented to avoid selecting potentially cytotoxic motifs.
FIG 3.

Screening of divalent peptides synthesized on a membrane based on binding to 1BH but not to 1BH-G62A. (A) The structure of the divalent peptide synthesized on a membrane was the same as that shown in Fig. 2A. The density of the peptides (y) was 100%, and the spacer length (n) was 1. The first three amino acids present in the XXA-RRRR or AAX-RRRR motif are shown at positions A5 to P5 or positions P6 to P23, respectively. The divalent peptide with the original motif, KRR-RRRR, was synthesized at position P24. The gray boxes indicate the 64 candidate motifs that were selected after the binding analysis. (B) The membrane was blotted with 125I-1BH or 125I-1BH-G62A (1 μg/ml), and the radioactivity bound to each peptide spot was quantitated and analyzed as shown in Table S1 in the supplemental material.
FIG 4.

Binding analysis of the divalent peptides with 64 candidate motifs synthesized on a membrane to 1BH or 1BH-G62A. (A) The structure of the divalent peptide synthesized on a membrane was the same as that shown in Fig. 3A. The 64 divalent peptides with candidate motifs, the first three amino acids of which are shown in the panel, were synthesized at the indicated positions. The gray boxes indicate the 11 candidate motifs selected after the binding analysis as shown in Fig. S1 in the supplemental material. (B) The membrane was blotted with the indicated amounts of 125I-1BH or 125I-1BH-G62A, and the radioactivity bound to each peptide spot was quantitated and analyzed (see Fig. S1 in the supplemental material).
Tetravalent peptides with the identified motifs bind to 1BH through site 2 with high affinities.
Tetravalent peptides with the 11 motifs identified above were synthesized using the same core structure as described in Materials and Methods and were referred to as NKA-tet, IIA-tet, KGA-tet, KMA-tet, KFA-tet, FRA-tet, PQA-tet, YTA-tet, VIA-tet, AAL-tet, and AAK-tet. The binding of these tetravalent peptides to 1BH or 1BH-G62A was examined (Fig. 5). All of the tetravalent peptides bound to 1BH with more potency than to 1BH-G62A, indicating that site 2 is involved in the binding of these peptides. Under the same conditions, MMA-tet, which binds to sites 1 and 3 of 1BH but not site 2 (22), bound to 1BH-G62A with similar affinity as that of 1BH (data not shown), further confirming the binding specificity of the identified tetravalent peptides. As shown in Table 1, all of these tetravalent peptides bound to 1BH with high affinities.
FIG 5.

Analysis of the binding of the identified tetravalent peptides with 1BH or 1BH-G62A. The binding of 1BH (closed diamonds) or 1BH-G62A (open triangles) (1 μg/ml) with the indicated amounts of the tetravalent peptides was examined using ELISA.
TABLE 1.
Binding kinetics of the identified tetravalent peptidesa
| Tetravalent peptide | KD (μM), mean ± SE (n = 3) | RUmax (AU), mean ± SE (n = 3 to 4) |
|---|---|---|
| NKA-tet | 0.28 ± 0.01 | 2,460 ± 57 |
| IIA-tet | 0.51 ± 0.05 | 3,580 ± 52 |
| KGA-tet | 0.21 ± 0.01 | 1,390 ± 77 |
| KMA-tet | 0.43 ± 0.03 | 2,510 ± 71 |
| KFA-tet | 0.55 ± 0.04 | 3,630 ± 44 |
| FRA-tet | 0.63 ± 0.11 | 3,910 ± 95 |
| PQA-tet | 0.57 ± 0.01 | 2,140 ± 19 |
| YTA-tet | 0.34 ± 0.03 | 2,170 ± 364 |
| VIA-tet | 0.40 ± 0.01 | 2,050 ± 202 |
| AAL-tet | 1.00 ± 0.14 | 2,400 ± 175 |
| AAK-tet | 0.35 ± 0.02 | 2,630 ± 35 |
The binding kinetics of the identified tetravalent peptides to immobilized 1BH were analyzed using the Biacore system. KD, dissociation constant; RUmax, maximum resonance unit.
Tetravalent peptides with the identified motifs efficiently inhibit the cytotoxicity of Stx1a.
The ability of the identified tetravalent peptides to inhibit the cytotoxicity of Stx1a in Vero cells was examined (Fig. 6). MMA-tet was used as a positive control. Among the 11 tetravalent peptides, KGA-tet, KFA-tet, FRA-tet, PQA-tet, YTA-tet, VIA-tet, and AAL-tet inhibited Stx1a cytotoxicity as efficiently as MMA-tet, although the inhibitory effects of KFA-tet and FRA-tet were reduced at 52 μM because of their own cytotoxicity (data not shown). NKA-tet, IIA-tet, KMA-tet, and AAK-tet exerted inhibitory effects with lower efficiency.
FIG 6.
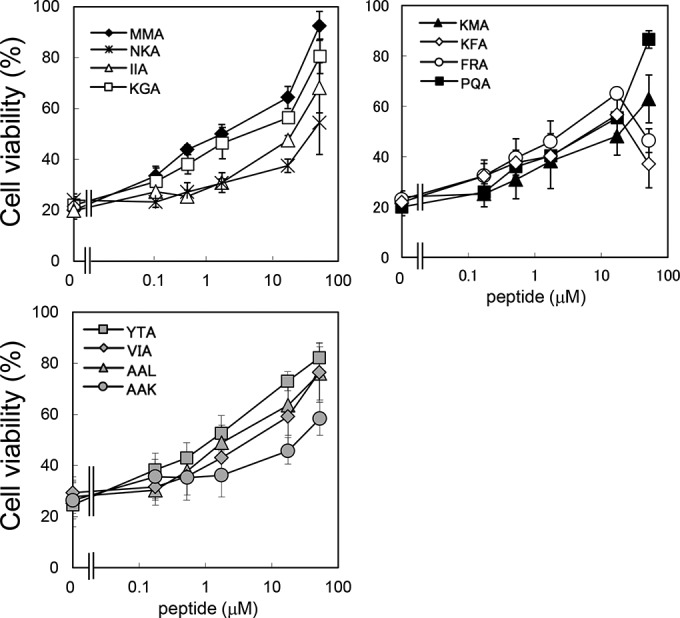
Inhibitory effects of the identified tetravalent peptides on the cytotoxicity of Stx1a in Vero cells. The effects of the identified tetravalent peptides on the cytotoxicity of Stx1a (1 pg/ml) in Vero cells were examined by the cytotoxicity assay. Data are presented as a percentage of the control value (mean ± standard error, n = 3).
DISCUSSION
In this study, we established a novel technique to synthesize, on a single cellulose membrane, divalent peptides that could exert the clustering effect on the Stx1a B subunit. By targeting one of the receptor-binding sites (site 2) of the B subunit, we screened divalent peptides whose sequences were designed based on information obtained by multivalent peptide library screening. We successfully identified 11 peptide-based neutralizers of Stx1a. Thus, the combination of these two techniques enables us to identify a wide range of biologically active neutralizers of Stx1a, whereas the multivalent peptide library technique yielded only one motif, which was found to be cytotoxic.
Previously, we developed a series of carbosilane dendrimers with clustered trisaccharides of Gb3, named SUPER TWIGs, as Stx neutralizers (27, 30). One of these compounds, SUPER TWIG (1)2, which has a divalent form of the trisaccharide, was found to sufficiently exert the clustering effect on the Stx B subunit to markedly increase binding affinity. Their KD (dissociation constant) values toward the Stx1a B subunit and Stx2a B subunit were determined to be 88 and 68 μM, respectively, using the Biacore system; no binding was observed with free trisaccharide up to 1.6 mM (20, 27). These observations provide a theoretical rationale for the use of membrane-synthesized divalent (but not monomeric) peptides during screening of high-affinity binding motifs against the B subunit. Furthermore, our finding that higher density and shorter spacer length of the divalent peptide resulted in higher binding efficacy clearly demonstrates that a spatially condensed configuration of the divalent peptide enables each motif to exert the clustering effect.
Here, we focused on site 2 as a target region to develop an Stx1a neutralizer because this site has already been shown to play an essential role in the receptor binding of Stx1a (26, 31, 32). One of the Stx neutralizers with clustered trisaccharides, the STARFISH compound, containing 10 trisaccharides assembled into a single glucose core through bifurcated spacers, was found to exclusively occupy site 2 of the Stx1a B subunit, further confirming the importance of site 2 as a target region (33). The 11 Stx1a neutralizers that we identified here are the first peptide-based neutralizers to target site 2, while PPP-tet and MMA-tet, which were identified by targeting site 3 of Stx2a and site 1 of Stx1a, respectively, did not touch site 2 on each B subunit (Fig. 5) (20, 22). Among these inhibitors, KGA-tet, PQA-tet, YTA-tet, VIA-tet, and AAL-tet inhibited the cytotoxicity of Stx1a with potency similar to that of MMA-tet (Fig. 6).
All the divalent peptides synthesized on a membrane have the RRRR motif at positions 4 to 7: this motif was introduced based on information obtained from multivalent peptide library screening. The addition of even a single acidic amino acid (Asp or Glu) to this motif at position 1, 2, or 3 markedly reduced the binding of 125I-1BH and 125I-1BH-G62A (positions C14 to D8, E4 to E22, F23, G16, G18, H11, H13, I6, I8, J1, J3, K15, K17, M5, M7, N2, and N21, in Fig. 3B), and surprisingly, no acidic amino acids were selected in the data shown in Table S1 in the supplemental material. This significant finding about the negative selectivity of acidic amino acids cannot be theoretically obtained by using the multivalent peptide library technique. In accordance with this observation, all the motifs with 1BH-binding values and 1BH-G62A values greater than 2.2 have at least one Lys in positions 1 to 3 (see Table S1 in the supplemental material), demonstrating that the basic amino acid cluster is required to efficiently bind to the B subunit, irrespective of the presence of Gly 62. It is possible that this basic amino acid cluster interacts with the acidic amino acids present on the receptor-binding surface of the B subunit (Asps 16, 17, and 18) (18) through electrostatic interactions. Thus, amino acids in other positions may contribute to binding site specificity. Specifically, 8 out of 11 identified Stx neutralizers have at least one hydrophobic amino acid in positions 1 to 3 (e.g., Ile, Met, Phe, Val, Pro, Tyr, or Leu) which might be involved in hydrophobic interactions at site 2, where hydrophobic interactions with the trisaccharide are prominent during receptor recognition compared to the other receptor-binding sites (18).
The novel membrane-screening technique established here substantially overcomes the problems of the multivalent peptide library technique, where only a limited number of binding motifs are determined and redundancy of amino acid selectivity may be observed in some positions in the motif. In this technique, hundreds of divalent peptides synthesized on a membrane with known sequences can be screened quantitatively, thus yielding a wide variety of binding motifs that could successfully distinguish a small change in amino acid sequence, such as Gly to Ala. Various closely related subtypes for Stx1 (Stx1a, -1c, and -1d) and Stx2 (Stx2a, -2b, -2c, -2d, -2e, -2f, and -2g) are present (12–14), but their modes of globosugar recognition are shown to be substantially different because of the minute differences present on the receptor-binding surface of the B subunits (34). Thus, the combination of the two techniques presented here will provide a powerful strategy to develop optimal neutralizers against specific subtypes, enabling customized therapy that targets individual Stx subtypes produced by various EHEC strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mika Fukumoto (Doshisha University) for her technical assistance.
This work was supported by a grant from the National Center for Global Health and Medicine, Japan; a grant from the MEXT-Supported Program for the Strategic Research Foundation at Private Universities; grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan; and a grant from the Ministry of Health, Labor and Welfare, Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03517-14.
REFERENCES
- 1.Karmali MA, Steele BT, Petric M, Lim C. 1983. Sporadic cases of haemolytic uraemic syndrome associated with fecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet i:619–620. [DOI] [PubMed] [Google Scholar]
- 2.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med 308:681–685. doi: 10.1056/NEJM198303243081203. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien AD, Holmes RK. 1987. Shiga and Shiga-like toxins. Microbiol Rev 51:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paton JC, Paton AW. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev 11:450–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 6.Trachtman H, Austin C, Lewinski M, Stahl RA. 2012. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol 8:658–669. doi: 10.1038/nrneph.2012.196. [DOI] [PubMed] [Google Scholar]
- 7.Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G, HUS Investigation Team . 2011. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 365:1771–1780. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- 8.Hauswaldt S, Nitschke M, Sayk F, Solbach W, Knobloch JK. 2013. Lessons learned from outbreaks of Shiga toxin producing Escherichia coli. Curr Infect Dis Rep 15:4–9. doi: 10.1007/s11908-012-0302-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda K, Ida O, Kimoto K, Takatorige T, Nakanishi N, Tatara K. 1999. Effect of early fosfomycin treatment on prevention of hemolytic uremic syndrome accompanying Escherichia coli O157:H7 infection. Clin Nephrol 52:357–362. [PubMed] [Google Scholar]
- 10.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. 2000. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 342:1930–1936. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong CS, Mooney JC, Brandt JR, Staples AO, Jelacic S, Boster DR, Watkins SL, Tarr PI. 2012. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: a multivariable analysis. Clin Infect Dis 55:33–41. doi: 10.1093/cid/cis299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyles CL. 2007. Shiga toxin-producing Escherichia coli: an overview. J Anim Sci 85:E45–E62. doi: 10.2527/jas.2006-508. [DOI] [PubMed] [Google Scholar]
- 13.Fuller CA, Pellino CA, Flagler MJ, Strasser JE, Weiss AA. 2011. Shiga toxin subtypes display dramatic differences in potency. Infect Immun 79:1329–1337. doi: 10.1128/IAI.01182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheutz F, Teel LD, Beutin L, Piérard D, Buvens G, Karch H, Mellmann A, Caprioli A, Tozzoli R, Morabito S, Strockbine NA, Melton-Celsa AR, Sanchez M, Persson S, O'Brien AD. 2012. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J Clin Microbiol 50:2951–2963. doi: 10.1128/JCM.00860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. 1985. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 151:775–782. doi: 10.1093/infdis/151.5.775. [DOI] [PubMed] [Google Scholar]
- 16.Melton-Celsa AR, O'Brien AD. 1998. Structure, biology, and relative toxicity of Shiga toxin family members for cells and animals, p 121–128. In Kaper JB, O'Brien AD (ed), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASM Press, Washington, DC. [Google Scholar]
- 17.DeGrandis S, Law H, Brunton J, Gyles C, Lingwood CA. 1989. Globotetraosylceramide is recognized by the pig edema disease toxin. J Biol Chem 264:12520–12525. [PubMed] [Google Scholar]
- 18.Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD, Brunton JL, Read RJ. 1998. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37:1777–1788. doi: 10.1021/bi971806n. [DOI] [PubMed] [Google Scholar]
- 19.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MNG. 2004. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem 279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 20.Nishikawa K, Watanabe M, Kita E, Igai K, Omata K, Yaffe MB, Natori Y. 2006. A multivalent peptide library approach identifies a novel Shiga toxin inhibitor that induces aberrant cellular transport of the toxin. FASEB J 20:2597–2599. doi: 10.1096/fj.06-6572fje. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe-Takahashi M, Sato T, Dohi T, Noguchi N, Kano F, Murata M, Hamabata T, Natori Y, Nishikawa K. 2010. An orally applicable Shiga toxin neutralizer functions in the intestine to inhibit the intracellular transport of the toxin. Infect Immun 78:177–183. doi: 10.1128/IAI.01022-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsutsuki K, Watanabe-Takahashi M, Takenaka Y, Kita E, Nishikawa K. 2013. Identification of a peptide-based neutralizer that potently inhibits both Shiga toxins 1 and 2 by targeting specific receptor-binding regions. Infect Immun 81:2133–2128. doi: 10.1128/IAI.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. 1989. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J Infect Dis 160:994–998. doi: 10.1093/infdis/160.6.994. [DOI] [PubMed] [Google Scholar]
- 24.Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O'Brien AD, Samuel JE. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immun 61:3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stearns-Kurosawa DJ, Collins V, Freeman S, Debord D, Nishikawa K, Oh SY, Leibowitz CS, Kurosawa S. 2011. Rescue from lethal Shiga toxin 2-induced renal failure with a cell-permeable peptide. Pediatr Nephrol 26:2031–2039. doi: 10.1007/s00467-011-1913-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soltyk AM, MacKenzie CR, Wolski VM, Hirama T, Kitov PI, Bundle DR, Brunton JL. 2002. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT. J Biol Chem 277:5351–5359. doi: 10.1074/jbc.M107472200. [DOI] [PubMed] [Google Scholar]
- 27.Nishikawa K, Matsuoka K, Watanabe M, Igai K, Hino K, Terunuma D, Kuzuhara H, Natori Y. 2005. Identification of the optimal structure for a Shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J Infect Dis 191:2097–2105. doi: 10.1086/430388. [DOI] [PubMed] [Google Scholar]
- 28.Frank R. 1992. Spot-synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48:9217–9232. doi: 10.1016/S0040-4020(01)85612-X. [DOI] [Google Scholar]
- 29.Frank R. 2002. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports—principles and applications. J Immunol Methods 267:13–26. doi: 10.1016/S0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa K, Matsuoka K, Kita E, Okabe N, Mizuguchi M, Hino K, Miyazawa S, Yamasaki C, Aoki J, Takashima S, Yamakawa Y, Nishijima M, Terunuma D, Kuzuhara H, Natori Y. 2002. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc Natl Acad Sci U S A 99:7669–7674. doi: 10.1073/pnas.112058999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolski VM, Soltyk AM, Brunton JL. 2001. Mouse toxicity and cytokine release by verotoxin 1B subunit mutants. Infect Immun 69:579–583. doi: 10.1128/IAI.69.1.579-583.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flagler MJ, Mahajan SS, Kulkarni AA, Iyer SS, Weiss AA. 2010. Comparison of binding platforms yields insights into receptor binding differences between Shiga toxins 1 and 2. Biochemistry 49:1649–1657. doi: 10.1021/bi902084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. 2000. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 34.Yosief HO, Iyerb SS, Weiss AA. 2013. Binding of Pk-trisaccharide analogs of globotriaosylceramide to Shiga toxin variants. Infect Immun 81:2753–2760. doi: 10.1128/IAI.00274-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.