

Abstract
The hindguts of lower termites and Cryptocercus cockroaches are home to a distinct community of archaea, bacteria, and protists (primarily parabasalids and some oxymonads). Within a host species, the composition of these hindgut communities appears relatively stable, but the evolutionary and ecological factors structuring community composition and stability are poorly understood, as are differential impacts of these factors on protists, bacteria, and archaea. We analyzed the microbial composition of parabasalids and bacteria in the hindguts of Cryptocercus punctulatus and 23 species spanning 4 families of lower termites by pyrosequencing variable regions of the small-subunit rRNA gene. Especially for the parabasalids, these data revealed undiscovered taxa and provided a phylogenetic basis for a more accurate understanding of diversity, diversification, and community composition. The composition of the parabasalid communities was found to be strongly structured by the phylogeny of their hosts, indicating the importance of historical effects, although exceptions were also identified. Particularly, spirotrichonymphids and trichonymphids likely were transferred between host lineages. In contrast, host phylogeny was not sufficient to explain the majority of bacterial community composition, but the compositions of the Bacteroidetes, Elusimicrobia, Tenericutes, Spirochaetes, and Synergistes were structured by host phylogeny perhaps due to their symbiotic associations with protists. All together, historical effects probably resulting from vertical inheritance have had a prominent role in structuring the hindgut communities, especially of the parabasalids, but dispersal and environmental acquisition have played a larger role in community composition than previously expected.
INTRODUCTION
Cryptocercus cockroaches and their sister lineage, the lower termites (1), are both dependent on diverse communities of microorganisms in their hindguts to gain nutrition from lignocellulose (reviewed in references 2 and 3). These are fascinating and complex communities dominated by parabasalid (Parabasalia) and oxymonad (Preaxostyla) protists (4) as well as numerous bacterial lineages, including Actinobacteria, Bacteroidetes, Elusimicrobia, Firmicutes, Proteobacteria, Spirochaetes, and Synergistes (5–7). Moreover, a large proportion of the bacteria form obligate symbiotic interactions with the protists (see, for example, references 8–14), so the community contains multiple levels of symbiosis. Because of the diversity of symbionts, the hindguts of Cryptocercus cockroaches and lower termites can be used as model systems to study the ecology and interactions between protists and bacteria. Here, we investigate the ecological and evolutionary differences between protists and bacteria in structuring community composition. These differences are critical to understanding the diversification and adaptation of microbes, the stability and resilience of community structure, and the maintenance of ecosystem functions, particularly in regard to understanding lignocellulose digestion and the ecology and evolution of the termites.
Protist diversity in the hindguts varies depending on the host species, but related hosts tend to harbor related protists, and the composition of the protist communities for each host species appears to be stable over space and time (4). The symbionts are transmitted among family or colony members through the consumption of proctodeal fluid, which limits the dispersal of symbionts to closely interacting individuals. These observations suggest that the composition of protists in the hindguts is strongly structured by historical (evolutionary) effects due to vertical transmission of the protists and codiversification within their hosts (15–17). However, this has never been tested using molecular surveys of diversity and phylogenetic approaches. Most of our knowledge on protist diversity in the hindguts is based on morphological descriptions and focused on individual taxa and not the community.
Unlike the protist communities, where over 100 years of morphological examination and decades of molecular characterization provided some expectation of the composition and even conservation between hosts, relatively little is known of the bacteria in lower termites and Cryptocercus cockroaches. Recent studies have described the bacterial diversity in the hindguts of Cryptocercus cockroaches and a limited representation of lower and higher termites (6, 18–21). Like the protists, the diversity of bacteria is distinct, unique, and consistent compared to other insect gut communities, and historical effects also appear to have played a role in their community structure (18–20). Bacterial diversity, however, has neither been surveyed across the lower termites nor in conjunction with the protists to compare the relative influence of historical effects on community structure in these two groups of microorganisms.
To investigate the similarities and differences regulating protist and bacterial community structure, and how each community affects the other at ecological and evolutionary scales, matched descriptions of protist and bacterial diversity are required. We used high-throughput sequencing of a variable region of the small-subunit rRNA gene to obtain comprehensive descriptions of the parabasalid and bacterial diversities in the hindguts of Cryptocercus cockroaches and 23 lower termite species. Using these data, we compare the extent by which historical effects have influenced the composition and diversification of parabasalids and bacteria and the role of additional factors in regulating community composition in the hindguts.
MATERIALS AND METHODS
Insect and hindgut collections and DNA extraction.
Twenty-three insect species, including representatives from 4 families of lower termites plus Cryptocercus cockroaches, were collected (see Table S1 in the supplemental material). The 3 Cryptocercus samples were from distinct populations of the Cryptocercus punctulatus species complex, many of which have different chromosome numbers (22). The paunch (P3) compartments from hindguts of live insects were dissected, and the contents were collected into Trager's solution U (23) and stored at −20°C. For Porotermes adamsoni and Coptotermes sp. TN2, the hindgut contents were collected into Trager's solution U and stored in 70% ethanol at −20°C. DNA was extracted from the hindgut contents using the MasterPure Complete DNA and RNA purification kit (Epicentre) by following the manufacturer's protocol. Host DNA was also extracted from either the head or leg of the insect.
Insect barcoding and phylogeny.
Host insects were identified based on morphology, and the identity was confirmed by DNA barcoding. For insect samples that were not previously barcoded, an approximately 380-bp fragment of the mitochondrial 16S (large subunit) rRNA gene was PCR amplified and sequenced from head, leg, or hindgut DNA (which contained DNA from both host and symbionts). Template DNA was added to a mixture of 1× EconoTaq Plus green (Lucigen) and 5 pmol of the primers LR-J (5′-TTACGCTGTTATCCCTAA-3′) and LR-N (5′-CGCCTGTTTATCAAAAACAT-3′) for a 25-μl reaction mixture. The following thermal profile was used: 94°C for 2 min, 35 cycles of 94°C for 30 s, 50°C for 1 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. The resulting PCR product was cleaned using a silica membrane spin column (Epoch Life Science) with the recommended buffers and Sanger sequenced using BigDye 3.1 (Applied Biosystems). Mitochondrial 16S rRNA sequences from termites previously barcoded were retrieved from GenBank.
The mitochondrial 16S rRNA sequences were aligned using MAFFT L-INS-i (24). The ends of the alignments were trimmed manually. Gblocks was used to remove highly variable and ambiguously aligned sites but allowed smaller final blocks, gap positions, and less strict flanking positions (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) (25). Phylogenetic trees were inferred from maximum likelihood (ML) analysis using RAxML 7.0.4 implementing a general-time-reversible (GTR) model of nucleotide substitution with the Γ (gamma) model of rate heterogeneity (26). Statistical support for the consensus tree was assessed from 1,000 bootstrap replicates.
The evolutionary relationships between the insects were also visualized by generating a pairwise distance matrix of the percent dissimilarity between the mitochondrial 16S rRNA sequences and conducting a principal coordinate analysis (PCoA). This PCoA plot was compared to the PCoA plots generated from the microbial communities (see below).
18S and 16S rRNA pyrotag sequencing, error correction, and operational taxonomic unit (OTU) clustering and curation.
The diversities of protist and bacterial hindgut microbes in each termite sample were determined separately by tag-encoded Roche 454 FLX+ titanium sequencing. For protists, the V4-to-V5 region of the 18S (small subunit) rRNA gene was amplified using the primers Euk560F (5′-CCAGCASCYGCGGTAATWCC-3′) and Euk1055R (5′-CGGCCATGCACCACC-3′). For bacteria, primers 28F (5′-GAGTTTGATCNTGGCTCAG-3′) and 519R (5′-GTNTTACNGCGGCKGCTG-3′) were used to amplify the V1-to-V2 region of the 16S (small subunit) rRNA gene. PCR amplification and sequencing were performed by the Research and Testing Laboratory (Lubbock, TX, USA) as described previously (27, 28).
The 16S rRNA and 18S rRNA sequences were analyzed separately, but by using identical methods, except where indicated below. As implemented in QIIME (29), AmpliconNoise and Perseus were used to remove sequencing errors, PCR errors, and reads that were chimeric (30). The denoised sequences were clustered using UClust (31) with a 97% similarity threshold to designate OTUs. OTUs represented by at least 5 sequence reads were retained for further analysis. The longest sequence of the cluster was chosen as the representative OTU. The OTUs were assigned a taxonomic designation using RDP Classifier (32). Reference taxonomies were obtained from the GreenGenes (v. 4_feb_2011) and Silva (v. r104) databases for 16S and 18S analyses, respectively.
From the 236 OTUs identified from the 18S rRNA sequence data, OTUs classified as Metazoa (17 OTUs, all of which corresponded to insect host sequences), Fungi (2 OTUs), Bacteria (8 OTUs), and Archaea (2 OTUs) were removed, as well as two potentially chimeric OTUs, identified based on divergent blastn hits along the length of the sequence to the nonredundant (nr) database in GenBank. To filter out cross-contamination between samples, OTUs were considered only if they were represented by at least 3 sequences in a sample.
Sequences from parabasalid protists dominated the 18S rRNA data set (97.9% of the sequences). Oxymonads are also known to be present in several hosts examined, but the 18S rRNA genes of oxymonads are unusually long and often refractory to molecular analysis (33–35), so we expected the oxymonads to be underrepresented. Accordingly, we removed oxymonad OTUs from the analyses (23 Saccinobaculus OTUs from Cryptocercus cockroaches and one Dinenympha OTU and one Pyrsonympha OTU from the hindgut of Reticulitermes okanaganensis) and also 4 potential diplomonad OTUs (found in Cryptocercus hindguts), to focus exclusively on the parabasalids which in any case dominate these communities.
For the OTUs identified from the 16S rRNA sequence data, a blastn search (E value cutoff = 1e−50, percent identity cutoff = 95%) to GenBank's nr database was used to identify potential contaminants in the data. This strict search revealed 7 OTUs that hit bacteria found on human or mouse skin as well as a single OTU corresponding to Blattabacterium, an endosymbiont found in Cryptocercus fat bodies, and 1 OTU matching a bacterium found in human blood. These OTUs were removed from further analysis. An additional chimeric OTU was also removed.
Phylogenetic and community analyses.
The parabasalid 18S rRNA OTU sequences were aligned using MAFFT L-INS-i. Gblocks was used to remove ambiguously aligned positions, and a phylogenetic tree was calculated using RAxML and a GTR-GAMMA nucleotide substitution model.
The bacterial 16S rRNA OTU sequences were aligned using MAFFT L-INS-i. Alignment positions that were gaps in greater than 80% of the sequences and the top 10% of positions with the most entropy (uncertainty) were removed using QIIME. RAxML with a GTR-GAMMA model was used to build a phylogenetic tree. Additional alignments and phylogenetic trees were made with sequences from OTUs belonging to specific bacterial phyla or classes.
Although the statistical support was not robust, 18S rRNA and 16S rRNA phylogenetic trees always recovered clusters containing OTUs with the same classification (as assigned by the RDP Classifier). Unclassified OTUs found within these clusters were manually assigned the same class-level taxonomy.
The OTU read abundances for each sample were subsampled (rarified) to normalize the total abundance across all samples. Rarefaction curves were calculated using the Chao-1 estimator as implemented in QIIME. Using total or normalized OTU abundances, unweighted UniFrac distances (36) between communities were calculated using QIIME, and beta diversity was examined using PCoA from the vegan package (http://CRAN.R-project.org/package=vegan) (37), as implemented in R v. 1.16 (www.r-project.org). PCoA plots were compared to each other using Procrustes analyses. PROTEST, a permutation test, was used to test the significance of the Procrustes analyses. Shannon-Wiener diversity index and Pielou's evenness index were also calculated using vegan. Topiary Explorer (38) was used to obtain subtrees and color branches from phylogenetic trees. OTU networks were visualized using Cytoscape v.2.8.2 (39).
Nucleotide sequence accession numbers.
Host mitochondrial 16S rRNA gene sequences were deposited in the NCBI nucleotide database (GenBank) under accession numbers KJ438360 to KJ438378. 454 sequence data have been deposited in the NCBI sequence read archive (SRA) under the BioProject accession number PRJNA238270 and study accession number SRP038994.
RESULTS
Insect phylogeny.
Host identifications were confirmed by DNA barcoding the mitochondrial 16S rRNA gene, and the barcode phylogeny was compared with other cockroach/termite phylogenies. Morphology and barcode data allowed most termites to be identified to the species level. For all termites except Neotermes jouteli, the phylogeny from the barcode data matched expectations predicated on morphology-based identifications (Fig. 1A). Phylogenetic analysis confirmed that each host insect fell into one of 6 distinct groups that were consistent with established phylogeny (40), essentially corresponding with their taxonomic family: Cryptocercidae, Kalotermitidae, Stolotermitidae, Archotermopsidae, Rhinotermitidae (excluding Reticulitermes), and Reticulitermes (Fig. 1A). The genus Reticulitermes branched as a sister to other members of its family, Rhinotermitidae, and, as other studies have shown, Heterotermes clustered with Coptotermes (40–42). The Reticulitermes and the rhinotermitids formed two distinct, strongly supported clusters, so we treated them as separate groups for analyzing the symbiont communities. Porotermes and Zootermopsis are members of different families but are considered close relatives and were once classified together within the same family, Termopsidae, albeit in different subfamilies. The kalotermitid termites grouped together, but with low support.
FIG 1.

(A) Phylogenetic tree of the insects sampled for this study based on an alignment of mitochondrial 16S rRNA sequences and calculated using maximum likelihood. Bootstrap values at nodes with greater than 60% support are shown. The color of the branches indicates the grouping of the insects into 6 lineages, which roughly correspond to their taxonomic families. GenBank accession numbers follow the species names. (B) Maximum likelihood tree from an alignment (255 positions) of parabasalid OTU sequences. The branches are colored corresponding to panel A, indicating the insect host lineage for the majority of sequences represented by an OTU. *, Trichonympha OTUs from Incisitermes species; #, Trichonympha OTUs from Reticulitermes species; ^, Spirotrichonymphea-like OTUs from Porotermes adamsoni that likely originated from horizontal transfer; +, Spirotrichonymphea OTUs from Paraneotermes simplicicornis that are possibly basal or horizontally transferred; x, divergent OTUs classified as “Trichomonadea?” in Reticulitermes.
Similar groupings were recovered using a PCoA of genetic distances (see Fig. S1 in the supplemental material). As other studies have shown, the phylogeny of the termites is not fully resolved, such as the placement of the Kalotermitidae relative to other families and the paraphyly of the Rhinotermitidae (40, 43, 44), so correlation of the PCoAs was used to assess the relationship between insect hosts and their hindgut communities instead of cocladogenesis (see below).
Alpha diversity of parabasalids and bacteria.
A total of 116,617 18S rRNA and 124,142 16S rRNA sequences were obtained, resulting in 236 18S rRNA OTUs and 1,902 16S rRNA OTUs after removing erroneous sequences, chimeras, and rare OTUs with less than 5 sequences. From these, manually curated data sets were established by removing potentially cross-contaminating OTUs or, from the 18S rRNA OTUs, nonparabasalid symbionts, resulting in an exclusively parabasalid data set of 172 OTUs (86,721 sequences) and a bacterial data set of 1,889 OTUs (122,516 sequences) (see Materials and Methods for details). From these final data sets, rarefaction curves were asymptotic, indicating that the parabasalid and bacterial communities were sufficiently sampled (see Fig. S2 in the supplemental material).
The rhinotermitids hosted hindgut communities with significantly lower bacterial diversity than the other host groups (one-way analysis of variance, both richness and evenness, P = 4.9 × 10−7; pairwise t tests, P values of < 0.0005) (see Table S1 in the supplemental material). Along with Porotermes adamsoni (Stolotermitidae), these hindgut communities were dominated by singular OTUs that were likely from symbiotic bacteria of their hindgut protists, as discussed below. Within the parabasalids, however, diversity indices did not correlate with insect host phylogenetic groups, tested by a one-way analysis of variance (richness, P = 0.142; evenness, P = 0.742).
Compared to each other, parabasalid and bacterial diversity indices also were not correlated, either positively or negatively, when tested by linear regression analysis (richness, r2 = 0.07, P = 0.19; evenness, r2 = 0.09, P = 0.13). Cryptocercus and termopsid species contained the highest diversity of parabasalid taxa, whereas bacterial diversity was highest in the kalotermitids and Reticulitermes species (see Table S1 in the supplemental material). Incisitermes banksi and Incisitermes schwarzi hosted the lowest diversity of parabasalids, harboring only 2 OTUs each.
Parabasalid endemicity and diversity.
The parabasalid taxa found in the hindguts mostly fell within one of five classes: Cristamonadea, Spirotrichonymphea, Trichomonadea, Trichonymphea, and Tritrichomonadea (Fig. 1B and 2A and B). Seventeen remaining OTUs were questionably assigned (marked with a question mark) or could not be assigned to an existing order. These OTUs likely represent lineages that lack molecular data, and their phylogenetic relationships are poorly understood. The vast majority of parabasalid OTUs were either unique to a host species (75% of OTUs) or restricted to their host group (Fig. 3A), suggesting that host species harbor highly endemic symbiont communities. Only a single OTU, belonging to the Trichomonadea, was shared in hindguts across host groups, between Porotermes adamsoni from the Stolotermitidae and Coptotermes testaceus from the Rhinotermitidae, but was the least abundant member in either community (0.03% and 3% of the sequence reads, respectively). In addition, only 45% of the parabasalid OTU sequences were at least 95% similar to known 18S rRNA sequences in GenBank, and the number of OTUs generally exceeded the number of known described species in a host (see Table S1 in the supplemental material).
FIG 2.

(A) Number of OTUs for a taxonomic class of parabasalids in the hindguts of Cryptocercus cockroaches and lower termites; (B) relative abundance of parabasalid sequence reads, classified by taxonomic class, with figure legend as shown in panel A; (C) number of OTUs for a taxonomic phylum or class (phylum) of bacteria in the hindguts of Cryptocercus cockroaches and lower termites; (D) relative abundance of bacterial sequence reads classified by taxonomic phylum or class (phylum). Figure legends for panels C and D are below the figures. For all figures, OTUs with questionable classifications are labeled with a question mark. The color of the bars next to the host names indicate the phylogenetic group of the insect corresponding to Fig. 1A: red, Cryptocercidae; blue, Kalotermitidae; pink, Stolotermitidae; purple, Archotermopsidae; orange, Reticulitermes; and green, Rhinotermitidae.
FIG 3.

(A) Occurrence network of parabasalid OTUs in the hindguts of Cryptocercus cockroaches and lower termites; (B) PCoA using unweighted UniFrac distances between parabasalid communities from the hindguts of Cryptocercus cockroaches and lower termites; (C and D) the bacterial communities as described for panels A and B. For panels B and D, similar results were obtained from subsampled, normalized OTU abundances. The communities are represented by symbols based on the phylogenetic grouping of the host insect: solid diamonds, Cryptocercidae; solid triangles, Kalotermitidae; open diamond, Stolotermitidae; solid circles, Archotermopsidae; open circles, Reticulitermes; and solid squares, Rhinotermitidae. Communities that did not cluster with communities from related hosts are labeled. I.imm, Incisitermes immigrans; I.sch, Incisitermes schwarzi; K.approx, Kalotermes approximatus; P.adam, Porotermes adamsoni.
Host phylogeny structures parabasalid community composition.
The similarities of the parabasalid communities were compared by multivariate analyses using a phylogenetic metric (UniFrac). The communities clustered based on the phylogenetic groupings of their hosts (Fig. 3B) and not by their geographic location (see Fig. S3 in the supplemental material). This indicates that historical effects, primarily vertical transmission and the diversification of parabasalids within host lineages, and not dispersal or environmental acquisition, were the likely explanations for this pattern.
Distinct clusters were evident for the parabasalid communities from the hindguts of Cryptocercus spp., Reticulitermes spp., Zootermopsis populations, and the rhinotermitids (Fig. 3B). The parabasalid communities from kalotermitid termites also clustered together, but not as tightly. This loose clustering was also observed for the genetic distances of the kalotermitids (see Fig. S1 in the supplemental material), which may reflect their being a more diverse group or that they were much better sampled.
A Procrustes analysis of the PCoA plots of the parabasalid communities with the mitochondrial 16S genetic distances of their hosts further demonstrates the congruence between parabasalid community structure and host evolution and the dominance of historical effects in structuring the community composition (Fig. 4A).
FIG 4.
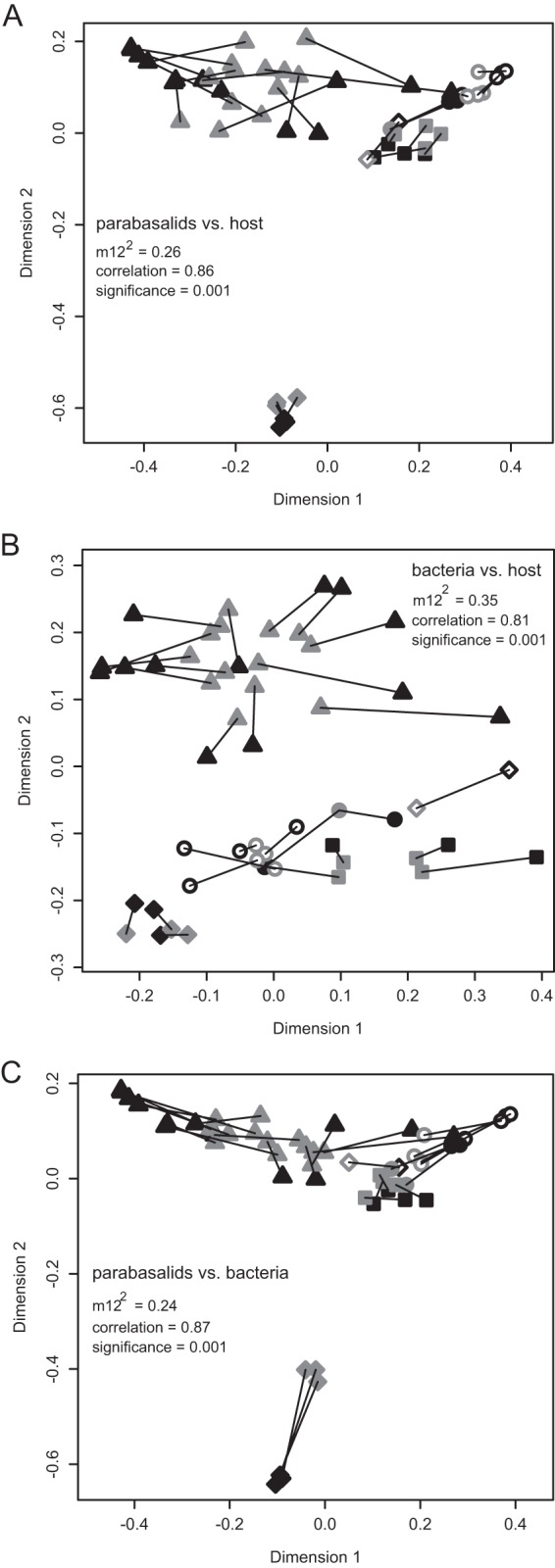
(A) Procrustes analysis comparing the PCoA plots of the parabasalid communities (black symbols) and the genetic distances of their hosts' mitochondrial 16S rRNA sequences (gray symbols); (B) Procrustes analysis comparing the bacterial communities (black symbols) and their hosts (gray symbols); (C) Procrustes analysis comparing the parabasalid (black symbols) and bacterial (gray symbols) communities. Symbols as described for Fig. 3.
A phylogenetic tree of the OTU sequences reveals the overall pattern of diversification and radiation of the parabasalid symbionts within host lineages (Fig. 1B). Of the major parabasalid taxa, the Trichonymphea appear to have radiated early, in the ancestor of Cryptocercus cockroaches and the termites, and also later diversified within specific termite lineages, such as Trichonympha within the archotermopsids and Pseudotrichonympha within the rhinotermitids. The Cristamonadea have almost exclusively diversified within the kalotermitids, and the Spirotrichonymphea likely originated and diversified alongside the rhinotermitids.
Supporting this pattern of diversification, Trichonymphea diversity was highest in Cryptocercus, while kalotermitids harbored the greatest diversity of Cristamonadea but very few Trichonymphea (Fig. 2A). Outside of the kalotermitids, Cristamonadea OTUs were very rare, being found in the stolotermitid Porotermes adamsoni and the rhinotermitid Coptotermes sp. Termites from the Rhinotermitidae hosted most of the diversity of Spirotrichonymphea, but the hindguts of Paraneotermes simplicicornis and Porotermes adamsoni also harbored spirotrichonymphid OTUs despite the fact that these were not found in the other kalotermitid and termopsid termites.
Exceptions to host phylogeny structuring parabasalid communities.
Not all of the parabasalid communities clustered alongside communities from related hosts, and examining symbiont phylogeny provided some potential explanations for these exceptions. For example, the communities from two kalotermitid species, Incisitermes immigrans and I. schwarzi, clustered closer to the communities from Zootermopsis than to other kalotermitids (Fig. 3B). From the phylogeny, these hosts can be seen to harbor Trichonympha taxa related to those found in Porotermes and Zootermopsis (Fig. 1B). The removal of these Trichonympha OTUs from the I. immigrans and I. schwarzi data resulted in these communities clustering with the rest of the kalotermitids (see Fig. S4A in the supplemental material). Likewise, the parabasalid community of Porotermes adamsoni clustered away from its relative Zootermopsis, which was due to a number of Spirotrichonymphea-like taxa in P. adamsoni (see Fig. S4B).
Other exceptions were noted that did not affect the clustering of the communities as a whole. The Paraneotermes simplicicornis community clustered with the rest of the kalotermitids as expected, but unlike the other kalotermitids, P. simplicicornis harbored Spirotrichonymphea taxa. These OTUs were on short branches basal to the rest of the Spirotrichonymphea (Fig. 1B).
Reticulitermes species also harbored parabasalids that were phylogenetically distinct from those in their sister lineage, the rhinotermitids (Fig. 1B). In particular, Reticulitermes species harbored Trichonympha symbionts that were closely related to those found in the Archotermopsidae, whereas the other rhinotermitids all harbored Pseudotrichonympha symbionts (except for a Trichonympha OTU that was found in Coptotermes sp.). Reticulitermes species also did not harbor Spirotrichonymphea taxa that were characteristic in the rhinotermitids, but both host lineages harbored Spirotrichonymphea-like OTUs. These OTUs, however, were highly divergent (2 were classified as “Trichomonadea?”), and the observed phylogenetic relationships may be due to long-branch attraction.
Bacterial diversity.
Similar to recent investigations of lower termites (6, 18–21), the bacterial communities were dominated by a combination of Bacteroidetes (34.7% of the sequences and 15.5% of the OTUs), Endomicrobia (Elusimicrobia) (14.7 and 4.0%), and Spirochaetes (18.4 and 28.4%) (Fig. 2C and D). For the Bacteroidetes and Endomicrobia in particular, a very high proportion of these sequences belonged to relatively few OTUs. In contrast, most of the diversity in the hindguts is represented by OTUs of low sequence abundance, including taxa from the Actinobacteria, Firmicutes (Clostridia and Bacilli), Planctomycetes, Proteobacteria, Synergistes, Tenericutes (Mollicutes), TM7, and Verrucomicrobia (Fig. 2C and D). A small proportion of the sequences (0.5%), representing 19.6% of the OTUs, were sufficiently divergent from known sequences that they could not be assigned to a phylum.
Bacterial community not as strongly influenced by historical effects.
Compared to the parabasalids, similar proportions of bacterial OTUs were unique to a host species (78%) or were shared among unrelated hosts (2%) (Fig. 3C). In contrast to the parabasalids, however, the taxonomic diversity of hindgut bacteria was not obviously consistent with host phylogeny. The rhinotermitids tended to lack Elusimicrobia and Synergistes, but this was not absolute (Fig. 2C). The other two dominant lineages, Spirochaetes and Bacteroidetes, were found in every hindgut sampled. Even upon examining the phylogenetic trees of individual bacterial phyla (which did not necessitate accurate classification), no termite lineages could be singled out as hosting a particular lineage of bacteria.
Despite the lack of obvious bacterial lineages occurring specifically within a host lineage, the bacterial communities did cluster based on the phylogeny of their hosts (Fig. 3D and 4B), indicating that historical effects had some influence on community composition. When the bacterial and parabasalid PCoA plots were compared with one another, the clustering of the bacterial communities also corresponded with that of the parabasalids (Fig. 4C).
Once again, the Porotermes adamsoni bacterial community stood out because it did not cluster with that of Zootermopsis as expected based on host phylogeny (Fig. 3D). Instead, its community clustered closer to that of Kalotermes approximatus, which also did not cluster as expected with the other kalotermitid termites. Although different sets of shared or unique taxa were tested, the diversity of the bacteria in the P. adamsoni and K. approximatus communities was sufficiently distinct that we could not identify specific taxa or OTUs that were causing these communities to cluster away from their relatives or driving them closer to nonrelatives.
The bacterial communities were split into separate data sets based on the assigned phylum or class of the OTUs to examine whether the clustering was driven by specific bacterial lineages. Endomicrobia, Mollicutes, and Spirochaetes communities had the strongest correspondence with host phylogeny, while Alphaproteobacteria, Bacilli, and TM7 communities had the least significant associations (Table 1).
TABLE 1.
Procrustes analysis of hindgut bacterial diversity compared to the host's mitochondrial 16S rRNA sequencea
| Bacterial class/phylum | m122 | Correlation | Significance |
|---|---|---|---|
| Actinobacteria | 0.452 | 0.741 | 0.007 |
| Alphaproteobacteria (Proteobacteria) | 0.489 | 0.715 | 0.033 |
| Bacilli (Firmicutes) | 0.495 | 0.711 | 0.015 |
| Bacteroidetes | 0.369 | 0.795 | 0.001 |
| Betaproteobacteria (Proteobacteria) | 0.447 | 0.744 | 0.001 |
| Clostridia (Firmicutes) | 0.424 | 0.759 | 0.002 |
| Deltaproteobacteria (Proteobacteria) | 0.400 | 0.775 | 0.006 |
| Endomicrobia (Elusimicrobia) | 0.320 | 0.824 | 0.001 |
| Mollicutes (Tenericutes) | 0.276 | 0.851 | 0.001 |
| Spirochaetes | 0.321 | 0.824 | 0.001 |
| Synergistes | 0.393 | 0.779 | 0.001 |
| TM7 | 0.516 | 0.695 | 0.118 |
| Verrucomicrobia | 0.467 | 0.730 | 0.008 |
Bacterial OTUs were split into their respective class (or phylum) before principal coordinate analyses were conducted. Only bacterial classes that had at least one sample with at least 5 OTUs were analyzed. Bolded are the bacterial classes that were the most significant in the Procrustes analysis (m122 values of <0.400, correlation values of >0.775, and significance values of ≤0.001).
Bacterial symbionts of protists.
Using a blastn search of GenBank's nonredundant nucleotide database, we identified OTUs closely related to those of confirmed protist symbionts and found that bacterial symbionts of protists comprised a major fraction of many of the hindgut communities. For the rhinotermitids, each hindgut was completely dominated by a single but different Bacteroidales (Bacteroidetes) OTU (94.4% of the 16S rRNA sequences in Coptotermes testaceus, 89.4% in Coptotermes sp., 81.7% in Heterotermes tenuis, and 95.6% in Prorhinotermes simplex). Three out of these 4 OTUs shared at least 95% similarity with endosymbionts of the protist Pseudotrichonympha known from termites of the same host genera (45, 46). Likewise, 8 out of 10 Endomicrobia OTUs in the hindguts of Zootermopsis angusticollis (comprising 83.0% of the 16S rRNA sequences) were at least 95% similar to endosymbionts of the parabasalid Trichonympha in the related termites Hodotermopsis sjoestedti and Zootermopsis nevadensis (47, 48). The Porotermes adamsoni hindgut was also dominated by Endomicrobia (4 OTUs representing 91.8% of the 16S rRNA sequences), and a single OTU related to Bacteroidetes ectosymbionts of Barbulanympha represented 51.3% of the 16S rRNA sequences from the hindguts of Cryptocercus cockroaches.
In total, 3.6% of the OTUs (25.3% of the sequences) shared at least 95% similarity with known bacterial symbionts of hindgut protists represented in GenBank. Many more OTUs were also likely symbionts of protists but have not been previously studied or were divergent from those in GenBank.
DISCUSSION
Weighing historical effects with horizontal transfer and ecological and stochastic effects on community composition.
The protist symbionts in the hindguts of Cryptocercus cockroaches and lower termites were long suspected to have codiversified with their hosts (15, 17, 49). The common ancestor of Cryptocercus cockroaches and termites is thought to have evolved a dependence on a specialized consortium of gut microbes that allowed it to gain nutrition from the consumption of wood (16, 50). The transmission of this gut consortium was ensured via coprophagy and by the development of proctodeal trophallaxis, which is the direct transfer of hindgut fluid from the rectum of a donor to the mouth of the recipient and dependent on some degree of social behavior in the host. Over evolutionary time, this mode of symbiont transmission further confined the dispersal of hindgut microbes to closely interacting individuals, such as within a family or colony, resulting in the diversification of the symbionts within their hosts from the ancestral gut consortia.
Using deep pyrotag sequencing to describe the parabasalid and bacterial diversity in the hindguts of Cryptocercus punctulatus and 23 species representing the major lineages of lower termites, we have provided molecular evidence that host phylogeny structures microbial community composition. The data suggest that host-symbiont codiversification not only took place but was the prevailing mode of evolution between the origin of termites and the diversification of the parabasalids. However, the composition of symbionts was not entirely due to these historical effects. Exceptions were observed, particularly for the Spirotrichonymphea, the Trichonymphea, and the rare bacteria. Instead of strict vertical transmission, these symbionts were likely transferred between distantly related hosts. Ecological and stochastic effects, such as selection for the most beneficial symbionts given the diet of their host or incomplete transmission of the symbiont community, may also partly explain the composition of communities that deviate from their expected distribution based on host phylogeny.
The occurrence of Spirotrichonymphea-like protists in the hindguts of Porotermes adamsoni and Paraneotermes simplicicornis was not consistent with diversification within their host lineage but possibly due to transfer from rhinotermitids. Alternatively, P. simplicicornis may be harboring slowly evolving basal spirotrichonymphids that were not retained in other kalotermitids. Spirotrichonymphea have also been documented in termites from the Hodotermitidae (4), which are close relatives of Porotermes adamsoni. Interestingly, P. simplicicornis shares nesting and feeding behaviors that are similar to those of rhinotermitids and hodotermitids. Unlike other kalotermitids which feed and nest in drywood, P. simplicicornis is subterranean and forages on dampwood at or near its nest (intermediate nester). Many rhinotermitids are also subterranean intermediate nesters that forage on dampwood, while other rhinotermitids and the hodotermitids are subterranean separate-piece nesters foraging for food that is distinct from the nesting substrate and includes wood, grass, or detritus (51, 52). The presence of Spirotrichonymphea in these hindguts might possibly play a role in these ecological similarities, or the similar habitats may have facilitated the movement of symbionts.
Reticulitermes species harbor protist communities that are phylogenetically distinct from their rhinotermitid relatives, including Trichonympha and divergent Spirotrichonymphea-like taxa but not typical Spirotrichonymphea. Furthermore, Reticulitermes species but not the other rhinotermitids are known to harbor oxymonad protists (53). The distinctness of the parabasalid community in the hindguts of Reticulitermes species, in particular the occurrence of Trichonympha closely related to those in Archotermopsidae, suggests that symbiont transfer has had a significant role in structuring these communities (49).
It is also likely that Trichonympha symbionts were transferred from stolotermitids to Incisitermes. However, because the evolutionary relationship of the Kalotermitidae relative to the termopsids and rhinotermitids is not resolved and the monophyly of the Rhinotermitidae is in dispute (40, 44), it is also possible that these Trichonympha symbionts have been retained in these host species from the ancestral consortium due to an ecological advantage or stochasticity but have gone extinct in their relatives.
The influence of symbiotic associations with protists, environmental acquisition, or dispersal on bacterial community composition.
Compared to the protists, the influence of historical effects on bacterial community composition was not as clearly evident. Host phylogeny corresponded with the diversity of the dominant bacterial phyla, the Bacteroidetes, Endomicrobia, and Spirochaetes, but, except for the Mollicutes and Synergistes, not with the more rare lineages. Most of the bacterial OTUs in the hindguts were unique to a host species, which suggests that these symbionts have become specialized to the Cryptocercus/termite hindgut environment and are unlikely to have been acquired from the external environment. However, without knowing the dispersal, evolutionary, and extinction rates of bacteria, the unique occurrence of a symbiont is not evidence of codiversification or host specificity. Differences in the bacterial communities from lower and higher termites are more likely driven by dietary changes and the environmental acquisition of symbionts (18, 19).
Bacteroidetes, Endomicrobia, Spirochaetes, and Synergistes, the bacterial lineages that showed the strongest correlation with host phylogeny, can be free-living in the hindguts, but many are protist symbionts (see, for example, references 9, 10, 13, 14, and 54–56). Bacteria from the Actinobacteria and Deltaproteobacteria are also known to have symbiotic associations with protists (48, 57). In previous studies (12, 45), and as we have shown here, greater than 80% of the bacteria can be symbionts of hindgut protists. The bacterial symbionts likely support the metabolic activities of their protist hosts by providing nitrogenous compounds through amino acid synthesis or nitrogen fixation or as a sink for hydrogen produced during lignocellulose fermentation (46, 58, 59). They have also been shown to assist in motility (14, 60).
It seems likely that the importance of historical effects on bacterial composition is driven by, or at least bolstered by, symbiotic associations between bacteria and protists, where the protists have codiversified with their insect host. If these associations in the hindguts were established early or prior to the evolution of Cryptocercus cockroaches and lower termites, three levels of coevolution and codiversification have occurred, between the insect, protist, and bacterium. Codiversification has been shown between species of rhinotermitid termites, one of their hindgut protists, Pseudotrichonympha, and their Bacteroidales endosymbionts (61). Codiversification has also been demonstrated between multiple species of the parabasalid Trichonympha and their Elusimicrobia endosymbionts (47) and between devescovinids and their Bacteroidales ectosymbionts (62). However, the three-tiered codiversification structure is broken if ancestral bacteria are replaced by a different symbiont or the protist-bacterium symbioses are more recently established (10, 63).
Host phylogeny does not seem to structure the less abundant bacteria, even those with relatively high diversity, such as the Firmicutes. Rare taxa can represent a major proportion of the diversity in the hindguts but may be part of a transient community of bacteria whose presence in the hindgut fluctuates (18). Indeed, it is not clear whether the composition of the bacterial community is stable and consistent in most or all individual insects in a colony, as is typical for the protists in termite hindguts. For the rare taxa, dispersal and environmental acquisition are probably more significant in structuring the communities. These bacteria may have more generalized or nonessential functions and can be absent or more easily replaced in the ecosystem by other bacteria (20). These bacteria may also be more resilient to survival outside the hindgut and more easily transferred among hosts.
Diversity and endemicity.
Parabasalid diversity in the hindguts of lower termites and Cryptocercus cockroaches is often incompletely described and largely based on morphology. The 18S rRNA pyrotag sequencing provided comprehensive and unbiased descriptions of parabasalid diversity in the hindguts and revealed many undiscovered taxa. The majority of the 18S rRNA OTUs were divergent (<95% similar) from known parabasalid sequences in molecular databases. In addition, the number of parabasalid OTUs often exceeded the number of known described species in the hindguts of lower termites and Cryptocercus cockroaches (see Table S1 in the supplemental material) or the taxonomic composition at the species or genus level differed. But in some rare cases, even if the overall number of OTUs observed exceeded the number of known species for a host, there was also evidence of overclassification within particular subgroups (where the number of OTUs was smaller than the number of described species). Nevertheless, for each host, we have likely uncovered novel and/or cryptic species or obtained molecular data from species known only through morphology. More importantly, these molecular surveys provide a phylogenetic basis for a more accurate understanding of parabasalid diversity, diversification, and community composition.
Although the hindguts and relevant environments have not been sampled exhaustively, it is likely that many of the hindgut microbes are endemic to a host species. Most of the symbiont OTUs were unique to a host species or shared only among closely related hosts (and the cutoff we applied might not capture sister species-level divergence). Especially for the parabasalids, the endemicity is due to diversification within specific host families. For example, most of the existing diversity of Cristamonadea, Spirotrichonymphea, and Trichonymphea results from radiation within the hindguts of specific host lineages, that is, the kalotermitids, rhinotermitids, and Cryptocercus, respectively. This diversification can result in several closely related species existing in a single host species (64).
The ecological niches of most species of symbionts have yet to be identified, so the factors driving this diversification are not known. Microenvironments, such as oxygen or pH gradients, within the different compartments of the hindgut or food particle size may have a role (65, 66). Moreover, the microbial community itself can create these microenvironments (67). Biochemical coordination of the symbiont community with their host to extract nutrition also likely influences symbiont diversification (68, 69). The biology of the symbionts must also complement the biology and behavior of the host. For example, symbionts of Cryptocercus punctulatus can encyst and are retained through molts, whereas in termites, symbionts die during molts and must be reacquired from nestmates (70). The gut structure, nesting style, and foraging behaviors of the termites may also impact the diversification of the symbiont community (71). Given that speciation has occurred within the environment of a host hindgut, these hindgut communities provide an interesting system to examine the mechanisms of sympatric speciation.
Conclusions.
The protist communities in the hindguts of Cryptocercus cockroaches and the lower termites are heavily structured by host phylogeny due to historical effects, but only a portion of the bacterial communities shared this characteristic. Correlation of the bacteria with host phylogeny may have occurred indirectly through their associations with protists, and direct interactions with their hosts were not a significant force. Even in these clear cases of vertical transmission, where historical effects were expected to dominate microbial community structure, a significant proportion of the bacterial community remained strongly influenced by dispersal and stochasticity compared to the protists.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant (227301) from the Natural Sciences and Engineering Research Council of Canada (NSERC). V. Tai was supported as a Global Scholar with the Canadian Institute for Advanced Research (CIFAR) and through a postdoctoral fellowship from NSERC. P. J. Keeling and S. J. Perlman are CIFAR fellows.
We thank Barbara Stay (University of Iowa) for providing live termite samples for this study. We also thank Phil Hugenholtz for an enjoyable and productive day of field work and for sharing data.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02945-14.
REFERENCES
- 1.Inward D, Beccaloni G, Eggleton P. 2007. Death of an order: a comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biol Lett 3:331–335. doi: 10.1098/rsbl.2007.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohkuma M. 2008. Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends Microbiol 16:345–352. doi: 10.1016/j.tim.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Brune A, Ohkuma M. 2011. Role of the termite gut microbiota in symbiotic digestion, p 439–475. In Bignell D, Roisin Y, Lo N (ed), Biology of termites: a modern synthesis. Springer, New York, NY. [Google Scholar]
- 4.Yamin MA. 1979. Flagellates of the orders Trichomonadida Kirby, Oxymonadida Grassé, and Hypermastigida Grassi and Foà reported from lower termites (Isoptera families Mastotermitidae, Kalotermitidae, Hodotermitidae, Termopsidae, Rhinotermitidae, and Serritermitidae) and from the wood-feeding roach Cryptocercus (Dictyoptera, Cryptocercidae). Sociobiology 4:3–119. [Google Scholar]
- 5.Hongoh Y, Ohkuma M, Kudo T. 2003. Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiol Ecol 44:231–242. doi: 10.1016/S0168-6496(03)00026-6. [DOI] [PubMed] [Google Scholar]
- 6.Boucias DG, Cai Y, Sun Y, Lietze V-U, Sen R, Raychoudhury R, Scharf ME. 2013. The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol Ecol 22:1836–1853. doi: 10.1111/mec.12230. [DOI] [PubMed] [Google Scholar]
- 7.Husseneder C. 2010. Symbiosis in subterranean termites: a review of insights from molecular studies. Environ Entomol 39:378–388. doi: 10.1603/EN09006. [DOI] [PubMed] [Google Scholar]
- 8.Brune A, Stingl U. 2006. Prokaryotic symbionts of termite gut flagellates: phylogenetic and metabolic implications of a tripartite symbiosis. Prog Mol Subcell Biol 41:39–60. doi: 10.1007/3-540-28221-1_3. [DOI] [PubMed] [Google Scholar]
- 9.Noda S, Inoue T, Hongoh Y, Kawai M, Nalepa CA, Vongkaluang C, Kudo T, Ohkuma M. 2006. Identification and characterization of ectosymbionts of distinct lineages in Bacteroidales attached to flagellated protists in the gut of termites and a wood-feeding cockroach. Environ Microbiol 8:11–20. doi: 10.1111/j.1462-2920.2005.00860.x. [DOI] [PubMed] [Google Scholar]
- 10.Noda S, Ohkuma M, Yamada A, Hongoh Y, Kudo T. 2003. Phylogenetic position and in situ identification of ectosymbiotic spirochetes on protists in the termite gut. Appl Environ Microbiol 69:625–633. doi: 10.1128/AEM.69.1.625-633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leander BS, Keeling PJ. 2004. Symbiotic innovation in the oxymonad Streblomastix strix. J Eukaryot Microbiol 51:291–300. doi: 10.1111/j.1550-7408.2004.tb00569.x. [DOI] [PubMed] [Google Scholar]
- 12.Berchtold M, Chatzinotas A, Schonhuber W, Brune A, Amann R, Hahn D, Konig H. 1999. Differential enumeration and in situ localization of microorganisms in the hindgut of the lower termite Mastotermes darwiniensis by hybridization with rRNA-targeted probes. Arch Microbiol 172:407–416. doi: 10.1007/s002030050778. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda-Ohtsubo W, Desai M, Stingl U, Brune A. 2007. Phylogenetic diversity of “Endomicrobia” and their specific affiliation with termite gut flagellates. Microbiology 153:3458–3465. doi: 10.1099/mic.0.2007/009217-0. [DOI] [PubMed] [Google Scholar]
- 14.Hongoh Y, Sato T, Dolan MF, Noda S, Ui S, Kudo T, Ohkuma M. 2007. The motility symbiont of the termite gut flagellate Caduceia versatilis is a member of the “Synergistes” group. Appl Environ Microbiol 73:6270–6276. doi: 10.1128/AEM.00750-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cleveland LR, Hall SR, Sanders EP, Collier J. 1934. The wood-feeding roach Cryptocercus, its protozoa, and the symbiosis between protozoa and roach. Memoirs of the American Academy of Arts and Sciences, New Series 17:185–342. [Google Scholar]
- 16.Nalepa C, Bignell D, Bandi C. 2001. Detritivory, coprophagy, and the evolution of digestive mutualisms in Dictyoptera. Insectes Soc 48:194–201. doi: 10.1007/PL00001767. [DOI] [Google Scholar]
- 17.Kirby HJ. 1937. Host-parasite relations in the distribution of protozoa in termites. Univ Calif Publ Zool 41:189–212. [Google Scholar]
- 18.Colman DR, Toolson EC, Takacs-Vesbach CD. 2012. Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol 21:5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- 19.Dietrich C, Köhler T, Brune A. 2014. The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Appl Environ Microbiol 80:2261–2269. doi: 10.1128/AEM.04206-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones RT, Sanchez LG, Fierer N. 2013. A cross-taxon analysis of insect-associated bacterial diversity. PLoS One 8:e61218. doi: 10.1371/journal.pone.0061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosengaus RB, Zecher CN, Schultheis KF, Brucker RM, Bordenstein SR. 2011. Disruption of the termite gut microbiota and its prolonged consequences for fitness. Appl Environ Microbiol 77:4303–4312. doi: 10.1128/AEM.01886-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everaerts C, Maekawa K, Farine JP, Shimada K, Luykx P, Brossut R, Nalepa CA. 2008. The Cryptocercus punctulatus species complex (Dictyoptera: Cryptocercidae) in the eastern United States: comparison of cuticular hydrocarbons, chromosome number, and DNA sequences. Mol Phylogenet Evol 47:950–959. doi: 10.1016/j.ympev.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Trager W. 1934. The cultivation of a cellulose-digesting flagellate, Trichomonas termopsidis, and of certain other termite protozoa. Biol Bull 66:182–190. doi: 10.2307/1537331. [DOI] [Google Scholar]
- 24.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 25.Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 26.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 27.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. 2008. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishak HD, Plowes R, Sen R, Kellner K, Meyer E, Estrada DA, Dowd SE, Mueller UG. 2011. Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb Ecol 61:821–831. doi: 10.1007/s00248-010-9793-4. [DOI] [PubMed] [Google Scholar]
- 29.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. 2011. Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heiss AA, Keeling PJ. 2006. The phylogenetic position of the oxymonad Saccinobaculus based on SSU rRNA. Protist 157:335–344. doi: 10.1016/j.protis.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Keeling PJ, Leander BS. 2003. Characterisation of a non-canonical genetic code in the oxymonad Streblomastix strix. J Mol Biol 326:1337–1349. doi: 10.1016/S0022-2836(03)00057-3. [DOI] [PubMed] [Google Scholar]
- 35.Hampl V, Horner DS, Dyal P, Kulda J, Flegr J, Foster PG, Embley TM. 2005. Inference of the phylogenetic position of oxymonads based on nine genes: support for metamonada and excavata. Mol Biol Evol 22:2508–2518. doi: 10.1093/molbev/msi245. [DOI] [PubMed] [Google Scholar]
- 36.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens M, Wagner H. 2013. vegan: community ecology package. R package version 2.0-9 http://CRAN.R-project.org/package=vegan.
- 38.Pirrung M, Kennedy R, Caporaso JG, Stombaugh J, Wendel D, Knight R. 2011. TopiaryExplorer: visualizing large phylogenetic trees with environmental metadata. Bioinformatics 27:3067–3069. doi: 10.1093/bioinformatics/btr517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cameron SL, Lo N, Bourguignon T, Svenson GJ, Evans TA. 2012. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol Phylogenet Evol 65:163–173. doi: 10.1016/j.ympev.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 41.Inward DJG, Vogler AP, Eggleton P. 2007. A comprehensive phylogenetic analysis of termites (Isoptera) illuminates key aspects of their evolutionary biology. Mol Phylogenet Evol 44:953–967. doi: 10.1016/j.ympev.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Legendre F, Whiting MF, Bordereau C, Cancello EM, Evans TA, Grandcolas P. 2008. The phylogeny of termites (Dictyoptera: Isoptera) based on mitochondrial and nuclear markers: implications for the evolution of the worker and pseudergate castes, and foraging behaviors. Mol Phylogenet Evol 48:615–627. doi: 10.1016/j.ympev.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 43.Ware JL, Grimaldi DA, Engel MS. 2010. The effects of fossil placement and calibration on divergence times and rates: an example from the termites (Insecta: Isoptera). Arthropod Struct Dev 39:204–219. doi: 10.1016/j.asd.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Lo N, Eggleton P. 2011. Termite phylogenetics and co-cladogenesis with symbionts, p 27–50. In Bignell D, Roisin Y, Lo N (ed), Biology of termites: a modern synthesis. Springer, New York, NY. [Google Scholar]
- 45.Noda S, Iida T, Kitade O, Nakajima H, Kudo T, Ohkuma M. 2005. Endosymbiotic Bacteroidales bacteria of the flagellated protist Pseudotrichonympha grassii in the gut of the termite Coptotermes formosanus. Appl Environ Microbiol 71:8811–8817. doi: 10.1128/AEM.71.12.8811-8817.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hongoh Y, Sharma VK, Prakash T, Noda S, Toh H, Taylor TD, Kudo T, Sakaki Y, Toyoda A, Hattori M, Ohkuma M. 2008. Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322:1108–1109. doi: 10.1126/science.1165578. [DOI] [PubMed] [Google Scholar]
- 47.Ikeda-Ohtsubo W, Brune A. 2009. Cospeciation of termite gut flagellates and their bacterial endosymbionts: Trichonympha species and “Candidatus Endomicrobium trichonymphae.” Mol Ecol 18:332–342. doi: 10.1111/j.1365-294X.2008.04029.x. [DOI] [PubMed] [Google Scholar]
- 48.Strassert JFH, Köhler T, Wienemann THG, Ikeda-Ohtsubo W, Faivre N, Franckenberg S, Plarre R, Radek R, Brune A. 2012. “Candidatus Ancillula trichonymphae,” a novel lineage of endosymbiotic Actinobacteria in termite gut flagellates of the genus Trichonympha. Environ Microbiol 14:3259–3270. doi: 10.1111/1462-2920.12012. [DOI] [PubMed] [Google Scholar]
- 49.Kitade O. 2004. Comparison of symbiotic flagellate faunae between termites and a wood-feeding cockroach of the genus Cryptocercus. Microbes Environ 19:215–220. doi: 10.1264/jsme2.19.215. [DOI] [Google Scholar]
- 50.Ohkuma M, Noda S, Hongoh Y, Nalepa CA, Inoue T. 2009. Inheritance and diversification of symbiotic trichonymphid flagellates from a common ancestor of termites and the cockroach Cryptocercus. Proc Biol Sci 276:239–245. doi: 10.1098/rspb.2008.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weesner FM. 1960. Evolution and biology of the termites. Annu Rev Entomol 5:153–170. doi: 10.1146/annurev.en.05.010160.001101. [DOI] [Google Scholar]
- 52.Eggleton P, Tayasu I. 2001. Feeding groups, lifetypes and the global ecology of termites. Ecol Res 16:941–960. doi: 10.1046/j.1440-1703.2001.00444.x. [DOI] [Google Scholar]
- 53.Kitade O, Matsumoto T. 1998. Characteristics of the symbiotic flagellate composition within the termite family Rhinotermitidae (Isoptera). Symbiosis 25:271–278. [Google Scholar]
- 54.Ohkuma M, Sato T, Noda S, Ui S, Kudo T, Hongoh Y. 2007. The candidate phylum “Termite Group 1” of bacteria: phylogenetic diversity, distribution, and endosymbiont members of various gut flagellated protists. FEMS Microbiol Ecol 60:467–476. doi: 10.1111/j.1574-6941.2007.00311.x. [DOI] [PubMed] [Google Scholar]
- 55.Strassert JFH, Desai MS, Radek R, Brune A. 2010. Identification and localization of the multiple bacterial symbionts of the termite gut flagellate Joenia annectens. Microbiology 156:2068–2079. doi: 10.1099/mic.0.037267-0. [DOI] [PubMed] [Google Scholar]
- 56.Carpenter KJ, Horak A, Chow L, Keeling PJ. 2011. Symbiosis, morphology, and phylogeny of Hoplonymphidae (Parabasalia) of the wood-feeding roach Cryptocercus punctulatus. J Eukaryot Microbiol 58:426–436. doi: 10.1111/j.1550-7408.2011.00564.x. [DOI] [PubMed] [Google Scholar]
- 57.Sato T, Hongoh Y, Noda S, Hattori S, Ui S, Ohkuma M. 2009. Candidatus Desulfovibrio trichonymphae, a novel intracellular symbiont of the flagellate Trichonympha agilis in termite gut. Environ Microbiol 11:1007–1015. doi: 10.1111/j.1462-2920.2008.01827.x. [DOI] [PubMed] [Google Scholar]
- 58.Hongoh Y, Sharma VK, Prakash T, Noda S, Taylor TD, Kudo T, Sakaki Y, Toyoda A, Hattori M, Ohkuma M. 2008. Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Natl Acad Sci U S A 105:5555–5560. doi: 10.1073/pnas.0801389105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desai MS, Brune A. 2011. Bacteroidales ectosymbionts of gut flagellates shape the nitrogen-fixing community in dry-wood termites. ISME J 6:1302–1313. doi: 10.1038/ismej.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wenzel M, Radek R, Brugerolle G, König H. 2003. Identification of the ectosymbiotic bacteria of Mixotricha paradoxa involved in movement symbiosis. Eur J Protistol 39:11–23. doi: 10.1078/0932-4739-00893. [DOI] [Google Scholar]
- 61.Noda S, Kitade O, Inoue T, Kawai M, Kanuka M, Hiroshima K, Hongoh Y, Constantino R, Uys V, Zhong J, Kudo T, Ohkuma M. 2007. Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol Ecol 16:1257–1266. doi: 10.1111/j.1365-294X.2006.03219.x. [DOI] [PubMed] [Google Scholar]
- 62.Desai MS, Strassert JFH, Meuser K, Hertel H, Ikeda-Ohtsubo W, Radek R, Brune A. 2010. Strict cospeciation of devescovinid flagellates and Bacteroidales ectosymbionts in the gut of dry-wood termites (Kalotermitidae). Environ Microbiol 12:2120–2132. doi: 10.1111/j.1462-2920.2009.02080.x. [DOI] [PubMed] [Google Scholar]
- 63.Noda S, Hongoh Y, Sato T, Ohkuma M. 2009. Complex coevolutionary history of symbiotic Bacteroidales bacteria of various protists in the gut of termites. BMC Evol Biol 9:158. doi: 10.1186/1471-2148-9-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tai V, James ER, Perlman SJ, Keeling PJ. 2013. Single-cell DNA barcoding using sequences from the small subunit rRNA and internal transcribed spacer region identifies new species of Trichonympha and Trichomitopsis from the hindgut of the termite Zootermopsis angusticollis. PLoS One 8:e58728. doi: 10.1371/journal.pone.0058728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brune A, Friedrich M. 2000. Microecology of the termite gut: structure and function on a microscale. Curr Opin Microbiol 3:263–269. doi: 10.1016/S1369-5274(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 66.Ebert A, Brune A. 1997. Hydrogen concentration profiles at the oxic-anoxic interface: a microsensor study of the hindgut of the wood-feeding lower termite Reticulitermes flavipes (Kollar). Appl Environ Microbiol 63:4039–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veivers PC, O'Brien RW, Slaytor M. 1982. Role of bacteria in maintaining the redox potential in the hindgut of termites and preventing entry of foreign bacteria. J Insect Physiol 28:947–951. doi: 10.1016/0022-1910(82)90111-1. [DOI] [Google Scholar]
- 68.Scharf ME, Karl ZJ, Sethi A, Boucias DG. 2011. Multiple levels of synergistic collaboration in termite lignocellulose digestion. PLoS One 6:e21709. doi: 10.1371/journal.pone.0021709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raychoudhury R, Sen R, Cai Y, Sun Y, Lietze VU, Boucias DG, Scharf ME. 2013. Comparative metatranscriptomic signatures of wood and paper feeding in the gut of the termite Reticulitermes flavipes (Isoptera: Rhinotermitidae). Insect Mol Biol 22:155–171. doi: 10.1111/imb.12011. [DOI] [PubMed] [Google Scholar]
- 70.Nutting WL. 1956. Reciprocal protozoan transfaunations between the roach, Cryptocercus, and the termite, Zootermopsis. Biol Bull 110:83–90. doi: 10.2307/1538895. [DOI] [Google Scholar]
- 71.Eggleton P. 2006. The termite gut habitat: its evolution and co-evolution, p 373–404. In Konig H, Varma A (ed), Soil biology—intestinal microorganisms of soil invertebrates. Springer-Verlag, Berlin, Germany. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.