

Abstract
Examining the population structure and the influence of recombination and ecology on microbial populations makes great sense for understanding microbial evolution and speciation. Streptomycetes are a diverse group of bacteria that are widely distributed in nature and a rich source of useful bioactive compounds; however, they are rarely subjected to population genetic investigations. In this study, we applied a five-gene-based multilocus sequence analysis (MLSA) scheme to 41 strains of Streptomyces albidoflavus derived from diverse sources, mainly insects, sea, and soil. Frequent recombination was detected in S. albidoflavus, supported by multiple lines of evidence from the pairwise homoplasy index (Φw) test, phylogenetic discordance, the Shimodaira-Hasegawa (SH) test, and network analysis, underpinning the predominance of homologous recombination within Streptomyces species. A strong habitat signal was also observed in both phylogenetic and Structure 2.3.3 analyses, indicating the importance of ecological difference in shaping the population structure. Moreover, all three habitat-associated groups, particularly the entomic group, demonstrated significantly reduced levels of gene flow with one another, generally revealing habitat barriers to recombination. Therefore, a combined effect of homologous recombination and ecology is inferred for S. albidoflavus, where dynamic evolution is at least partly balanced by the extent that differential distributions of strains among habitats limit genetic exchange. Our study stresses the significance of ecology in microbial speciation and reveals the coexistence of homologous recombination and ecological divergence in the evolution of streptomycetes.
INTRODUCTION
Despite various currently available approaches to defining prokaryotic species and a growing recognition of their vast diversity in nature, the concept of microbial species has long been under dispute due to their widespread genomic heterogeneity and variable levels of gene exchange (1–3). Genetic clusters that emerge in molecular sequence analyses of populations are often regarded as bearing the status of species, and multiple theoretical models to resolve the evolutionary mechanisms that cause the creation and maintenance of the genetic clusters have been proposed (3, 4). Until now, two major parallel concepts of microbial species have been developed, focused on barriers to recombination and ecological divergence, respectively (4): the neutral model, raised by Fraser and colleagues (3, 5), highlights the role of recombination in converging and diverging clusters with various recombination rates, while the ecotype model, developed by Cohan and colleagues (6, 7), defines an ecotype as a group of ecologically similar strains which is purged of its diversity by periodic selection and/or genetic drift. Consequently, recombination and divergent selection have emerged as two key processes driving the increasing discussion concerning theoretical models of microbial speciation. Moreover, coexistence of these processes has been suggested for several bacteria. For example, a mechanism of ecological differentiation analogous to that in sexual eukaryotes was revealed in two newly diverged Vibrio populations, in which recombination was strong enough relative to selection to allow genome regions rather than whole genomes to sweep through populations in a habitat-specific manner (8). However, most of the current relevant studies are concentrating upon pathogenic bacteria (9), which represent a rather limited spectrum of microorganisms.
Members of the genus Streptomyces are aerobic, Gram-positive bacteria characterized by high genome G+C contents and a complex lifestyle. They are well known as a rich source of multifarious secondary metabolites and are responsible for about one-third of the currently known microbial bioactive compounds, notably antibiotics (10). Streptomycetes are widely distributed in nature, inhabiting a variety of habitats, including deserts, ice in the South Pole, insects, plants, and sea, as well as their primary habitat, soil (11, 12). They also play a significant role in biodegradation and bioremediation by decomposing insoluble polymers, such as lignin, and synthetic insecticides (13, 14). Given these notable features, streptomycetes have been a focus in microbial research and development for decades.
In virtue of various genetic and molecular methods, much progress has been made in evolutionary studies of streptomycetes (15, 16); however, their population genetics has rarely been investigated. Only recently has a population genetic study on Streptomyces flavogriseus, which was later transferred to Streptomyces pratensis (17), detected a much higher recombination rate within species than between them, indicating the dominance of recombination in shaping the evolution of streptomycetes (18). This finding sheds new light on the evolutionary history of streptomycetes and reminds us of the importance and necessity to examine population structure and recombination in Streptomyces species. As one of the early described species of the genus Streptomyces, Streptomyces albidoflavus has long been investigated for bioactive metabolites (19, 20) and tackled taxonomically using multiple approaches (21, 22). Here we obtained dozens of strains of this species from diverse sources, which provide an interesting sample with which to examine the influence of habitat and recombination in streptomycetes.
Multilocus sequence analysis (MLSA) has proved to be feasible for population genetic analyses of many microbial groups (9, 23). Our previously established streptomycete MLSA scheme (24), relying on five housekeeping genes, atpD (ATP synthase F1, β subunit), gyrB (DNA gyrase, B subunit), recA (recombinase A), rpoB (RNA polymerase, β subunit), and trpB (tryptophan synthase, β subunit), has been successfully applied to systematic analyses of four Streptomyces 16S rRNA gene clades at inter- and intraspecies levels (17, 22, 24–26) and has also shown great potential in population genetics analysis of streptomycetes (18). In the present study, a collection of 41 S. albidoflavus strains from diverse sources was subjected to the five-gene-based MLSA scheme to describe the population structure of S. albidoflavus, followed by various analyses to examine the roles homologous recombination and habitat played in shaping such structure. The evidence obtained from this study is expected to provide more insights into the evolution of streptomycetes for a better understanding of the origin and persistence of Streptomyces species.
MATERIALS AND METHODS
Bacterial strains and nucleotide polymorphism.
A total of 41 S. albidoflavus strains were tested in this study (Table 1), including 26 isolates from diverse sources and 10 reference strains from our previous study (22), as well as 5 whose genome sequences are available in GenBank. These strains were isolated from various habitats and geographic locations (Table 1), and most of them fell into three habitat-associated groups: edaphic, entomic, and marine (oceanic). The edaphic group encompassed five strains isolated from soil, two from potatoes, and one from Muschelkalk (a sequence of sedimentary rock strata in the geology of central and western Europe); the genome-sequenced strain Streptomyces albus J1074, which was recently proposed to be a member of S. albidoflavus (27), was also included. The entomic group consisted of 20 strains, of which 15 were isolated from imperial moths (Eacles imperialis, Saturniidae), three from owl butterflies (Caligo sp., Nymphalidae), and two genome-sequenced strains from leaf-cutting ants (Acromyrmex octospinosus) and an unknown insect, respectively. The marine group was composed of eight strains, of which six were isolated from deep seawater (at depths from 2,800 to 4,069 m) and two genome-sequenced strains from neritic sediments and sponges, respectively. The remaining five strains, which were isolated from air, ceiling filler, contaminated plates, and medicinal herbs, stayed uncategorized.
TABLE 1.
Strains used in this study
| Strain_habitat-associated groupa | Habitatb | Origin | Geographic location | ST | Allelic profilec | Date of isolationd |
|---|---|---|---|---|---|---|
| CGMCC 4.1615_Ed | Soil | Champavathi River, Andhra Pradesh, India | — | 1 | 1-1-1-1-1 | 1959* |
| FXJ2.339_Ed | Soil | Kaifeng, Henan Province, China | 34°49′N, 114°18′E | 2 | 2-2-2-2-2 | 08/2011 |
| J1074_Ed | Soil | —e | — | 3 | 3-2-1-1-2 | — |
| NBRC 100770_Ed | Soil | Caucasus | — | 4 | 1-1-3-1-2 | 1957* |
| NBRC 12790_Ed | Riverbank slime | Böhmischbruck, Upper Palatinate, Germany | — | 5 | 2-3-1-1-2 | 1952* |
| DSM 40233_Ed | Potato | Germany | — | 6 | 2-1-3-3-3 | 1908* |
| NBRC 13083_Ed | Potato scab | United Kingdom | — | 7 | 2-3-2-1-4 | 1926* |
| CGMCC 4.1677_Ed | Muschelkalk | Werra, Germany | — | 5 | 2-3-1-1-2 | 1952* |
| CR4_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 8 | 4-1-4-1-5 | 06/2012 |
| CR10_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 8 | 4-1-4-1-5 | 06/2012 |
| CR11_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 9 | 5-2-4-4-5 | 06/2012 |
| CR13_En | Imperial moth (adult body of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 10 | 2-1-5-5-5 | 08/2012 |
| CR15_En | Imperial moth (adult wing of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 11 | 6-2-6-1-5 | 09/2012 |
| CR16_En | Imperial moth (adult body of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 12 | 6-2-4-1-5 | 09/2012 |
| CR17_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 8 | 4-1-4-1-5 | 06/2012 |
| CR19_En | Imperial moth (adult wing of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 13 | 7-4-1-1-6 | 09/2012 |
| CR21_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 8 | 4-1-4-1-5 | 06/2012 |
| CR25_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 14 | 5-2-4-6-7 | 12/2011 |
| CR33_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 14 | 5-2-4-6-7 | 12/2011 |
| CR35_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 14 | 5-2-4-6-7 | 12/2011 |
| Ma1_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 15 | 5-2-4-1-5 | 10/2011 |
| Ma24_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 16 | 1-5-7-1-5 | 12/2011 |
| Ma25_En | Imperial moth (pupa of EI) | Guanacaste Conservation Area, Costa Rica | 10°51′N, 85°37′W | 14 | 5-2-4-6-7 | 12/2011 |
| CR46_En | Owl butterfly (pupa of CT) | San José, Costa Rica | 9°56′N, 84°03′W | 17 | 7-2-1-7-5 | 05/2011 |
| CR47_En | Owl butterfly (pupa of CE) | San José, Costa Rica | 9°56′N, 84°03′W | 18 | 6-2-8-1-7 | 12/2010 |
| CR49_En | Owl butterfly (pupa of CT) | San José, Costa Rica | 9°56′N, 84°03′W | 17 | 7-2-1-7-5 | 05/2011 |
| LaPpAH-202_En | Unknown insect | — | — | 19 | 1-6-1-1-8 | — |
| S4_En | Leaf-cutting ant (AO) | Gamboa, Panama | — | 20 | 8-2-4-1-7 | — |
| FXJ8.002_M | Deep seawater | Southern Indian Ocean | 37°49′S, 62°00′E (depth, 3,500 m) | 21 | 1-2-1-2-7 | 10/2008 |
| FXJ8.008_M | Deep seawater | Southern Indian Ocean | 37°49′S, 72°00′E (depth, 3,838 m) | 21 | 1-2-1-2-7 | 10/2008 |
| FXJ8.011_M | Deep seawater | Southern Indian Ocean | 37°49′S, 72°00′E (depth, 3,838 m) | 22 | 2-3-1-2-2 | 10/2008 |
| FXJ8.026_M | Deep seawater | Southwest Indian Ocean | 35°13′S, 50°48′E (depth, 3,000 m) | 21 | 1-2-1-2-7 | 11/2008 |
| FXJ8.028_M | Deep seawater | Southeast Indian Ocean | 37°49′S, 109°59′E (depth, 4,069 m) | 21 | 1-2-1-2-7 | 10/2008 |
| FXJ8.031_M | Deep seawater | Southwest Indian Ocean | 37°46′S, 49°38′E (depth, 2,800 m) | 1 | 1-1-1-1-1 | 10/2008 |
| CNY228_M | Neritic sediment | Coastal California, USA | — | 23 | 9-4-1-1-1 | — |
| SM8_M | Sponge (HS) | Kilkieran Bay, west coast of Galway, Ireland | 9°40′S, 53°18′E | 24 | 1-7-3-8-2 | - |
| CGMCC 4.1681 | Contaminated fungus plate | Commercial Solvents Corporation, USA | — | 25 | 2-1-1-1-5 | 1957* |
| CGMCC 4.1845 | Air of the laboratory | Princeton, NJ, USA | — | 3 | 3-2-1-1-2 | 1970* |
| DSM 40455T | Contaminated plate | Rome, Italy | — | 26 | 2-1-3-1-2 | 1891* |
| NBRC 13365 | Ceiling filler | Germany | — | 27 | 1-1-3-5-5 | 1895* |
| D62 | Medicinal herb (leaf of CJ) | Xishuangbanna, Yunnan Province, China | 22°01′N, 100°47′E | 28 | 1-2-1-5-2 | 12/2004 |
Strains in bold are those whose genome sequences are available in GenBank. Ed, edaphic; En, entomic; M, marine.
AO, Acromyrmex octospinosus; CE, Caligo eurilochus; CJ, Cirsium japonicum DC; CT, Caligo telamonius; EI, Eacles imperialis; HS, Haliclona simulans. Homogenates of the rinsed whole samples, i.e., pupa, adult body, adult wing, and leaf, were used for isolation.
In the order atpD, gyrB, recA, rpoB, and trpB.
Dates are given as month/year. *, year when the strain was validly published (11).
—, information unavailable.
The sequence data for MLSA were obtained using the methods of Guo et al. (24) and Rong et al. (22). Sequences of gene fragments were aligned using the ClustalW algorithm of the software program MEGA 5.2 (28) and trimmed manually at the same position. Statistics for single genes, such as the proportion of polymorphic sites, nucleotide diversity (π), and mean G+C content, etc., were calculated by using MEGA 5.2 (28). Pairwise distances between sequences of loci were calculated using the Kimura two-parameter (K2) model (29). Average nucleotide identity (ANI) values between whole-genome sequences were calculated using the program Jspecies v1.2.1 (30) based on the BLASTN method. To detect footprints of natural selection on housekeeping genes, the ratios of nonsynonymous to synonymous substitutions (dN/dS) were calculated by using the method of Nei and Gojobori (31) in the program DnaSP 5.10 (32). Employing the Web tool of nonredundant databases (NRDB; http://pubmlst.org/analysis/), allele numbers at each locus were assigned and combined into an allelic profile, designated as a sequence type (ST), for each strain.
Phylogenetic analysis.
Phylogenetic trees were reconstructed from the concatenation of five gene loci in MEGA 5.2 (28) with the option of complete deletion of gaps, using three tree-making algorithms, i.e., neighbor-joining (NJ), maximum-parsimony (MP), and maximum-likelihood (ML). The K2 model was chosen as a substitution model for NJ. ML tree construction was performed with parameters selected by the software program ModelTest using the Bayesian information criterion as a basis for model selection (33). The tree topologies were evaluated in bootstrap analyses (34) of 1,000 resamplings. Concatenated sequences of all five housekeeping gene loci were joined head-to-tail in-frame. ML trees for single genes were also inferred from heuristic searches on 1,000 random replicates using the tree bisection reconnection algorithm in the software program PAUP 4.0b10 (35). Three closest neighbors, Streptomyces argenteolus CGMCC 4.1693T and two Streptomyces isolates labeled FXJ6.047 and SCA2-2, were used as outgroups.
Assessment of population structure.
Five independent runs of the ClonalFrame software program (36) were performed with the concatenated data set, each consisting of 500,000 burn-in iterations followed by 500,000 more updates with the scaled mutational rate θ set equal to Watterson's moment estimator (37). Tests for convergence were performed using the Gelman-Rubin statistic (38). Further genetic structure was examined using the Bayesian clustering approach of the Structure 2.3.3 program (39), in which individuals were assigned to K predefined populations according to their allele frequency. The Structure procedure was run using the linkage model with a K value of 2 to 10 for 100,000 iterations after a burn-in period of 100,000 iterations, 10 replicates per K.
Tests for recombination.
The consistency of phylogenetic topologies of single genes was examined by using the Shimodaira-Hasegawa (SH) test (40) in the software program PAUP 4.0b10 (35) with the fully optimized model, and the likelihood scores (−lnL) of a given data set and each single-gene ML phylogeny were compared. Two parameters, ρ/θ and r/m, were calculated using ClonalFrame (36) to assess the contribution of recombination relative to mutation: ρ/θ is the ratio of rates at which recombination and mutation occur and thus is a measure of the frequency of recombination relative to that of mutation; r/m is the ratio of probabilities that a certain site gets altered through recombination and mutation and thus is a measure of how important the effect of recombination is in diversification of the population relative to mutation. LIAN 3.6 software (41) was used to calculate the standardized index of association (ISA), which tests for the null hypothesis of linkage equilibrium: a value significantly different from zero indicates a tendency of linkage disequilibrium, whereas a nonsignificant value indicates a recombining population structure. The program SplitsTree 4.12 (42) was employed to perform the pairwise homoplasy index test (Φw test) (43) with the concatenated sequences and to create the phylogenetic network with the neighbor-net algorithm. To determine the level of gene flow between groups, pairwise FST (44) values were calculated and tested for significance against 1,000 bootstrap replicates using the software program Arlequin 3.1 (45).
Nucleotide sequence accession numbers.
All the nucleotide sequences obtained in this study have been submitted to the GenBank database. The accession numbers are KM189916 to KM190071 (see Table S1 in the supplemental material).
RESULTS
Strains, genes, and alleles.
The 41 analyzed strains morphologically resembled each other, forming an abundant off-white to light-yellow aerial spore mass on whitish-yellow to brown substrate mycelium on ISP medium 2, the same as with the type strain S. albidoflavus DSM 40455T (11, 22). Their 16S rRNA gene sequences were highly similar, with only one variable site along 1,375 nucleotide positions, showing a close relationship in the phylogenetic tree (see Fig. S1 in the supplemental material). The whole-genome ANI values between the five genome strains that covered all three habitat-associated groups were greater than 98%, revealing an intraspecific relationship among them (30).
Five housekeeping gene loci of the 41 strains were characterized and are summarized in Table 2. These five loci had comparable numbers of alleles and G+C contents but differed in distance ranges and nucleotide diversity. gyrB displayed the widest distance range, and atpD had the highest nucleotide diversity, while rpoB showed both the narrowest distance range and lowest nucleotide diversity. The concatenated alignment of the five loci showed that MLSA distances between the strains ranged from 0 to 0.006 (mean, 0.003), values below the 0.007 cutoff point recommended for the delineation of genomic species of Streptomyces (25, 26). Regarding alleles, many were observed more than once, and the ones most frequently found were atpD1, gyrB2, recA1, rpoB1, and trpB5, observed 12, 20, 18, 23, and 14 times, respectively (Table 1). The 41 strains defined 28 STs, of which the vast majority (21 STs) occurred only once. The most frequently found STs were 8, 14, and 21, each occurring in four strains. It was noticeable that half of the entomic strains, including CR11, CR15, CR16, CR25, CR33, CR35, Ma1, and Ma25, isolated from imperial moths, CR47, from owl butterflies, and S4, from leaf-cutting ants, owned merely a quarter of the total polymorphic sites of the entomic group and shared exactly the same gyrB sequences (Table 1); they also shared an exclusive transition in the first two bases of the atpD locus, from common double guanine to double adenine, creating a glycine-to-asparagine amino acid change along with an enhancement of hydrophilicity in AtpD. All dN/dS values of the loci were much lower than 1 (Table 2), with recA lacking nonsynonymous substitutions, indicating a strong purifying selection on these gene loci.
TABLE 2.
Properties of loci
| Locus or method of analysis | Length (bp) | No. of alleles | No. (%) of polymorphic sites | π value | Mean G+C content (mol%) | K2 distance (range) | dN/dS |
|---|---|---|---|---|---|---|---|
| atpD | 495 | 9 | 8 (1.62) | 0.0036 | 64.7 | 0–0.010 | 0.225 |
| gyrB | 411 | 7 | 7 (1.70) | 0.0035 | 69.9 | 0–0.017 | 0.049 |
| recA | 504 | 8 | 7 (1.39) | 0.0020 | 69.9 | 0–0.008 | 0 |
| rpoB | 540 | 8 | 7 (1.30) | 0.0016 | 68.5 | 0–0.006 | 0.020 |
| trpB | 567 | 8 | 8 (1.41) | 0.0024 | 73.3 | 0–0.011 | 0.027 |
| MLSA | 2,517 | 28a | 37 (1.47) | 0.0026 | 69.3 | 0–0.006 |
This is the number of combined allelic profiles.
Distribution of alleles and STs between habitat-associated groups.
With the exception of trpB, all other loci had one or two alleles found in strains of all three habitat-associated groups (Fig. 1). Altogether, the edaphic and marine groups shared 11 alleles with each other but only 6 and 8 alleles, respectively, with the entomic group. In terms of STs, one was present in both the edaphic and marine groups, whereas no common STs were found in the entomic group and either of the other two. It was obvious that more alleles and STs coexisted in the edaphic and marine groups, which seemed to indicate that the strains residing in soil and sea were more similar to each other than to those associated with insects.
FIG 1.
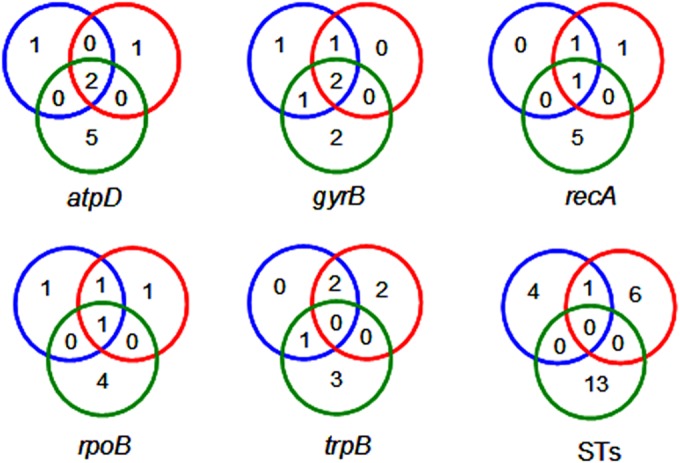
Venn diagrams of allele and ST distributions. Red, blue, and green circles represent the edaphic, marine, and entomic groups, respectively. Numbers inside the circles indicate the numbers of alleles or STs in each set.
Phylogeny and population structure of S. albidoflavus.
The phylogenetic tree in the upper part of Fig. 2 was constructed based on the concatenated alignment of the five loci. Despite a very close relationship among all the strains, they were grouped into four clusters, designated I to IV. The five genome strains spanned the phylogeny and fell into clusters I, II, and IV. Cluster I, dominated by the edaphic strains, lumped together all the soil-derived strains and five uncategorized strains, exhibiting a close relationship among them; notably, it also contained a few strains from insects and sea, suggesting frequent individual migrations or genetic exchange between strains in soil and other environments. Cluster II incorporated most of the remaining marine strains into one of its three subclusters, supported by all three tree-making algorithms and a high bootstrap value; and the other two subclusters were mainly comprised of entomic strains. Clusters III and IV were constituted purely of strains from insects, which made up to 70% (14/20) of all the entomic strains in the sample, with cluster IV formed exactly by the special half of the entomic strains that had reduced genetic polymorphism and an exclusive amino acid change. These clusters and subclusters were largely consistent with the habitats, tending to group together strains from similar sources.
FIG 2.

Phylogenetic NJ tree and Structure analyses of S. albidoflavus MLSA data. P and L indicate branches of the tree that were also determined using MP and ML algorithms; asterisks indicate branches that were supported by all three tree-making algorithms. Only bootstrap support values above 50% are shown. Each strain in the Structure results is represented by a vertical histogram partitioned into K colored segments that represent the proportion of ancestry from the major ancestral sources. The five genome strains are displayed in bold.
Given that habitat might matter in the relationship among strains, further analysis of population structure was performed, employing the Structure procedure in terms of strains' habitats. The outputs assuming K = 2, 3, and 5 are presented in the lower part of Fig. 2. At a K value of 2, most of the entomic strains, those in clusters III and IV, emerged as a distinct population, strikingly different from the others; in contrast, strains derived from sea and soil were similar to each other. An increase in the K value split most of the marine strains together with several entomic strains as a different population. A higher and optimal resolution was achieved at a K value of 5, in which strains of insects, sea, and soil were generally recognized as different populations, successively displayed in green, purple, yellow, blue, and red, with further divergence in the entomic strains (green, purple, and yellow). The correspondence between habitats and the Structure-defined populations of strains indicated a strong influence of habitat in the evolution of S. albidoflavus. Moreover, strain D62 and the other four uncategorized strains showed a high level of genetic overlap with the edaphic strains independently of the K value used.
The Structure results agreed well with the phylogenetic tree in depicting relationships among strains and also provided more insights into interactions between strains of distinct populations. For example, evidence of shared genetic variants by diverse populations is consistently found, independently of the K value, suggesting frequent gene flow; and much higher mixing arose in cluster I at K = 5, with multiple contributions from other populations, correlating well with the topology of this cluster in the phylogeny. Furthermore, cluster IV, which formed an independent lineage in the clonal genealogy inferred by ClonalFrame (see Fig. S2 in the supplemental material), was preserved as a separate and distinct population with all tested K values.
Evidence and extent of recombination in S. albidoflavus.
In order to examine the role of recombination in S. albidoflavus, a series of analyses were performed based on single and concatenated gene alignments. ML trees were generated for each single gene (see Fig. S3 in the supplemental material), resulting in all five individual phylogenies differing in topology. For instance, strains DSM 40455T and DSM 40233, which clustered in the atpD, gyrB, and recA phylogenies, occupied completely different positions in the rpoB and trpB phylogenies. The SH test results confirmed this topological incongruence, detecting a significant disagreement between single-gene topologies and all data sets with the exception of recA and a highly significant disagreement for the reciprocal comparison between atpD and gyrB (Table 3). The extensive discordance between phylogenies indicated frequent occurrences of recombination. To evaluate recombination among the five housekeeping genes in a visual way, a NeighborNet network was reconstructed based on the concatenated sequences (Fig. 3). The network showed a reticulate structure, and recombination represented by lines between splits was widespread within single habitat-associated groups and also occurred between them, resulting in strains of different habitats slightly mixed instead of forming clearly distant splits. Furthermore, the Φw test provided statistically strong evidence of recombination by rejecting the null hypothesis of no recombination (P = 0.003).
TABLE 3.
Results of SH test of alternative tree topologies for individual genes in this samplea
| Tree topology | Result for comparison with data set |
||||
|---|---|---|---|---|---|
| atpD | gyrB | recA | rpoB | trpB | |
| atpD | 767.84 | 881.08** | 862.15 | 911.84* | 1,010.40* |
| gyrB | 1,038.94** | 691.87 | 958.00 | 905.97* | 1,020.90* |
| recA | 992.21* | 892.05* | 783.30 | 899.24* | 1,030.43* |
| rpoB | 1,016.83* | 914.54* | 917.64 | 799.73 | 995.08* |
| trpB | 1,042.29** | 915.41* | 969.28 | 911.84* | 835.91 |
Values indicate the likelihood score (−lnL) of a given data set across its own ML tree (in boldface) and each of the alternative trees. *, P < 0.05; **, P < 0.01.
FIG 3.

Phylogenetic network inferred with the concatenation of five housekeeping gene loci (atpD-gyrB-recA-rpoB-trpB). Lines between splits represent uncertainty in the phylogeny and are expected if recombination has occurred. Strains from insects, sea, soil, and other sources are in green, blue, red and black, respectively. Clusters identified in the phylogenetic analysis are labeled, and cluster IV, which may represent an incipient species in S. albidoflavus, is green coded. Bar = 0.1% sequence divergence.
One way to examine the extent of recombination within natural populations is to evaluate the degree of linkage between alleles using the standardized index of association (ISA) (46). If there is perfect linkage equilibrium (alleles are independently distributed at all loci analyzed, and recombination events occur freely), the expected value of ISA is 0; if there is complete linkage disequilibrium (nonrandom association of alleles), the expected value of ISA is 1. As shown in Table 4, in the whole species, the value of ISA was positively significant but close to 0, indicating a high level of gene exchange despite being insufficient to generate complete linkage equilibrium. Positively significant but relatively low ISA values also occurred within the entomic and the marine groups; while a nonsignificant ISA value was observed within the edaphic group, suggesting a recombining structure therein. Together with the low ISA or nonsignificant values in the Structure-defined populations when the K value is 5 (see Table S2 in the supplemental material), these findings revealed frequent homologous recombination within S. albidoflavus.
TABLE 4.
Population parameters calculated from the concatenated five-gene sequences
| Group | No. of strains | No. of alleles | ISA (P value) | Recombination ratea |
FST and mean K2 distanceb |
|||
|---|---|---|---|---|---|---|---|---|
| ρ/θ | r/m | Edaphic | Entomic | Marine | ||||
| Edaphic | 8 | 7 | 0.0015 (0.520) | 10.72 ± 1.18 | 4.51 ± 0.61 | 0.003 | 0.002 | |
| Entomic | 20 | 13 | 0.1275 (<0.001) | 0.06 ± 0.01 | 1.48 ± 0.08 | 0.3125** | 0.003 | |
| Marine | 8 | 5 | 0.3699 (<0.001) | 0.09 ± 0.05 | 0.38 ± 0.08 | 0.1324* | 0.2012** | |
| All | 41 | 28 | 0.0678 (0.001) | 0.68 ± 0.09 | 0.25 ± 0.03 | |||
The mean values from five independent runs are shown as means ± standard errors of the means.
FST values are shown in the lower left quadrant, and the mean K2 distances between groups are in the upper right quadrant. *, P < 0.05, **, P < 0.01.
In order to quantify the contribution of recombination relative to that of mutation, values of ρ/θ and r/m were calculated for all 41 strains of the species, the habitat-associated groups, and the Structure-defined populations (Table 4; see also Table S2 in the supplemental material) in ClonalFrame. The results showed that within all strains, recombination occurred less frequently than mutation and acted one-fourth as importantly as mutation in introducing genetic variation. The marine group displayed a similar case with a low recombination rate, in striking contrast to the edaphic strains, where the role recombination played was far more salient than that of point mutation, since both ρ/θ and r/m were much higher than 1. For the entomic group, however, a third case occurred, in which recombination was 1.5-fold more likely to alter a nucleotide site than mutation despite its very low frequency. Recombination rates of the Structure-defined populations were largely comparable to those of respective habitat-associated groups (Table 4; see also Table S2), especially for the couples of population in green and the entomic group and population in red and the edaphic group.
Genetic exchange across habitats.
In addition to analyzing recombination within individual habitat-associated groups, we also evaluated the level of intergroup gene flow by using Weir and Cockerham's FST parameter (44) based on variance in genetic diversity within and between groups. According to Wright's F-statistics model, a zero FST value implies complete panmixis within populations, whereas a value of one indicates complete isolation of populations. As shown in Table 4, FST between the edaphic and marine groups was significant but relatively low, while the entomic group exhibited much higher FST values with either the edaphic or marine group, both significantly different from zero at the 0.01 level. This observation accords well with the distribution pattern of alleles and STs between the three habitat-associated groups and generally reveals habitat barriers to genetic exchange in S. albidoflavus, showing slightly reduced gene exchange between the soil- and sea-dwelling strains and a greater differentiation of the insect-associated strains, with definitely reduced gene exchange with the others.
DISCUSSION
Recombination in S. albidoflavus.
Frequent recombination was inferred and supported by multiple lines of evidence: (a) the Φw test strongly rejected the null hypothesis of no recombination; (b) clear topological discrepancies between single-gene phylogenies were observed by visual inspections and verified by the SH test; (c) homologous recombination both within and between the habitat-associated groups was found in the network; and (d) the low ISA or nonsignificant values calculated in the whole species or single groups and populations revealed the sexual and recombining nature of this species. Actually, the significance of recombination in Streptomyces has been reported for S. pratensis (18), where homologous recombination was first suggested to shape the evolution of streptomycetes. In addition, it was recently reported that homologous recombination is responsible for the amino acid changes in RpoB of Salinispora arenicola, which confer resistance to compounds in the rifamycin class (47). Our analyses affirm the predominance of homologous recombination in Streptomyces species, which may serve as a cohesive force slowing down the process of genetic divergence in species (5).
It appears discordant that the recombination rate detected in the whole species was fairly low (ρ/θ = 0.68; r/m = 0.25), contrasting with the extensive gene exchange in S. pratensis (18). However, this may be ascribed primarily to barriers of diverse habitats to genetic exchange, for recombination rates are supposed to be underestimated in structured populations with different ecotypes inhabiting different niches, because the presence of population structure violates the assumption of an unstructured population of the model used to estimate recombination rates in ClonalFrame (9, 36). This is supported by the observation that the entire species gave the lowest value of r/m compared with each single habitat-associated group and Structure-defined population, in accordance with habitat isolation and a structured population. Moreover, the comparable r/m values of S. albidoflavus and Salinispora (47), a related actinomycete genus, further suggest an existence of great hindrance to genetic exchange within S. albidoflavus. Therefore, the distinct habitats may preclude genetic exchange between the inhabiting strains and be responsible for the apparent low recombination rate detected in the S. albidoflavus species.
Interestingly, we observed great variation in recombination rates among the habitat-associated groups and the Structure-defined populations. In terms of habitats, recombination acts as a powerful force in shaping the genetic diversity of the edaphic and entomic strains, since both of these groups had an r/m value of >1. Strains in soil are much more recombinogenic, with a quite high recombination rate, comparable to that of Haemophilus influenzae (r/m = 3.7) (9), and this may be bound up with the complex signal exchange in soil (16). In contrast, the entomic strains present a high contribution but fairly low frequency of recombination relative to that of mutation, probably implying a preference for exchanging large nucleotide segments in recombination events among them. The low frequency and contribution of recombination observed in the marine group seem incongruent with the suggested high chance of frequent genetic exchange in aquatic bacteria (9). However, this finding may be partially explained by the presence of four marine isolates, FXJ8.002, FXJ8.008, FXJ8.026, and FXJ8.028, which could have caused an inflated estimate of clonality of the population by sharing the same genotype despite having disparate geographic origins (9). Taken together, it is possible that recombination plays uneven roles in different S. albidoflavus groups and populations, as has been observed in several other species (48, 49). However, there is also a possibility that these recombination rates were artificially skewed due to the limited numbers of strains from each habitat type and potentially uneven sampling across spatial scales.
Influence of ecology on the population structure of S. albidoflavus.
We acknowledge our possible limitations due to the loose categorization that assigned most of the specific sources into insects, sea, and soil. However, the analyses based on these three roughly defined habitat types did reveal the significance of ecology in structuring S. albidoflavus population on a large scale, supported by the strong habitat signal in both phylogenetic and Structure analyses.
Effects of habitat on population structure are often reported for macroorganisms, especially animals because of their excellent ability to disperse in nature. Microorganisms, in contrast, are tiny and much more sensitive to environmental changes but are also commonly discussed regarding their interactions with ecological environments or niches and are even found to undergo more adaptive evolution, inferred from protein-coding gene sequences (50). Compared with the meadow soil-inhabiting S. pratensis (18), S. albidoflavus is present in a variety of ecological environments, and more information on habitat is available and is considered here. Their sources, including deep seawater, imperial moths, leaf-cutting ants, medical herbs, owl butterflies, sponges, and soil, may differ in a spectrum of ecological factors, including nutrient contents, temperature, and pH, as well as other biotic factors. It is likely that natural selection based on differences between these factors promotes adaptations to local environments and causes strains to be distributed differentially among habitats. The further divergence observed within the entomic strains in Structure (Fig. 2) suggests that the differential distribution of bacterial strains among environments can arise not only during invasion of new habitats but also within individual habitat types with differentiated environmental conditions, which can be equivalent to niches. Therefore, ecological divergence should be a nonnegligible factor in causing genetic divergence in bacterial populations, even those residing in various habitats or with a worldwide distribution.
On the other hand, the closer relationship found between the edaphic and marine strains than to the entomic strains might be attributed to both sea and soil representing open environments, while insects provide isolated and more constant conditions for microbial inhabitants, with frequent interaction with hosts yet less disturbance from the surroundings. It is also reasonable that the single endophytic strain D62 exhibited more genetic overlap with the edaphic strains in Structure results at all K values, since endophytic strains can also act as saprophytes and subsist in the soil when the leaves decay (51). Additionally, all of the other four uncategorized strains with obscure sources (CGMCC 4.1681, CGMCC 4.1845, DSM 40455T, and NBRC 13365) (Table 1) showed a close relationship with the soil-derived strains and a major contribution from the population in red in Structure when the K value was 5 (Fig. 2), suggesting their probable edaphic origin. These results demonstrate that genetic relationships and ecological information can be correlated with each other. To figure out the specific ecological factors that lead to the differential distribution of S. albidoflavus strains, more investigations with detailed ecological and physiological information are needed.
Combined effects of homologous recombination and ecological divergence.
It is likely that frequent homologous recombination serves as a cohesive force maintaining convergence of species, akin to its role in sexual organisms, and that when local adaptation occurs during invasion of new habitats or niches, if the differential distribution of strains among habitats is strong enough to depress gene flow sufficiently between related genotypes (5, 52), certain strains will escape from being homogenized by recombination, and subsequent accumulations of mutations and/or genetic drift within them will eventually lead to independent evolutionary clusters. This trigger-like action of ecological divergence among frequent recombination events in the process of speciation has been observed in some other bacterial populations, especially Vibrionaceae (8, 53, 54). To some extent, the way S. albidoflavus strains occupy and adapt to a new habitat resembles the widely discussed formation of new ecotypes (6, 52), therefore, the observed habitat barriers to recombination in S. albidoflavus clarify the coexistence of homologous recombination and ecological divergence in the diversification of streptomycetes.
The complex and specific environmental conditions in insect-associated niches may enable further genetic differentiation within the entomic strains, especially those in cluster IV (Fig. 2; see also Fig. S2 in the supplemental material). Since most (8/10) of the cluster IV strains were isolated from the same species of imperial moths, Eacles imperialis, there is a high possibility that their reduced genetic diversity and exclusive amino acid change resulted from recent selection events (55) within imperial moths and reflected traits specific to certain conditions in the host. Taken together with the much greater recombination within it than with the others in the network (Fig. 3), it seems that cluster IV has emerged as a distinct and cohesive population that resulted at least partly from effects of both ecological selection and recombination, which exemplifies the scenario of dynamic speciation we suggested above, and it may even represent an incipient species. More evidence from population genomics and physiological and biochemical analyses is needed for further verification of this possibility.
Since most of the analyzed habitats were distributed in different geographic regions, it is difficult to completely tell geographic isolation apart from barriers to recombination in shaping the S. albidoflavus population structure. A thorough examination of the correlation between geographic distance and genetic distance was also unavailable due to the limited numbers of sites per habitat and strains per site and their wide spatial distribution. Nevertheless, it is obvious that all the edaphic strains clustered together in cluster I (Fig. 2 and 3) despite being sampled from disparate regions, as was the case with the four marine isolates mentioned above, while most entomic strains were distinctly diverged into two clusters (III and IV; Fig. 2 and 3) regardless of their having the same geographic location. In addition, a relaxed Mantel test (56) conducted on all strains with available geographic location information implied a weak correlation between geographic and genetic distances (data not shown). To further determine the exact geographic role in population structure, studies with more strains and sites are needed.
Implications for streptomycete evolution.
So far, the predominant recombination detected in both S. pratensis (18) and S. albidoflavus seems to picture Streptomyces species as strain groups characterized by frequent genetic exchange. This phenomenon may have its roots in multiple unique characteristics of streptomycetes, including their large and unstable linear chromosomes (57), the specialized conjugative system relying on a single plasmid-encoded protein for transferring double-stranded DNA (58), and their complex lifestyle (11, 18). The vast recombination in Streptomyces will certainly facilitate the rapid sweep of clones bearing advantageous traits, promote local adaption, and benefit a fast spread of resistance genes (57, 58). However, as a strong mixing force, it may also obscure the genuine evolutionary history of species by decaying clonality. Meanwhile, the impact of habitat divergence on S. albidoflavus seems to be profound. Although our data are insufficient to fully address the cause of the population structure, they indicate that habitat barriers to recombination are one of the drivers; S. albidoflavus is probably structured in a combined scenario of both environmental selection and geographic isolation, which has proved to be common within eukaryotes (59). Since streptomycetes are very environmentally versatile and adaptable, our findings emphasize the importance and necessity of incorporating ecology in unraveling the evolutionary history of these microorganisms.
What we observed in S. albidoflavus suggests that the evolution of streptomycetes is a long-term and complicated process affected by multiple factors, including homologous recombination, ecological divergence, and potentially geographic isolation and genetic drift. The recommended unity of homologous recombination and ecology in this work sheds light on the Streptomyces species concept. Nevertheless, to get a comprehensive examination of the driving forces of speciation, more population genetic analyses of Streptomyces species with larger population sizes and more available ecological information are indispensable.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Xi (China University of Petroleum) for her assistance in isolation and DNA sequence amplification of the marine strains and J. Wang (Institute of Microbiology, Chinese Academy of Sciences) for his help in acquiring the ecological information of multiple reference strains. We are also grateful to the Ministry of Environment, Energy and Telecommunications and the Guanacaste Conservation Area for providing collection permits in Costa Rica (resolution no. ACG-PI-018-2012).
This work was funded by the National Science Foundation of China (NSFC) (no. 31100003, 31470140, and 31470142), the External Cooperation Program of Chinese Academy of Sciences (no. GJHZ1108), and the program of the China Ocean Mineral Resources R & D Association (no. DY125-15-R-02).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02925-14.
REFERENCES
- 1.Rosselló-Mora R, Amann R. 2001. The species concept for prokaryotes. FEMS Microbiol Rev 25:39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x. [DOI] [PubMed] [Google Scholar]
- 2.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL, Swings J. 2005. Re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 3.Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. 2009. The bacterial species challenge: making sense of genetic and ecological diversity. Science 323:741–746. doi: 10.1126/science.1159388. [DOI] [PubMed] [Google Scholar]
- 4.Barraclough TG, Balbi KJ, Ellis RJ. 2012. Evolving concepts of bacterial species. Evol Biol 39:148–157. doi: 10.1007/s11692-012-9181-8. [DOI] [Google Scholar]
- 5.Fraser C, Hanage WP, Spratt BG. 2007. Recombination and the nature of bacterial speciation. Science 315:476–480. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohan FM, Perry EB. 2007. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol 17:R373–R386. doi: 10.1016/j.cub.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 7.Koeppel A, Perry EB, Sikorski J, Krizanc D, Warner A, Ward DM, Rooney AP, Brambilla E, Connor N, Ratcliff RM, Nevo E, Cohan FM. 2008. Identifying the fundamental units of bacterial diversity: a paradigm shift to incorporate ecology into bacterial systematics. Proc Natl Acad Sci U S A 105:2504–2509. doi: 10.1073/pnas.0712205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, Polz MF, Alm EJ. 2012. Population genomics of early events in the ecological differentiation of bacteria. Science 336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vos M, Didelot X. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J 3:199–208. doi: 10.1038/ismej.2008.93. [DOI] [PubMed] [Google Scholar]
- 10.Berdy J. 2012. Thoughts and facts about antibiotics: where we are now and where we are heading. J Antibiot 65:385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]
- 11.Kämpfer P. 2012. Genus I. Streptomyces Waksman and Henrici 1943, 339,AL emend. Witt and Stackebrandt 1990, 370, emend. Wellington, Stackebrandt, Sanders, Wolstrup and Jorgensen 1992, 159, p 1455–1767. In Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB (ed), Bergey's manual of systematic bacteriology, 2nd ed, vol 5 Springer, New York, NY. [Google Scholar]
- 12.Bull AT. 2011. Actinobacteria of the extremobiosphere, p 1203–1240. In Horikoshi K. (ed), Extremophiles handbook, vol 2 Springer, Tokyo, Japan. [Google Scholar]
- 13.Bugg TD, Ahmad M, Hardiman EM, Singh R. 2011. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol 22:394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Geng P, Xiao Y, Hu M. 2012. Bioremediation of β-cypermethrin and 3-phenoxybenzaldehyde contaminated soils using Streptomyces aureus HP-S-01. Appl Microbiol Biotechnol 94:505–515. doi: 10.1007/s00253-011-3640-5. [DOI] [PubMed] [Google Scholar]
- 15.Challis GL, Hopwood DA. 2003. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A 100:14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vetsigian K, Jajoo R, Kishony R. 2011. Structure and evolution of Streptomyces interaction networks in soil and in silico. PLoS Biol 9:e1001184. doi: 10.1371/journal.pbio.1001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rong X, Doroghazi JR, Cheng K, Zhang L, Buckley DH, Huang Y. 2013. Classification of Streptomyces phylogroup pratensis (Doroghazi and Buckley, 2010) based on genetic and phenotypic evidence, and proposal of Streptomyces pratensis sp. nov. Syst Appl Microbiol 36:401–407. doi: 10.1016/j.syapm.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Doroghazi JR, Buckley DH. 2010. Widespread homologous recombination within and between Streptomyces species. ISME J 4:1136–1143. doi: 10.1038/ismej.2010.45. [DOI] [PubMed] [Google Scholar]
- 19.Skinner FA. 1953. Inhibition of Fusarium culmorum by Streptomyces albidoflavus. Nature 172:1191. doi: 10.1038/1721191a0. [DOI] [PubMed] [Google Scholar]
- 20.Seipke RF, Hutchings MI. 2013. The regulation and biosynthesis of antimycins. Beilstein J Org Chem 9:2556–2563. doi: 10.3762/bjoc.9.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hain T, Ward-Rainey N, Kroppenstedt RM, Stackebrandt E, Rainey FA. 1997. Discrimination of Streptomyces albidoflavus strains based on the size and number of 16S-23S ribosomal DNA intergenic spacers. Int J Syst Bacteriol 47:202–206. doi: 10.1099/00207713-47-1-202. [DOI] [PubMed] [Google Scholar]
- 22.Rong X, Guo Y, Huang Y. 2009. Proposal to reclassify the Streptomyces albidoflavus clade on the basis of multilocus sequence analysis and DNA-DNA hybridization, and taxonomic elucidation of Streptomyces griseus subsp. solvifaciens. Syst Appl Microbiol 32:314–322. doi: 10.1016/j.syapm.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Nunney L, Vickerman DB, Bromley RE, Russell SA, Hartman JR, Morano LD, Stouthamer R. 2013. Recent evolutionary radiation and host plant specialization in the Xylella fastidiosa subspecies native to the United States. Appl Environ Microbiol 79:2189–2200. doi: 10.1128/AEM.03208-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Y, Zheng W, Rong X, Huang Y. 2008. A multilocus phylogeny of the Streptomyces griseus 16S rRNA gene clade: use of multilocus sequence analysis for streptomycete systematics. Int J Syst Evol Microbiol 58:149–159. doi: 10.1099/ijs.0.65224-0. [DOI] [PubMed] [Google Scholar]
- 25.Rong X, Huang Y. 2010. Taxonomic evaluation of the Streptomyces griseus clade using multilocus sequence analysis and DNA-DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int J Syst Evol Microbiol 60:696–703. doi: 10.1099/ijs.0.012419-0. [DOI] [PubMed] [Google Scholar]
- 26.Rong X, Huang Y. 2012. Taxonomic evaluation of the Streptomyces hygroscopicus clade using multilocus sequence analysis and DNA-DNA hybridization, validating the MLSA scheme for systematics of the whole genus. Syst Appl Microbiol 35:7–18. doi: 10.1016/j.syapm.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Labeda DP, Doroghazi JR, Ju K-S, Metcalf WW. 2014. Taxonomic evaluation of Streptomyces albus and related species using multilocus sequence analysis and proposals to emend the description of Streptomyces albus and describe Streptomyces pathocidini sp. nov. Int J Syst Evol Microbiol 64:894–900. doi: 10.1099/ijs.0.058107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 30.Richter M, Rosselló-Mora R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. [DOI] [PubMed] [Google Scholar]
- 32.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 33.Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 34.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 35.Swofford DL. 2003. PAUP*: phylogenetic analysis using parsimony (* and other methods). Version 4.0b10. Sinauer Associates, Sunderland, MA. [Google Scholar]
- 36.Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watterson GA. 1978. The homozygosity test of neutrality. Genetics 88:405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gelman A, Rubin DB. 1992. Inference from iterative simulation using multiple sequences. Stat Sci 7:457–472. doi: 10.1214/ss/1177011136. [DOI] [Google Scholar]
- 39.Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimodaira H, Hasegawa M. 1999. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114–1116. doi: 10.1093/oxfordjournals.molbev.a026201. [DOI] [Google Scholar]
- 41.Haubold B, Hudson RR. 2000. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16:847–849. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- 42.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 43.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- 45.Excoffier L, Laval G, Schneider S. 2005. Arlequin version 3.0: an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 46.Smith JM, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc Natl Acad Sci U S A 90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freel KC, Millán-Aguiñaga N, Jensen PR. 2013. Multilocus sequence typing reveals evidence of homologous recombination linked to antibiotic resistance in the genus Salinispora. Appl Environ Microbiol 79:5997–6005. doi: 10.1128/AEM.00880-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Didelot X, Bowden R, Street T, Golubchik T, Spencer C, McVean G, Sangal V, Anjum MF, Achtman M, Falush D, Donnelly P. 2011. Recombination and population structure in Salmonella enterica. PLoS Genet 7:e1002191. doi: 10.1371/journal.pgen.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Didelot X, Nell S, Yang I, Woltemate S, van der Merwe S, Suerbaum S. 2013. Genomic evolution and transmission of Helicobacter pylori in two South African families. Proc Natl Acad Sci U S A 110:13880–13885. doi: 10.1073/pnas.1304681110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyre-Walker A. 2006. The genomic rate of adaptive evolution. Trends Ecol Evol 21:569–575. doi: 10.1016/j.tree.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 51.Bacon CW, White JF. 2000. Physiological adaptations in the evolution of endophytism in the Clavicipitaceae, p 237–261. In Bacon CW, White JF (ed), Microbial endophytes. Marcel Dekker, Inc., New York, NY. [Google Scholar]
- 52.Cohan FM. 2002. Sexual isolation and speciation in bacteria. Genetica 116:359–370. doi: 10.1023/A:1021232409545. [DOI] [PubMed] [Google Scholar]
- 53.Hunt DE, David LA, Gevers D, Preheim SP, Alm EJ, Polz MF. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 54.Szabo G, Preheim SP, Kauffman KM, David LA, Shapiro J, Alm EJ, Polz MF. 2013. Reproducibility of Vibrionaceae population structure in coastal bacterioplankton. ISME J 7:509–519. doi: 10.1038/ismej.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cohan FM. 2002. What are bacterial species? Annu Rev Microbiol 56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- 56.Mantel N. 1967. The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220. [PubMed] [Google Scholar]
- 57.Hopwood DA. 2006. Soil to genomics: the Streptomyces chromosome. Annu Rev Genet 40:1–23. doi: 10.1146/annurev.genet.40.110405.090639. [DOI] [PubMed] [Google Scholar]
- 58.Thoma L, Muth G. 2012. Conjugative DNA transfer in Streptomyces by TraB: is one protein enough? FEMS Microbiol Lett 337:81–88. doi: 10.1111/1574-6968.12031. [DOI] [PubMed] [Google Scholar]
- 59.Orsini L, Vanoverbeke J, Swillen I, Mergeay J, Meester L. 2013. Drivers of population genetic differentiation in the wild: isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Mol Ecol 22:5983–5999. doi: 10.1111/mec.12561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.