

Abstract
Hepatitis B virus (HBV) is a deadly pathogen that has killed countless people worldwide. Saccharomyces cerevisiae-derived HBV vaccines based upon hepatitis B surface antigen (HBsAg) is highly effective. However, the emergence of vaccine escape mutants due to mutations on the HBsAg and polymerase genes has produced a continuous need for the development of new HBV vaccines. In this study, the “a” determinant within HBsAg was displayed on the recombinant capsid protein of Macrobrachium rosenbergii nodavirus (MrNV), which can be purified easily in a single step through immobilized metal affinity chromatography (IMAC). The purified protein self-assembled into virus-like particles (VLPs) when observed under a transmission electron microscope (TEM). Immunization of BALB/c mice with this chimeric protein induced specific antibodies against the “a” determinant. In addition, it induced significantly more natural killer and cytotoxic T cells, as well as an increase in interferon gamma (IFN-γ) secretion, which are vital for virus clearance. Collectively, these findings demonstrated that the MrNV capsid protein is a potential carrier for the HBV “a” determinant, which can be further extended to display other foreign epitopes. This paper is the first to report the application of MrNV VLPs as a novel platform to display foreign epitopes.
INTRODUCTION
Hepatitis B virus (HBV) is a partially double-stranded DNA virus that belongs to the family Hepadnaviridae. It is known to be the major cause of liver-associated diseases, including liver cirrhosis and hepatocellular carcinoma. Approximately 2 billion people worldwide have been infected by HBV, among which about 350 million chronically infected people serve as a reservoir for the virus (1, 2). To date, there is no effective treatment for HBV infection despite intensive studies on antiviral drugs. Thus, preventive measures, including immunization with HBV vaccine, remain necessary.
HBV surface antigens (HBsAg) purified from the sera of infected patients have been used for immunization against HBV infection since 1981. The full-length HBsAg gene has three different start codons and a common stop codon and encodes HBsAg of different lengths (3). The longest is the large HBsAg (L-HBsAg), followed by the middle HBsAg (M-HBsAg), and the smallest is the small HBsAg (S-HBsAg). L-HBsAg is composed of 389 or 400 amino acids (aa), depending on the virus genotypes. It contains the pre-S1 region (108 or 119 aa), followed by the pre-S2 region (55 aa) and S-HBsAg (226 aa). The N-terminal end of M-HBsAg harbors the pre-S2 region at its S-HBsAg. The S-HBsAg contains 226 aa in the absence of the pre-S1 and pre-S2 regions.
The HBsAg in serum self-assemble into noninfectious 22-nm spherical or filamentous particles, along with lipid components (4), which are capable of inducing neutralizing antibodies against HBV infection. Although highly effective, the use of plasma-derived HBsAg for vaccination is limited by the production cost, as well as the possibility of contamination by other infectious pathogens present in human sera. These problems have been solved by advances in recombinant DNA technology, which culminated in the production of recombinant HBV vaccine in Saccharomyces cerevisiae based upon HBsAg (5, 6). A highly immunodominant region known as the “a” determinant is located in aa 121 to 149 of the S-HBsAg (7). The region was demonstrated to induce the production of neutralizing antibodies against HBV infection in humans (7).
HBV vaccines containing the “a” determinant often induce protective antibodies with high cross-reactivity against HBV of various subtypes (8, 9). Despite the effectiveness of these yeast-derived vaccines, HBV variants with mutations across the “a” determinant have been shown to escape the immune responses generated by the yeast-derived HBsAg (10–13). In addition, vaccine escape mutants with nucleotide changes in their polymerase and HBsAg genes have been reported widely due to continuous treatment of patients with chronic hepatitis B with nucleoside analogs (14). These life-threatening mutants and the genetic diversity of the HBV genome strongly justify a continuing need for the development of new HBV vaccines (15). Therefore, the “a” determinant of HBV was selected as a foreign epitope to be displayed on Macrobrachium rosenbergii nodavirus (MrNV) virus-like particles (VLPs) in this study.
MrNV was isolated from M. rosenbergii (16), which is commonly known as the freshwater prawn (17). The recombinant MrNV capsid protein expressed in Escherichia coli self-assembled into VLPs in the absence of other viral proteins (18, 19). VLPs have been widely used for various purposes, including drug delivery (20, 21), gene therapy (22), vaccine development (23–27), and display of epitopes (28). Therefore, it is hypothesized that the VLPs of MrNV capsid protein can be used to display foreign epitopes. In the present study, the VLPs of MrNV capsid protein were established as a novel display system for the “a” determinant of HBV. MrNv VLPs harboring the “a” determinant, namely, NvC-aD, were then produced in bacteria, and their immune responses in mice were studied. A commercial HBV vaccine, Engerix B (GlaxoSmithKline, Middlesex, United Kingdom), containing S-HBsAg produced in yeast (S. cerevisiae), was included in this study as a positive control.
MATERIALS AND METHODS
Fusion of the “a” determinant of HBV to the C-terminal end of MrNV capsid protein.
The coding sequence of the “a” determinant was amplified from pMDHBs3 (29) by PCR with DreamTaq DNA polymerase (Thermo Scientific, Waltham, MA) in the presence of 25 μM forward primer (SAD-NCF; 5′-GT CCT GAA TTC CCA GGA TCA TCA AC-3′ [the EcoRI restriction site is underlined]) and reverse primer (SAD-NCR; 5′-CC ATA AAG CTT CTC CGA AAG CCC AG-3′; the HindIII restriction site is underlined). The initial denaturation step was carried out at 94°C for 10 min, followed by 25 cycles of 94°C (45 s), 52°C (45 s), and 72°C (1 min) and an extension of 72°C for 10 min. The PCR product was purified with phenol-chloroform-isoamyl alcohol (25:24:1) prior to EcoRI and HindIII digestion.
The plasmid pTrcHis-TARNA2 (18), encoding the recombinant MrNV capsid protein (NvC), was first digested with HindIII, followed by EcoRI. The digested plasmid was analyzed by 1% agarose gel electrophoresis in TAE (40 mM Tris-acetate, 1 mM EDTA, pH 8.0) buffer. A band of 5.5 kb was excised from the gel and purified with the Qiaquick gel extraction kit (Qiagen, Hilden, Germany). The digested PCR product containing the “a” determinant coding sequence was then ligated with the linearized pTrcHis-TARNA2 via EcoRI and HindIII sites by using T4 DNA ligase (Promega, Madison, WI, USA) at 25°C for 3 h.
The ligation mixture was introduced into E. coli TOP10 competent cells via the heat shock method, and positive transformants were selected on Luria-Bertani (LB) agar plates containing ampicillin (100 μg/ml). The positive transformants were screened by PCR using PNCx-Forward (5′-CAG GCC AAC AAT ATT GGT GAA GC-3′) as the forward primer and SAD-NCR (see above) as the reverse primer. Recombinant plasmids were extracted using the alkaline lysis method (30) from the positive transformants, and the presence of the insert was verified by restriction enzyme digestion and DNA sequencing prior to protein expression.
Protein expression and purification.
A single colony of the transformant carrying the recombinant plasmid (namely, pNvC-aD) harboring the coding sequence of the fusion protein was inoculated into LB broth (50 ml) containing ampicillin (100 μg/ml) and incubated at 37°C at 220 rpm overnight. The overnight culture (10 ml) was then transferred into fresh LB broth (500 ml) and incubated at 37°C at 220 rpm for 2 h until an A600 of 0.6 to 0.8 was reached. IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM) was added to the culture as an inducer for the expression of the fusion protein, NvC-aD. Incubation of the culture at 25°C and 220 rpm was continued for another 4 h.
The induced cells were pelleted by centrifugation at 8,000 × g for 5 min. The pellet was then resuspended in lysis buffer (25 mM HEPES, 500 mM NaCl, pH 7.4; 15 ml), followed by the addition of MgCl2 (4 mM) and freshly prepared lysozyme (0.2 mg/ml). The mixture was incubated at room temperature (RT) for 2 h prior to sonication. Sonication was done at 30 MHz in an ice bath (30 s for 12 cycles, with 30-s intervals of cooling) (31). After sonication, the cell lysate was centrifuged at 12,000 × g for 10 min. The supernatant was filtered through a syringe filter (0.45 μm; Millipore, Billerica, MA, USA).
The filtered sample was loaded onto a His-Trap HP 1-ml column (GE Healthcare, Buckinghamshire, United Kingdom). A total of 10 column volumes (CV) of washing buffer A (25 mM HEPES, 500 mM NaCl, 50 mM imidazole, pH 7.4) and washing buffer B (25 mM HEPES, 500 mM NaCl, 200 mM imidazole, pH 7.4) flowed through the column. The bound proteins were then eluted from the column by 3 CV of elution buffer (25 mM HEPES, 500 mM NaCl, 500 mM imidazole, pH 7.4). The purified sample was then analyzed by SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting.
Protein samples were mixed with loading buffer (100 mM Tris-HCl, pH 6.8, 20% [vol/vol] glycerol, 4% [wt/vol] SDS, 0.2% [wt/vol] bromophenol blue, 200 mM mercaptoethanol) and immersed in a boiling-water bath for 15 min before being loaded onto 12% SDS-polyacrylamide gels. The gels were then electrophoresed at 16 mA for 1 h. Proteins on the polyacrylamide gels were electrotransferred onto nitrocellulose membranes and blocked with 10% skim milk (Anlene, Auckland, New Zealand) for 1 hour. The membranes were incubated in anti-His monoclonal antibody (1:5,000 dilution; Invitrogen, San Diego, CA, USA) or anti-HBsAg antibody (1:2,500 dilution; MP Biomedicals, California, USA) separately in TBS buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) for 2 h. The membranes were then rinsed 3 times with TBST buffer (50 mM Tris-HCl, 150 mM NaCl, 0.01% [vol/vol] Tween 20, pH 7.4). TBS buffer containing anti-mouse (1:5,000 dilution; KPL, Gaithersburg, MD) or anti-guinea pig (1:5,000 dilution; KPL) antibody conjugated to alkaline phosphatase (AP) was added as the secondary antibody and incubated for 1 h. The membranes were washed 3 times with TBST buffer. Finally, BCIP-NBT (5-bromo-4-chloro-3′-indolylphosphate p-toluidine salt–Nitro Blue Tetrazolium chloride) was added as the substrate for color development.
Enzyme-linked immunosorbent assay (ELISA).
Purified NvC-aD and NvC (negative control) were diluted to 20, 10, 5, and 2 μg/ml with HEPES buffer (25 mM HEPES, 500 mM NaCl, pH 7.4). The samples (100 μl) were loaded in triplicate into each well of a 96-well microtiter plate and incubated overnight at 4°C. The wells were then rinsed 3 times with HEPES buffer containing Tween 20 (0.05% [vol/vol]). Blocking was done by adding milk diluents (200 μl; KPL, Gaithersburg, MD) to each well and incubated for 2 h. The wells were then rinsed 3 times with HEPES-Tween buffer and incubated for another 2 h upon the addition of anti-HBsAg (100 μl; 1:2,500 dilution). After that, the wells were rinsed 3 times with HEPES-Tween buffer, anti-guinea pig-AP conjugate (100 μl; 1:5,000 dilution) was added, and the plate was incubated for another 2 h. Then, the wells were washed 4 times with HEPES-Tween buffer with 5 min of incubation between the washes. p-Nitrophenyl phosphate (p-NPP) (100 μl) was added to each well as a substrate. The microtiter plate was incubated for 45 min, and the A405 was determined.
Transmission electron microscopy.
Purified samples (15 ng/μl; 15 μl) were dropped onto a parafilm, and a carbon-coated copper grid was placed on top of the sample droplet for 5 min. Excess solution on the grid was removed by touching blotting paper to the side of the grid, and the grid was then placed on top of a drop of freshly prepared uranyl acetate solution (1% [wt/vol]) for another 5 min. The excess solution was removed from the grid.
For immunogold staining, purified NvC and NvCaD (15 ng/μl; 15 μl) were mounted onto carbon-coated grids as described above. The grids were blocked with phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 1% [wt/vol])–bovine serum albumin (BSA) (15 μl; pH 7.4) for 5 min after the removal of excess sample with filter paper. The grids were then incubated with anti-HBsAg (15 μl; 1:25 dilution in PBS-BSA) for 1 h. After incubation with anti-HBsAg, the grids were rinsed thrice with PBS-BSA prior to incubation with protein A conjugated to 5-nm gold particles (MP Biomedicals, California, USA; 15 μl; 1:25 dilution in PBS-BSA) for another 1 h. The grids were then rinsed 4 times with PBS-BSA, followed by negative staining with uranyl acetate (1% [wt/vol]) for 5 min. The samples were dried and viewed under a transmission electron microscope (TEM) (Hitachi H7700, Japan).
Immunization of BALB/c mice.
All animal experiments were approved by the Institutional Animal Care and Use Committee, Universiti Putra Malaysia (AUP no. R058/2013). Four- to 5-week-old female BALB/c mice were acclimatized for 2 weeks. After the 2nd week, the mice (n = 7) were injected with 100 μl of samples subcutaneously. The samples included purified NvC and NvC-aD (0.34 mg/ml) emulsified in Freund's adjuvant (complete Freund's adjuvant, F 5881; incomplete Freund's adjuvant, F 5506; Sigma-Aldrich, St. Louis, MO, USA), HEPES buffer, and Engerix B. In addition, 2 boosters were given at 3-week intervals. Serum samples were collected by submandibular bleeding before every injection and 1 week after the second booster.
Immunogenicity of the recombinant proteins.
Serum samples collected during the 2nd week, 5th week, 8th week, and 9th week were analyzed by ELISA. Ninety-six-well microtiter plates were coated with the synthetic “a” determinant peptide (0.5 μg; GL Biochem, Shanghai, China) or plasma-purified HBsAg (0.2 μg; MP Biomedicals, California, USA). Upon blocking with 200 μl of milk diluents, 100 μl of serum samples diluted in TBS (1:500 dilution) was added and incubated for 1 h at RT. After washing with TBST, anti-mouse antibody conjugated to alkaline phosphatase (100 μl; 1:5,000 dilution) was added, and the plates were incubated for 1 h. The plates were then washed again with TBST, and color development was performed by adding p-NPP. The plates were incubated at RT for 20 min, and the A405 was determined.
Immunophenotyping of splenocytes.
Spleens of mice (n = 2) from each group were harvested at week 9 (1 week after the 2nd booster injection). PBS (pH 7.4) was added to the spleens, and they were passed through a cell strainer (70 μm). Single-cell suspensions were centrifuged at 300 × g for 10 min. The cell pellet was then resuspended in erythrocyte lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4) and incubated at 4°C for 10 min. The splenocytes were washed and resuspended in PBS. Two sets of antibodies were added to the splenocytes: anti-CD3-fluorescein isothiocyanate (FITC), CD4-phycoerythrin (PE), and CD8-allophycocyanin (APC) and NK1.1-APC (Abcam; Cambridge, United Kingdom). The mixtures were incubated for 2 h on ice in the dark. Fixation was done with 1% (wt/vol) paraformaldehyde in PBS for 24 h at 4°C. Samples were then analyzed with a flow cytometer (FACSCalibur; BD Biosciences, California, USA).
Cytokine quantification through sandwich ELISA.
The concentrations of interferon gamma (IFN-γ) and interleukin-10 (IL-10) present in the peripheral blood were analyzed with the ELISA Max (BioLegend, California, USA) according to the manufacturer's protocol. A 96-well microtiter plate was coated with cytokine capture antibodies (100 μl) overnight at 4°C. The wells were rinsed 4 times with PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 0.05% [vol/vol] Tween 20, pH 7.4), followed by blocking with PBS-BSA (200 μl) for 2 h at RT. The plate was then rinsed again with PBST. Standards (IFN-γ and IL-10) and serum samples (100 μl) were added to the wells and incubated for another 2 h at RT. The wells were rinsed with PBST. Cytokine detection antibodies (100 μl) were added and incubated for 2 h at RT. The wells were rinsed with PBST before the addition of avidin conjugated with horseradish peroxidase (HRP) (100 μl). The plate was then incubated for 30 min at RT, followed by 5 washes with PBST. 3,3′,5,5′-Tetramethylbenzidine (TMB) (100 μl) was added as a substrate and incubated for 5 min. Sulfuric acid (100 μl; 1 M) was added as a stop solution. The A450 was determined immediately.
Statistical analysis.
Variations among different immunization groups in terms of antibody responses, splenocyte immunophenotyping, and cytokine concentration in peripheral blood were analyzed using one-way analysis of variance (ANOVA). The significant differences between different groups were discriminated using Duncan's multiple-range tests, where P values of less than 0.05 were considered significant, P values of less than 0.001 were very significant, and P values of less than 0.0001 were extremely significant. All data were analyzed using IBM SPSS statistics software for Windows version 19.
RESULTS
Fusion of the “a” determinant of HBV to the C-terminal end of the MrNV capsid protein.
The coding sequence of the HBV “a” determinant (148 bases), which encodes 49 amino acids, was inserted into pTrcHis-TARNA2 at the EcoRI and HindIII restriction sites. The amino acid sequence of the recombinant MrNV capsid protein fused with the “a” determinant of HBV (namely, NvC-aD) is depicted in Fig. 1.
FIG 1.

Primary structure of the MrNV fusion proteins. The “a” determinant was inserted at the C-terminal end of the MrNV capsid protein and the N-terminal ends of the myc epitope and His tag. The positions of amino acids are shown below the diagram. The amino acid sequences for each part of the fusion proteins are shown. NvC, MrNV capsid protein with a myc epitope and His tag; NvC-aD, fusion protein of MrNV capsid protein and “a” determinant with a myc epitope and His tag.
Purification of the fusion protein.
Cell lysate was purified by immobilized metal affinity chromatography (IMAC). Washing buffer containing 50 mM imidazole removed most of the host proteins from the column (Fig. 2A, lane 3). The remaining host proteins could be removed fully only by washing with 200 mM imidazole (Fig. 2A, lane 4). The NvC-aD eluted with 500 mM imidazole was approximately 95% pure (Fig. 2A, lane 5).
FIG 2.
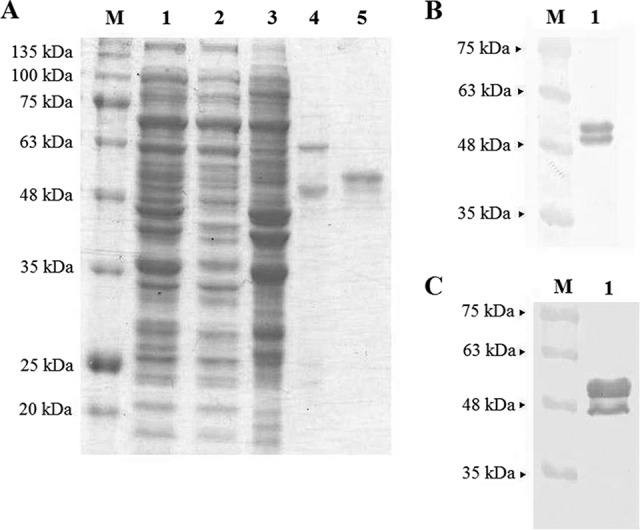
SDS-PAGE and Western blotting of the fusion protein. (A) Purification of NvC-aD with a His trap HP column. Lane 1, cell lysate; lane 2, flow through; lane 3, elution with 50 mM imidazole; lane 4, elution with 200 mM imidazole; lane 5, purified NvC-aD eluted with 500 mM imidazole. (B) Western blotting with anti-His antibody. (C) Western blotting with anti-HBsAg antibody. SDS-PAGE and the Western blot of the purified protein show both the full-length NvC-aD (51 kDa) and the N-terminal degraded NvC-aD (48 kDa).
The purified NvC-aD was analyzed by Western blotting. A band of approximately 51 kDa was detected with anti-His and anti-HBsAg antibodies, which corresponded well to the calculated molecular mass of 50.6 kDa. Another, smaller band of approximately 48 kDa was also detected with both antibodies, suggesting that NvC-aD was proteolytically degraded at the N-terminal end (Fig. 2B and C).
Transmission electron microscopy.
TEM analysis revealed that the purified NvC-aD assembled into spherical VLPs approximately 30 nm in diameter (Fig. 3A). In order to study whether the “a” determinant is exposed on the surfaces of VLPs, the purified NvC-aD was treated with anti-HBsAg antibody, followed by protein A conjugated to 5-nm gold particles, and analyzed under a TEM. The gold particles, which appeared as black granules, were predominantly observed on the surfaces of VLPs, suggesting that the “a” determinant is exposed on the surfaces of VLPs (Fig. 3B). NvC, in the absence of the “a” determinant, was used as a negative control for immunogold labeling. Only a few gold particles were observed in the electron micrograph (Fig. 3C).
FIG 3.

Transmission electron micrographs of the purified fusion proteins. (A) TEM analysis of NvC-aD. (B) NvC-aD probed with anti-HBsAg, followed by protein A conjugated to 5-nm gold particles. The arrows indicate gold particles. (C) NvC probed with anti-HBsAg, followed by protein A conjugated to 5-nm gold particles. The samples were stained negatively with uranyl acetate (1% [wt/vol]) and viewed under ×100,000 magnification.
Antigenicity of the fusion protein.
ELISA was performed to study the antigenicity of the “a” determinant fused to the MrNV capsid protein. Microtiter plate wells were coated with different concentrations of the purified NvC-aD. Purified MrNV capsid protein without the “a” determinant, NvC, served as a negative control. Anti-HBsAg reacted strongly with NvC-aD, suggesting the “a” determinant fused to NvC was antigenic (Fig. 4).
FIG 4.
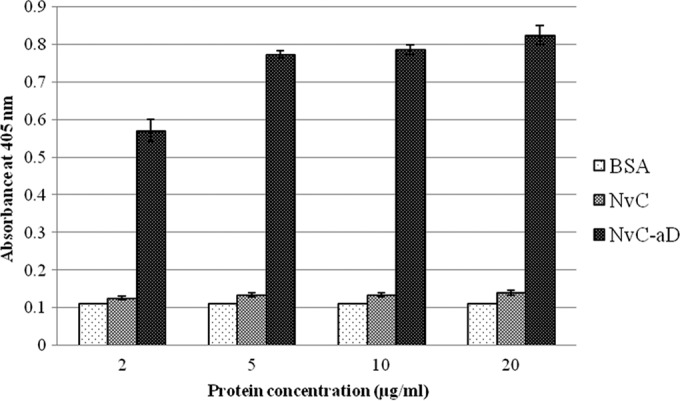
Antigenicity of the “a” determinant fused to MrNV capsid protein. Microtiter plate wells were coated with each protein diluted to 20, 10, 5, and 2 μg/ml. BSA was the negative control. NvC, MrNV capsid protein without the “a” determinant; NvC-aD, fusion protein of MrNV capsid protein and the “a” determinant. The error bars indicate standard deviations.
Immunogenicity of the fusion protein.
Serum samples from mice immunized with HEPES buffer, NvC, NvC-aD, and Engerix B were collected before every injection and 1 week after the 2nd booster injection. Total antibodies against the “a” determinant and plasma-purified HBsAg were studied. The results showed that both NvC-aD and Engerix B elicited the production of antibodies against the plasma-purified HBsAg (Fig. 5A) and the “a” determinant peptide (Fig. 5B). In general, the maximum antibody level was achieved after the 1st booster injection.
FIG 5.

Immunogenicity of the fusion protein in mice. Total antibodies against the plasma-purified HBsAg (A) and the “a” determinant peptide (B) in serum samples collected at week 2 (preinjection), week 5 (3 weeks after 1st injection), week 8 (3 weeks after 1st booster injection), and week 9 (1 week after 2nd booster injection). Serum samples from mice were pooled into groups injected with HEPES buffer, NvC, NvC-aD, and Engerix B. The serum samples were then reacted with the plasma-purified HBsAg and the “a” determinant peptide immobilized on microtiter plate wells. The letters and Roman numerals above the bars represent the statistical significance of results within each time point; a and b, w and x, x and y, y and z, I and II, II and III, and III and IV each represent a P value of <0.001. **, results are extremely significant (P < 0.0001) in comparison to a group of mice injected with NvC (control). The error bars indicate standard deviations.
Immunophenotyping.
The spleens of mice were harvested 1 week after the 2nd booster injection to study the presence of helper T lymphocytes (Th), cytotoxic T lymphocytes (CTL), and natural killer (NK) cells via immunophenotyping (Table 1). Overall, Engerix B and NvC-aD induced a significantly higher CD8+/CD4+ ratio (P < 0.05), indicating CTL activities. In addition, Engerix B and NvC-aD also induced a high level of NK cells in mice, especially those immunized with NvC-aD. Interestingly, NvC also induced NK cells.
TABLE 1.
Immunophenotyping of mouse splenocytes
| Group | % cells gateda |
CD8+/CD4+ ratio | ||
|---|---|---|---|---|
| CD3+ CD4+ | CD3+ CD8+ | NK1.1+ | ||
| HEPES | 27.44 ± 0.28d | 15.73 ± 0.24k | 6.74 ± 1.67x | 0.57 ± 0.01q |
| NvC | 16.29 ± 0.56b | 8.37 ± 0.21i | 10.72 ± 0.72y | 0.51 ± 0.02p |
| NvC-aD | 14.84 ± 0.17a | 10.98 ± 0.47j | 16.91 ± 0.95z | 0.74 ± 0.03s |
| Engerix B | 23.34 ± 0.51c | 15.03 ± 0.83k | 8.75 ± 1.58x,y | 0.64 ± 0.02r |
Numbers followed by different letters are significantly different (P < 0.05).
Cytokine concentrations in mouse serum samples.
The concentrations of IFN-γ and IL-10 in serum samples were analyzed by sandwich ELISA. NvC-aD induced IFN-γ production but suppressed IL-10 secretion at a significant level (P < 0.001) (Table 2), indicating a type 1 helper T cell (Th1)-biased immune response.
TABLE 2.
Cytokine concentrations in pooled serum samples
| Group | Cytokine concn (pg/ml) |
|
|---|---|---|
| IFN-γa | IL-10b | |
| HEPES | 498.312 ± 84.619a | 421.099 ± 0z |
| NvC | 620.540 ± 131.630a | 432.181 ± 11.082z |
| NvC-aD | 1777.001 ± 122.228b | 321.365 ± 33.245y |
| Engerix B | 338.476 ± 0a | 421.099 ± 0z |
Numbers followed by different letters are significantly different (P < 0.001).
Numbers followed by different letters are significantly different (P < 0.001).
DISCUSSION
VLPs formed by the capsid proteins of viruses such as HBV (25, 27, 32) and human papillomavirus (33) and bacteriophages (8, 34) have been widely used for displaying foreign epitopes for vaccine development. The carriers enhance the antigenicity of the fused epitopes (35–37), which are often found to be inefficient in eliciting immune responses. In addition, VLPs allow the display of more than a single epitope, enabling the development of multivalent vaccines (25, 38, 39). MrNV capsid protein self-assembles into VLPs (18, 19); therefore, we hypothesize that MrNV VLPs can be used to display a foreign epitope and enhance B cell and T cell responses.
In this experiment, the “a” determinant of HBV was fused to the C-terminal end of NvC, forming a fusion recombinant protein, namely, NvC-aD. The production of NvC-aD can be scaled up easily, and it can be purified rapidly in a single-step IMAC purification, with approximately 95% purity. NvC-aD provides an alternative to the yeast-derived HBsAg vaccine. The former is produced in bacteria, which reduces the cost of production and thus promotes vaccination programs in developing countries. NvC-aD with a molecular mass of about 51 kDa was detected with anti-HBsAg and anti-His antibodies by Western blotting, which corresponded well to the calculated molecular mass of 50.6 kDa.
The self-assembly property of the MrNV capsid protein was not impaired by the addition of 49 amino acid residues at the C-terminal end of the protein. NvC-aD formed spherical VLPs approximately 30 nm in diameter when observed under a TEM, showing that the addition of extra amino acid residues did not alter the shape of the NvC particles. Anti-HBsAg antibody, which recognizes the “a” determinant of HBsAg, was used to detect the “a” determinant displayed on the surfaces of NvC-aD particles through immunogold TEM analysis and ELISA. The results indicate that the “a” determinant fused at the C-terminal end of NvC is antigenic and is displayed on the surfaces of the VLPs.
The “a” determinant of HBsAg induces HBV-neutralizing antibodies regardless of the virus subtype (40), and they provide up to 2,000-fold-higher specific activities than the polyclonal antibodies induced by whole HBV (41). Immunization of BALB/c mice with the purified NvC-aD resulted in the production of antibodies specifically against both the “a” determinant peptide and the “a” determinant present in the plasma-purified HBsAg. These findings suggest that NvC-aD is a potential HBV vaccine candidate.
In addition to antibody production, immunization of BALB/c mice with NvC-aD resulted in a significant increase of NK cells, which are part of the innate immunity. NK cells are among the major IFN-γ-secreting cells, which provide protection through various pathways, including recruitment of immune cells, such as other NK cells, macrophages, CTL, and antigen-presenting cells (42, 43). During an infection, NK cells are often the first to react while adaptive immune responses develop. Moreover, NK cells were shown to provide protection against HBV infection in mice depleted of CD4+ and CD8+ T cells (44).
Administration of NvC-aD in mice also resulted in an increase of CTL. CTL are believed to play a vital role during HBV infection by killing virus-infected hepatocytes. Thimme et al. (45) showed that CTL are the main effector cells for viral clearance, as well as disease pathogenesis, in CD8-deficient chimpanzees. However, Bertoletti et al. (46) demonstrated the inhibition of HBV replication by CTL without causing liver damage. Phillips et al. (47) concluded that the cytolytic effect of CTL during HBV infection is minimal, and the main mechanisms in play were secretion of IFN-γ and of tumor necrosis factor alpha (TNF-α). This conclusion is supported by Guidotti et al. (48), who demonstrated that passive transfer of virus-specific CTL strongly inhibits HBV replication through mediation by IFN-γ and TNF-α. Despite the potential role of TNF-α, both NvC-aD and Engerix B based upon HBsAg did not induce TNF-α secretion in mice (data not shown). Instead, HBV core antigen (HBcAg) was demonstrated to induce TNF-α secretion in vitro and in vivo (49–51).
It is widely accepted that IFN-γ plays an important role in noncytolytic virus clearance in vivo and in vitro (47, 48, 52–56). Several mechanisms have been proposed, such as IFN-γ-induced proteosome-dependent degradation of viral or cellular proteins needed for viral replication (55), destabilization of viral RNA (48), and inhibition of HBV-mediated nuclear factor kappa B activation, which signals liver diseases, including hepatocellular carcinoma (53). Thus, an increase in IFN-γ production following NvC-aD administration is believed to provide additional protection against HBV infection in a noncytolytic way.
In summary, the “a” determinant of HBV was fused to the C-terminal end of the MrNV capsid protein. The fusion protein NvC-aD self-assembled into VLPs and induced the production of specific anti-“a” determinant antibodies, CTL, NK cells, and IFN-γ in mice. Collectively, this study demonstrated the potential of MrNV VLPs as an epitope display system for the development of vaccines that enhance B cell and T cell responses.
ACKNOWLEDGMENT
This study was supported by ERGS grant no. 1-2012/5527078 from the Ministry of Higher Education of Malaysia.
REFERENCES
- 1.Lavanchy D. 2005. Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J Clin Virol 34:S1–S3. doi: 10.1016/S1386-6532(05)00384-7. [DOI] [PubMed] [Google Scholar]
- 2.Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. 2006. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev 28:112–125. doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- 3.Bruss V, Ganem D. 1991. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci U S A 88:1059–1063. doi: 10.1073/pnas.88.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dane DS, Cameron CH, Briggs M. 1970. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet i:695–698. [DOI] [PubMed] [Google Scholar]
- 5.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. 1984. Human hepatitis B vaccine from recombinant yeast. Nature 307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 6.Murray K, Bruce SA, Wingfield P, van Eerd P, De Reus A, Schellekens H. 1984. Hepatitis B virus antigen made in microbial cells immunise against viral infection. EMBO J 3:645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howard CR, Allison LM. 1995. Hepatitis B surface antigen variation and protective immunity. Intervirology 38:35–40. [DOI] [PubMed] [Google Scholar]
- 8.Tan GH, Yusoff K, Seow HF, Tan WS. 2005. Antigenicity and immunogenicity of the immunodominant region of hepatitis B surface antigen displayed on bacteriophage T7. J Med Virol 77:475–480. doi: 10.1002/jmv.20479. [DOI] [PubMed] [Google Scholar]
- 9.Ottone S, Nguyen X, Bazin J, Berard C, Jimenez S, Letourneur O. 2007. Expression of hepatitis B surface antigen major subtypes in Pichia pastoris and purification for in vitro diagnosis. Protein Expr Purif 56:177–188. doi: 10.1016/j.pep.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Coleman PF. 2006. Detecting hepatitis B surface antigen mutants. Emerg Infect Dis 12:198–203. doi: 10.3201/eid1203.050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawlotsky JM. 2005. The concept of hepatitis B virus mutant escape. J Clin Virol 34:S125–S129. doi: 10.1016/S1386-6532(05)80021-6. [DOI] [PubMed] [Google Scholar]
- 12.Zanetti AR, Tanzi E, Manzillo G, Maio G, Sbreglia C, Caporaso N, Thomas H, Zuckerman AJ. 1988. Hepatitis B variant in Europe. Lancet ii:1132–1133. [DOI] [PubMed] [Google Scholar]
- 13.Zuckerman JN, Zuckerman AJ. 2003. Mutations of the surface protein of hepatitis B virus. Antiviral Res 60:75–78. doi: 10.1016/j.antiviral.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Lapiński TW, Pogorzelska J, Flisiak R. 2012. HBV mutations and their clinical significance. Adv Med Sci 57:18–22. doi: 10.2478/v10039-012-0006-x. [DOI] [PubMed] [Google Scholar]
- 15.Tan WS, Ho KL. 2014. Phage display creates innovative applications to combat hepatitis B virus. World J Gastroenterol 20:11650–11670. doi: 10.3748/wjg.v20.i33.11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arcier JM, Herman F, Lightner DV, Redman RM, Mari J, Bonami JR. 1999. A viral disease associated with mortalities in hatchery-reared postlarvae of the giant freshwater prawn Macrobrachium rosenbergii. Dis Aquat Organ 38:177–181. doi: 10.3354/dao038177. [DOI] [Google Scholar]
- 17.Saedi TA, Moeini H, Tan WS, Yusoff K, Daud HM, Chu KB, Tan SG, Bhassu S. 2012. Detection and phylogenetic profiling of nodavirus associated with white tail disease in Malaysian Macrobrachium rosenbergii de Man. Mol Biol Rep 39:5785–5790. doi: 10.1007/s11033-011-1389-7. [DOI] [PubMed] [Google Scholar]
- 18.Goh ZH, Tan SG, Bhassu S, Tan WS. 2011. Virus-like particles of Macrobrachium rosenbergii nodavirus produced in bacteria. J Virol Methods 175:74–79. doi: 10.1016/j.jviromet.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 19.Goh ZH, Mohd NA, Tan SG, Bhassu S, Tan WS. 2014. RNA-binding region of Macrobrachium rosenbergii nodavirus capsid protein. J Gen Virol 95:1919–1928. doi: 10.1099/vir.0.064014-0. [DOI] [PubMed] [Google Scholar]
- 20.Lee KW, Tey BT, Ho KL, Tan WS. 2012. Delivery of chimeric hepatitis B core particles into liver cells. J Appl Microbiol 112:119–131. doi: 10.1111/j.1365-2672.2011.05176.x. [DOI] [PubMed] [Google Scholar]
- 21.Lee KW, Tey BT, Ho KL, Tejo BA, Tan WS. 2012. Nanoglue: an alternative way to display cell-internalizing peptide at the spikes of hepatitis B virus core nanoparticles for cell-targeting delivery. Mol Pharm 9:2415–2423. doi: 10.1021/mp200389t. [DOI] [PubMed] [Google Scholar]
- 22.Petry H, Goldmann C, Ast O, Luke W. 2003. The use of virus-like particles for gene transfer. Curr Opin Mol Ther 5:524–528. [PubMed] [Google Scholar]
- 23.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. 1992. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A 89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murata K, Lechmann M, Qiao M, Gunji T, Alter HJ, Liang TJ. 2003. Immunization with hepatitis C virus-like particles protects mice from recombinant hepatitis C virus-vaccinia infection. Proc Natl Acad Sci U S A 100:6753–6758. doi: 10.1073/pnas.1131929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray K, Shiau AL. 1999. The core antigen of hepatitis B virus as a carrier for immunogenic peptides. Biol Chem 380:277–283. [DOI] [PubMed] [Google Scholar]
- 26.Quan FS, Compans RW, Nguyen HH, Kang SM. 2008. Induction of heterosubtypic immunity to influenza virus by intranasal immunization. J Virol 82:1350–1359. doi: 10.1128/JVI.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yap WB, Tey BT, Alitheen NB, Tan WS. 2012. Display of the antigenic region of Nipah virus nucleocapsid protein on hepatitis B virus capsid. J Biosci Bioeng 113:26–29. doi: 10.1016/j.jbiosc.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Chackerian B, Caldeira JDC, Peabody J, Peabody DS. 2011. Peptide epitope identification by affinity selection on bacteriophage MS2 virus-like particles. J Mol Biol 409:225–237. doi: 10.1016/j.jmb.2011.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan WS, Dyson MR, Murray K. 1999. Two distinct segments of the hepatitis B virus surface antigen contribute synergistically to its association with the viral core particles. J Mol Biol 286:797–808. doi: 10.1006/jmbi.1998.2525. [DOI] [PubMed] [Google Scholar]
- 30.Birnboim HC, Doly J. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho CW, Chew TK, Ling TC, Kamaruddin S, Tan WS, Tey BT. 2006. Efficient mechanical cell disruption of Escherichia coli by an ultrasonicator and recovery of intracellular hepatitis B core antigen. Process Biochem 41:1829–1834. doi: 10.1016/j.procbio.2006.03.043. [DOI] [Google Scholar]
- 32.Ibanez LI, Roose K, De Filette M, Schotsaert M, De Sloovere J, Roels S, Pollard C, Schepens B, Grooten J, Fiers W, Saelens X. 2013. M2e-displaying virus-like particles with associated RNA promote T helper 1 type adaptive immunity against influenza A. PLoS One 8:e59081. doi: 10.1371/journal.pone.0059081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matic S, Rinaldi R, Masenga V, Noris E. 2011. Efficient production of chimeric human papillomavirus 16 L1 protein bearing the M2e influenza epitope in Nicotiana benthamiana plants. BMC Biotechnol 11:106. doi: 10.1186/1472-6750-11-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashemi H, Pouyanfard S, Bandehpour M, Noroozbabaei Z, Kazemi B, Saelens X, Mokhtari-Azad T. 2012. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza A virus challenge. PLoS One 7:e45765. doi: 10.1371/journal.pone.0045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sominskaya I, Skrastina D, Dislers A, Vasiljev D, Mihailova M, Ose V, Dreilina D, Pumpens P. 2010. Construction and immunological evaluation of multivalent hepatitis B virus (HBV) core virus-like particles carrying HBV and HCV epitopes. Clin Vaccine Immunol 17:1027–1033. doi: 10.1128/CVI.00468-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vietheer PT, Boo I, Drummer HE, Netter HJ. 2007. Immunizations with chimeric hepatitis B virus-like particles to induce potential anti-hepatitis C virus neutralizing antibodies. Antivir Ther 12:477–487. [PubMed] [Google Scholar]
- 37.Wang YS, Ouyang W, Liu XJ, He KW, Yu SQ, Zhang HB, Fan HJ, Lu CP. 2012. Virus-like particles of hepatitis B virus core protein containing five mimotopes of infectious bursal disease virus (IBDV) protect chickens against IBDV. Vaccine 30:2125–2130. doi: 10.1016/j.vaccine.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 38.Jackwood DJ. 2013. Multivalent virus-like-particle vaccine protects against classic and variant infectious bursal disease viruses. Avian Dis 57:41–50. doi: 10.1637/10312-080212-Reg.1. [DOI] [PubMed] [Google Scholar]
- 39.Kanai Y, Athmaram TN, Stewart M, Roy P. 2013. Multiple large foreign protein expression by a single recombinant baculovirus: a system for production of multivalent vaccines. Protein Expr Purif 91:77–84. doi: 10.1016/j.pep.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Pride MW, Shi H, Anchin JM, Linthicum DS, LoVerde PT, Thakur A, Thanavala Y. 1992. Molecular mimicry of hepatitis B surface antigen by an anti-idiotype-derived synthetic peptide. Proc Natl Acad Sci U S A 89:11900–11904. doi: 10.1073/pnas.89.24.11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryu CJ, Gripon P, Park HR, Park SS, Kim YK, Guguen-Guillouzo C, Yoo OJ, Hong HJ. 1997. In vitro neutralization of hepatitis B virus by monoclonal antibodies against the viral surface antigen. J Med Virol 52:226–233. doi:. [DOI] [PubMed] [Google Scholar]
- 42.Arase H, Arase N, Saito T. 1996. Interferon gamma production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. J Exp Med 183:2391–2396. doi: 10.1084/jem.183.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. 2012. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol 91:299–309. doi: 10.1189/jlb.0611308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. 2000. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med 192:921–930. doi: 10.1084/jem.192.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. 2003. CD8+ T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertoletti A, Maini M, Williams R. 2003. Role of hepatitis B virus specific cytotoxic T cells in liver damage and viral control. Antiviral Res 60:61–66. doi: 10.1016/j.antiviral.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Phillips S, Chokshi S, Riva A, Evans A, Williams R, Naoumov NV. 2010. CD8+ T cell control of hepatitis B virus replication: direct comparison between cytolytic and noncytolytic functions. J Immunol 184:287–295. doi: 10.4049/jimmunol.0902761. [DOI] [PubMed] [Google Scholar]
- 48.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36. doi: 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 49.Cao T, Meuleman P, Desombere I, Sällberg M, Leroux-Roels G. 2001. In vivo inhibition of anti-hepatitis B virus core antigen (HBcAg) immunoglobulin G production by HBcAg-specific CD4+ Th1-type T-cell clones in a hu-PBL-NOD/SCID mouse model. J Virol 75:11449–11456. doi: 10.1128/JVI.75.23.11449-11456.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper A, Tal G, Lider O, Shaul Y. 2005. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol 175:3165–3176. doi: 10.4049/jimmunol.175.5.3165. [DOI] [PubMed] [Google Scholar]
- 51.Tzeng HT, Tsai HF, Chyuan IT, Liao HJ, Chen CJ, Chen PJ, Hsu PN. 2014. Tumor necrosis factor-alpha induced by hepatitis B virus core mediating the immune response for hepatitis B viral clearance in mice model. PLoS One 9:e103008. doi: 10.1371/journal.pone.0103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kan Q, Li D, Yu Z. 2012. Specific expression of human interferon-gamma controls hepatitis B virus replication in vitro in secreting hepatitis B surface antigen hepatocytes. J Virol Methods 180:84–90. doi: 10.1016/j.jviromet.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 53.Park SG, Ryu HM, Lim SO, Kim YI, Hwang SB, Jung G. 2005. Interferon-γ inhibits hepatitis B virus-induced NF-κB activation through nuclear localization of NF-κB-inducing kinase. Gastroenterology 128:2042–2053. doi: 10.1053/j.gastro.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV. 2002. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J Virol 76:5646–5653. doi: 10.1128/JVI.76.11.5646-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robek MD, Wieland SF, Chisari FV. 2002. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J Virol 76:3570–3574. doi: 10.1128/JVI.76.7.3570-3574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wieland S, Thimme R, Purcell RH, Chisari FV. 2004. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A 101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]