

Abstract
Mathematical modeling of the electrical activity of the pancreatic β-cell has been extremely important for understanding the cellular mechanisms involved in glucose-stimulated insulin secretion. Several models have been proposed over the last 30 y, growing in complexity as experimental evidence of the cellular mechanisms involved has become available. Almost all the models have been developed based on experimental data from rodents. However, given the many important differences between species, models of human β-cells have recently been developed. This review summarizes how modeling of β-cells has evolved, highlighting the proposed physiological mechanisms underlying β-cell electrical activity.
Keywords: action potentials, β-cell, bursting, calcium, electrical activity, ion channels, mathematical model
Abbreviations
- T2D
Type 2 Diabetes
- GSIS
glucose-stimulated insulin secretion
- Ca2+
calcium ions
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- KATP
ATP-sensitive K+ channels
- [ATP]i
cytosolic ATP
- Vm
membrane potential
- VDCC
voltage-dependent Ca2+ channels
- [Ca2+]i
intracellular calcium concentration
- TRP
transient receptor potential
- PMCA
plasma membrane Ca2+-ATPase
- NCX
Na+/Ca2+ exchanger
- ER
endoplasmic reticulum
- SERCA
sarco-endoplasmic reticulum Ca2+-ATPase
- IP3R
inositol-1,4,5-trisphosphate receptors
- RyR
ryanodine receptors
- MCU
mitochondrial Ca2+ uniporter
- mNCX
mitochondrial Na+/Ca2+ exchanger
- [Ca2+]m
mitochondrial calcium
- KCa
Ca2+-dependent K+ channels
- Kv
voltage-dependent K+ channels
- CK
Chay-Keizer
- CRAC
calcium release-activated current
- [Na+]
Na+ concentration
- DOM
dual oscillator model
- HERG
human eter à-go-go related gene
- TCA
trycarboxylic acid cycle
- FBP
fructose-1,6-bisphosphate
- PFK
phosphofructokinase
- F6P
fructose-6-phosphate
- cAMP
cyclic AMP
- ROS
reactive oxygen species
- GLUT
glucose transporter
Introduction
Insulin, synthesized and secreted by the pancreatic β-cells of the islets of Langerhans, is the only hormone responsible for lowering blood glucose levels. Under normal conditions, blood insulin is pulsatile both in humans and in rodents.1,2 Interestingly, insulin oscillations are disrupted in patients with Type 2 Diabetes (T2D).3 It has been shown that single β-cells contribute to the oscillations of insulin at higher levels of organization,4 underscoring the importance of studying β-cell functioning at the cellular level.
In β-cells, glucose-stimulated insulin secretion (GSIS) is mediated by the increase of intracellular calcium concentration ([Ca2+]i), driven by a well-established sequence of events (Fig. 1), beginning with the transport of glucose into the cell through the glucose transporters (GLUT), accelerating the metabolism and therefore the production of adenosine triphosphate (ATP) at the expense of adenosine diphosphate (ADP). This induces an increase in the ATP/ADP ratio, causing the closure of the ATP-sensitive K+ channels (KATP), and consequently, promoting the slow depolarization of the membrane potential (Vm) upon the threshold value at which the voltage-dependent Ca2+ channels (VDCCs) are activated, allowing the influx of calcium ions (Ca2+). It is the increase in [Ca2+]i that finally promotes insulin secretion. This is the main pathway of GSIS, and it is referred to as the triggering or KATP-dependent pathway (Fig. 1). As a complement to the triggering pathway, GSIS is regulated by the amplifying pathway, also known as the KATP-independent pathway, which enhances the effects of [Ca2+]i on the exocytotic machinery.5–7
Figure 1.
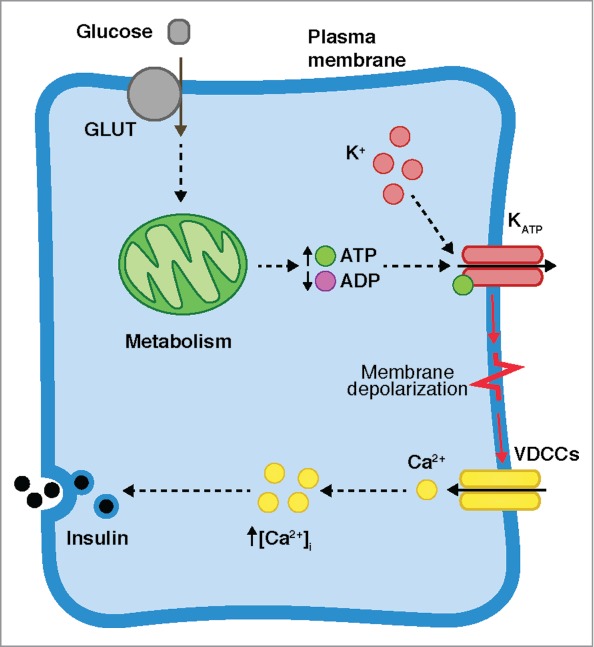
Glucose-stimulated insulin secretion (GSIS). After glucose is transported into the cell by the GLUT transporters, it is metabolized, potentiating the production of ATP and the closure of the ATP-sensitive K+ channels (KATP). The membrane is depolarized and voltage-dependent Ca2+ channels (VDCCs) are activated, allowing the influx of Ca2+. The increase of the intracellular Ca2+ concentration ([Ca2+]i) stimulates Ca2+-dependent insulin secretion.
Alterations in β-cells are highly related to impaired fasting glucose and/or impaired glucose tolerance, which eventually progress to T2D,8 a disease characterized by insulin resistance and β-cell dysfunction. Several factors are known to impair the proper secretion of insulin at the cellular level. For example, mutations in ionic channels have been associated with a higher diabetes risk.9–11 Moreover, it has also been demonstrated that defective β-cell sensitivity and impaired metabolism could result in hyperglycemia and eventually in T2D.12,13
As a complement to experimental work, mathematical models of β-cells have been used to elucidate how the cellular mechanisms involved in GSIS interact, providing feasible explanations and hypotheses to experimental observations in β-cells. Models have grown in complexity as new experimental evidence has emerged: from the early minimal models that included a few ionic channels and a basic representation of Ca2+ handling and metabolism, to the current complex models that incorporate detailed representations of glycolysis and ATP production, and the recent appearance of models of human β-cells.
The aim of this work is to give a general overview of the progress in the field of β-cell modeling from a physiological perspective; thus, a detailed discussion about the mathematical aspects of the models is beyond the scope of this review. Interested readers are referred to other works on the subject.14–16
This review is structured as follows. First, a brief introduction to the physiology of the pancreatic β-cells considered in mathematical models is presented. In the following section, we address the evolution of mathematical models of β-cells, including the most recent models developed specifically for human β-cells. Finally, we briefly discuss the applications and limitations of the mathematical models of the pancreatic β-cells.
General Overview of the Cellular Mechanisms Involved in the Electrical Activity of β-cells
Electrophysiology of the β-cell
The changes in membrane potential needed to allow the influx of Ca2+ through the VDCCs are generated through the concerted action of ionic transport mechanisms (ionic channels, pumps, and exchangers), which are regulated by ligands (e.g., Ca2+ or ATP) or by the Vm itself. Several ionic channels participate in the formation of the electrical activity pattern, including voltage-dependent Ca2+, Na+, and K+ channels and Ca2+-dependent K+ and Ca2+ channels.17–20 Of special interest is the KATP channel, which links the changes in glucose metabolism to the electrical activity in β-cells.21 Nonselective cationic channels, like the transient receptor potential channels (TRP) have also been found in β-cells.17,20 The detailed electrophysiological properties of ionic channels in β-cells can be found elsewhere.17,18,20
The expression of specific ionic channels differs between species,22,23 which is reflected in the corresponding pattern of electrical activity (for recent reviews see refs. 10, 17, 20, 24). In rodents, β-cells exhibit a characteristic electrical pattern, composed of slow oscillations in Vm, above which action potentials are superimposed.17–19 This is known as bursts of action potentials, or bursting electrical activity. Heterogeneous bursting patterns have been reported in rodents,25,26 which can be classified as “fast” (period of <60 s), “slow” (period from 1 to several minutes), and “mixed” or “compound” oscillations (fast oscillations superimposed on slow oscillations). Simulations of the distinct electrical patterns observed experimentally in rodent cells are shown in Fig. 2A (experimental recordings can be seen in Fig. 1 in ref. 25). On the other hand, in human β-cells, action potential firing is the most common electrical behavior, although bursting has been observed occasionally.10,22,27,28 Simulations reproducing the electrical patterns observed in the human β-cell (see Figs. 1, 2 and 6 in ref. 28 for the experimental recordings) are shown in Fig. 2B.
Figure 2.
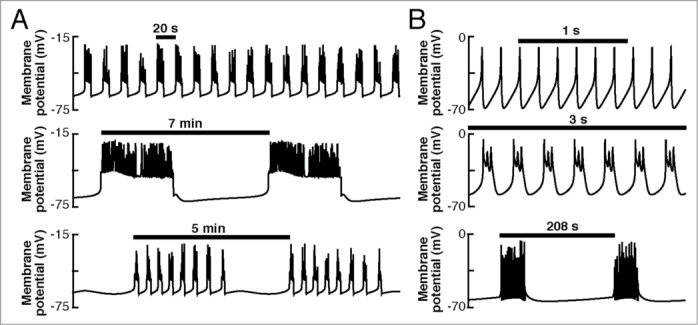
Electrical activity patterns in pancreatic β-cells. (A) Simulated fast (top), slow (middle), and compound (bottom) bursting behavior in rodent cells (simulations made with the Dual Oscillator Model120). (B) Simulations of action potential firing (top), fast (middle), and slow (bottom) bursting in human cells (simulated with the model of the human β-cell of Riz et al.28).
Calcium handling and metabolism
An increase in cytosolic Ca2+ concentration is used as a signal to control insulin exocytosis. It has been observed that [Ca2+]i oscillates in synchrony with membrane potential29–33 and that insulin exocytosis occurs when Ca2+ channels are active.34–36 Several mechanisms are responsible for the oscillations of [Ca2+]i. Calcium entry is mediated by the activity of voltage-gated Ca2+ channels, whereas Ca2+ is extruded from the β-cell mainly by the plasma membrane Ca2+-ATPase (PMCA)37 and the Na+/Ca2+ exchanger (NCX).38,39
Once in the intracellular space, [Ca2+]i is regulated by internal stores, namely the endoplasmic reticulum (ER) and the mitochondria. The ER captures Ca2+ through the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) during the rise in [Ca2+]i caused by the effects of depolarization on the VDCCs. This limits the amplitude of [Ca2+]i oscillations. Upon membrane repolarization, Ca2+ is released from the ER, preventing an abrupt drop in [Ca2+]i.40,41 Efflux of Ca2+ from the ER through channels such as inositol-1,4,5-trisphosphate receptors (IP3Rs) or ryanodine receptors (RyRs) is controlled by Ca2+ itself or by intracellular messengers (e.g., IP3).41–43 This enables the β-cells to respond to muscarinic agonists by releasing Ca2+ from the ER to the cytoplasm.44 In addition, it has been shown that the ER is relevant for both the oscillations of [Ca2+]i and the control of Vm.45
On the other hand, mammalian mitochondria have a high capacity for Ca2+ uptake, although it is known that under resting conditions mitochondria do not play an important role as Ca2+ deposits as there is not a significant gradient between cytosolic and mitochondrial Ca2+ ([Ca2+]m).12,46 It has been proposed that mitochondria serve as a buffer of Ca2+ that limits the amplitude of the cytosolic Ca2+ transients.47,48 A rise in [Ca2+]i is relayed to the mitochondria where Ca2+ influx is mediated by the mitochondrial uniporter (MCU), which transports Ca2+ from the cytosol into the mitochondrial matrix. Ca2+ is then regulated by the mitochondrial Na+/Ca2+ exchanger (mNCX), responsible for Ca2+ efflux from the mitochondria.49,50
Mitochondria and glucose metabolism play an extremely important role in the control of GSIS (extensively reviewed in refs. 12, 51–54). In β-cells the first stage of metabolism is glycolysis, where glucose is metabolized to pyruvate by means of a complex cascade of enzymatic reactions. Pyruvate is then processed during the TCA (trycarboxylic acid) cycle, resulting in the electron carriers NADH and FADH2, which are then used to generate a proton gradient across the mitochondrial membrane. The resulting proton flux through the ATP synthase finally drives the phosphorylation of ADP to ATP. ATP then regulates the activity of the KATP channels21 and drives several ATP-consuming processes of the cell, such as those involving Ca2+-ATPases (e.g., PMCA and SERCA),38,45,55 and insulin exocytosis.56 In spite of the higher demand for ATP due to the activation of the ATP-consuming processes, a net increase in the cytosolic ATP/ADP ratio has been observed.49,57 This overcompensation can be explained by 2 factors: the greater availability of glucose to be metabolized in the first place and the subsequent effect of the increase of [Ca2+]m in the oxidation of glucose,52 which is known to involve the activation of the mitochondrial dehydrogenases and other enzimes.58–60 Consistent with this proposal, a biphasic behavior was observed in measurements of the ATP/ADP ratio and oxygen consumption.49,57 In addition, it has been proposed that [Ca2+]m also has a negative effect on ATP production by reducing the proton motive force,48,61,62 although there is a large body of evidence that the overall effect of mitochondrial Ca2+ is to potentiate rather than to inhibit ATP production.49,52,63 For example, it has been demonstrated that glucose oxidation is considerably reduced when Ca2+ influx is prevented.64,65 In addition, recent studies49,57 have demonstrated that increases of mitochondrial Ca2+ are required for normal changes in the ATP/ADP ratio to occur in response to glucose stimulation. Detailed models addressing the role of the main processes involved in energy metabolism in β-cells have been developed recently.66,67 Besides the role of mitochondria in the triggering pathway of insulin secretion, it has been proposed that mitochondrial-derived metabolites are involved in the amplifying pathway of insulin secretion.51,68 However, the role of mitochondria in this KATP-independent pathway is still poorly understood.
Insulin secretion is pulsatile with a period of several minutes.2 Given that metabolic oscillations with a similar period have been observed,61,62,64,69–75 it has been proposed that they are involved in the generation of the pulsatile behavior of insulin secretion. For example, oscillations both in the cytosolic ATP ([ATP]i) and ATP/ADP ratio have been linked to oscillatory changes in the conductance of the KATP channels.69,70,72 In addition, oscillations in NAD(P)H,64,73 O2,74 mitochondrial membrane potential,61,62 and cAMP75 have been reported. The origin of these oscillations is still matter of debate, though at least 2 hypotheses have been proposed. On the one hand, it has been suggested that metabolic oscillations are generated during glycolysis by the positive feedback of the product FBP onto the PFK activity.76,77 On the other hand, other authors have proposed that the interplay between the production and consumption of ATP is responsible for the observed oscillations in [ATP]i.55,72,78 Interestingly, in a recent study, Tanaka et al.79 failed to observe significant oscillations in cytosolic ATP in mouse islets during GSIS, in contrast to the oscillations of sub-membrane ATP reported by Li et al.55
It is likely that mitochondrial dysfunction is involved in the onset of T2D and its related complications (reviewed in refs. 80–82). For example, in islets from diabetic subjects it has been shown that mitochondria have an altered morphology.83 In addition, reduced glucose oxidation, oxygen consumption and ATP production have been also reported.83,84 Moreover, it has been proposed that alterations in the free radicals derived from metabolism (reactive oxygen species, ROS) could have negative effects on glucose oxidation.51,63 It is thought that novel therapies to treat diabetes may involve pharmacologic agents targeting mitochondria whether to enhance mitochondrial function or to reduce negative effects of alterations in metabolism.82
Mathematical Models of Pancreatic β-Cells
Models of β-cells have been proposed as a tool to explain how the cellular mechanisms involved in GSIS interact. Most of the cellular processes mentioned in the previous section have been included in models of β-cells. In this section, several models based on both mouse and human β-cells are described in terms of the physiological mechanisms involved. Model descriptions are accompanied with a schematic diagram of the corresponding physiological hypothesis. Simulations showing the behavior of key variables are also presented. Implementation of the models and simulations were performed in Mathematica 9.0 (Wolfram Research, Inc., Champaign, IL).
Models of rodent β-cells
Dean and Matthews85,86 provided the first evidence of changes in the membrane potential of β-cells induced by glucose, consisting of fast bursting electrical activity. This fast pattern has been observed in isolated single mouse β-cells and isolated islets.26,87,88 Several hypotheses have been proposed and analyzed theoretically in order to elucidate the cellular mechanisms responsible for the fast bursting behavior. In their pioneering model, Chay and Keizer89 (CK model) were able to reproduce fast bursting electrical behavior (Fig. 3B, top panel). The CK model (Fig. 3A) includes Ca2+-dependent K+ channels (KCa) and voltage-dependent Ca2+ and K+ channels (VDCCs and Kv, respectively). Intracellular calcium handling was modeled in a minimal manner. As proposed by Atwater et al.,90 the CK model uses the effects of [Ca2+]i in the large conductance KCa channels as the mechanism to initiate or terminate the bursts of action potentials (Fig. 3B, bottom panel). During the active phase, sustained by Kv channels and VDCCs, [Ca2+]i increases slowly, activating the KCa channels and leading to membrane repolarization. During the silent phase, Ca2+ entry through VDCCs is inhibited, resulting in a decrease in [Ca2+]i due to the extrusion of Ca2+ from the cytosol. The KCa channels are then gradually closed, inducing depolarization of the membrane potential at which VDCCs and Kv are activated, initiating a new burst of action potentials. In this model, bursting depends entirely on one pacemaker variable ([Ca2+]i). The hypothesis proposed by the CK model was discarded when [Ca2+]i was measured in β-cells, revealing more rapid dynamics than predicted by the model.29,30 Moreover, blocking KCa channels with charybdotoxin produced no significant effect on the electrical activity.91 Recently, Houamed et al.92 showed that the BK channels do contribute to the repolarization of the action potentials in mouse β-cells, without a relevant role in the duration of the active and silent phases of the bursting electrical pattern. In spite of the evidence against this hypothesis, practically all the existing models of β-cells are based on the minimal CK model. Subsequent models were able to generate fast bursting using the same mathematical principle as the CK model, only changing the identity of the slow pacemaker variable.
Figure 3.
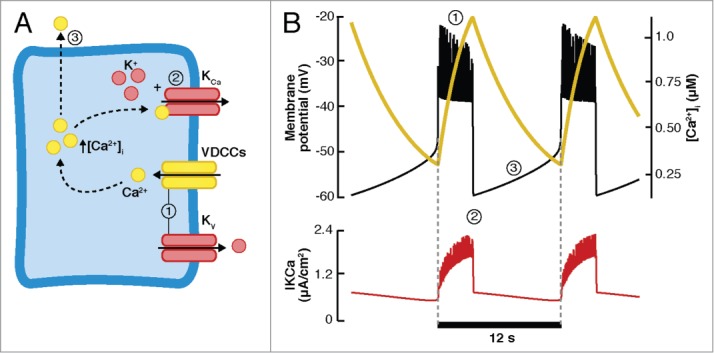
Minimal model of Chay and Keizer (CK model). A. Scheme of the CK model. The active phase (1) is sustained by the VDCCs and Kv channels, slowly increasing [Ca2+]i. The KCa channels are activated, eventually repolarizing the membrane (2). During the silent phase (3), the VDCCs and Kv channels are closed and Ca2+ is extruded from the cell, inhibiting the activity of the KCa channels. The slow depolarization eventually activates the VDCCs and Kv channels, initiating a new burst. B. Fast bursting simulated with the CK model. Top: Membrane potential (black curve) and intracellular Ca2+ concentration ([Ca2+]i, yellow curve). Bottom: Ca2+-dependent K+ (KCa) current.
Motivated by electrophysiological studies by Rorsman and Trube,93 Chay et al.94 replaced the KCa channels in the CK model with voltage-activated Ca2+-inactivated Ca2+ channels. In contrast to the CK model, in which the KCa channels are activated by an increase of [Ca2+]i, in this proposal the Ca2+ channels are inactivated by the changes in [Ca2+]i itself, allowing the K+ current to repolarize the membrane potential at the end of the burst of action potentials. Although it is well known that Ca2+ currents are extremely important for the electrical activity and insulin secretion both in mouse and human cells,22,31,95 their role as a pacemaker variable lacks sufficient experimental support.26
In 1984, KATP channels were identified in rodent β-cells,96,97 emerging as a feasible link between metabolism and electrical activity. In short, the activity of the KATP channels is inhibited by ATP and stimulated by ADP. The KATP channels are extremely important for β-cells, being responsible for the resting membrane potential of β-cells. In addition, the closure of the KATP channels due to an increase of the cytosolic ATP allows inward currents carried by Na+ and/or Ca2+ to depolarize, thus triggering electrical activity.10,21,97,98
Keizer and Magnus99-101 and Smolen and Keizer102 introduced KATP channels to the models of β-cells in order to analyze the role of the cyclical changes in the ATP/ADP ratio in β-cell electrical activity. In general, these models follow the hypothesis (see Fig. 4) that stipulates that during the active phase of the electrical activity, the cytosolic ATP concentration decreases due to the inhibiting effects of Ca2+ on the production of ATP (i.e., increasing ADP). As a consequence, KATP channels are activated, repolarizing the membrane potential. Closure of VDCCs during the silent phase inhibits Ca2+ entry and its negative effects on ATP production, allowing [ATP]i to increase, inhibiting KATP channels and initiating the slow depolarization to the threshold potential of activation of the VDCCs and Kv channels, once again initiating the active phase.
Figure 4.
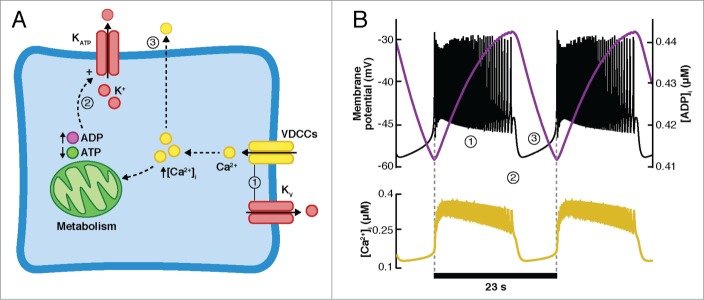
Oscillations in ATP regulate the conductance of the KATP channels. (A) During the active phase (1), sustained by the VDCC and the Kv channels, [Ca2+]i increases, exerting a negative effect on the production of ATP, reflected in the increase in ADP and the corresponding decrease in the ATP/ADP ratio. The KATP channels are slowly opened, eventually repolarizing the membrane (2). During the silent phase, VDCCs are inhibited, and the influx of Ca2+ is ceased as Ca2+ is also extruded from the cell. As [Ca2+]i decreases, the production of ATP is potentiated, closing the KATP channels and initiating slow depolarization (3). (B) Simulations with the model of Smolen-Keizer. Top: Vm (black curve) and [ADP]i (purple curve). Bottom: the fast dynamics of [Ca2+]i resembles the experimental observations.
The model of Keizer and Magnus99 uses the changes in [ADP], following the slow oscillations in [Ca2+]i as the pacemaker variable that triggers the transition between the active and silent phase of electrical activity by regulating the conductance of the KATP channels. One important drawback of this model is that, as in other models described above, the slow dynamics of [Ca2+]i contradicts the fast dynamics observed experimentally.29,30 However, Keizer and Magnus provided an equation for the KATP current that is still used in recent models. On the other hand, the Smolen-Keizer102 model (SK model) was able to reproduce the fast dynamics of [Ca2+]i oscillations including an improved model of the Ca2+ currents. As can be seen in Figure 4B, where simulations performed with the SK model are shown, ADP concentration rises slowly during the active phase and [Ca2+]i closely follows the dynamics of Vm. Assuming a constant nucleotide concentration, the latter means that ATP is declining during the active phase, thus activating the KATP channels and repolarizing the membrane potential. As mentioned above, these models assume a negative influence of Ca2+ in ATP production. Given the importance of metabolism on GSIS, Magnus and Keizer48 developed a minimal model of β-cell mitochondrial Ca2+ handling, considering only the negative effects of Ca2+ in ATP production and neglecting the activation of the dehydrogenases by Ca2+. Later, they extended their model to include a more refined representation of glucose metabolism (including, for example, the activation of dehydrogenases) and combined it with a model of the electrical activity induced by glucose.100,101 With this complex model, they explored the role of mitochondrial Ca2+-handling mechanisms during glucose-stimulated electrical activity.
There is experimental evidence of oscillations both in cytosolic ATP55,72 and KATP channel conductance during glucose stimulation,103 which supports this hypothesis. However, others have reported the persistence of electrical activity in β-cells that lack functional KATP channels,104 possibly indicating that the modulation of KATP channel conductance by the ATP/ADP ratio is not the only pacemaker mechanism for bursting electrical activity. In addition, Ravier et al.105 have suggested that KATP channels are not the only mechanism linking glucose metabolism with Ca2+-dependent insulin release via changes in membrane potential. The models based on the oscillations of the ATP/ADP ratio to produce bursting electrical activity by regulating the conductance of the KATP channels are unable to reproduce these observations, although it should be noted that the identity of the mechanism driving bursting electrical activity in KATP deficient β-cells is still unclear.
It has been proposed that ATP-consuming processes activated due to an increase of [Ca2+]i (e.g., Ca2+-pumps) could be the origin of the observed oscillations in cytosolic ATP.26,55,78,106 Recent ATP measurements in the sub-membrane compartment in β-cells suggest that Ca2+ extrusion mechanisms are responsible for the observed oscillations in ATP,55 giving support to this proposal. Whether the changes in the conductance of the KATP channels are mediated by the influence (negative or positive) of Ca2+ in ATP production or by the interplay between ATP production and consumption is still a matter of debate. Other complex models that include a detailed description of glucose metabolism were developed later,77,107,108 though based on the hypothesis of intrinsic glycolytic oscillations as the origin of the oscillatory behavior of β-cells (described below).
In contrast to the fast oscillations observed by Dean and Mathews,85,86 Smith et al.109 reported slow bursting activity with a periodicity of minutes. In order to explain the origin of the slow oscillations observed experimentally in single cells, clusters of β-cells, and isolated islets,26,88,109 Bertram et al.110 and Chay et al.111 included the endoplasmic reticulum (ER) as a second Ca2+ compartment in β-cell models (Fig. 5A). As observed experimentally, in these models, Ca2+ is transported into the ER by the SERCA pumps during the active phase of the electrical activity and is released during the silent phase, mainly through the IP3 receptor channels and the ryanodine receptor channels.41-43 One important aspect of these models is the presence of non-specific calcium release-activated currents (CRAC) in the β-cells. The main idea (depicted schematically in Fig. 5A) is that during the silent phase, Ca2+ is slowly released from the ER, preventing an abrupt drop of [Ca2+]i (Figs. 5B and C, bottom panel). As [Ca2+]i is extruded from the cell, the inactivation of the Ca2+-inactivating Ca2+ current is removed. Simultaneously, as the Ca2+ concentration in the ER ([Ca2+]ER) declines, the CRAC current increases. Eventually, the combination of these 2 currents becomes large enough to initiate a new burst. Then, [Ca2+]i is increased, driving the transport of Ca2+ into the ER, promoting inactivation of both the Ca2+ and CRAC currents. Finally, when these currents are sufficiently small, the active phase terminates. In terms of periodicity, models including [Ca2+]ER as a second slow process were able to generate both fast and slow bursting (Figs. 5B and C), in contrast to models that depend on a single slow process (e.g., [Ca2+]i in the CK model or [ADP] in the SK model), which only generated bursting with a periodicity of seconds (fast oscillations).16,112 The period of the oscillations in models including the ER is determined by the release rate of Ca2+ from the ER. When the release rate is low, [Ca2+]ER reaches a high level during the active phase, and because Ca2+ is released from the ER slowly, [Ca2+]i stays elevated (thus making the Ca2+-dependent Ca2+ channels inactive), preventing the initiation of a new burst of action potentials. By including the ER, it was possible to simulate the effects of muscarinic agonists (e.g., acetylcholine) in the electrical activity of β-cells, which are known to mediate Ca2+ release from the ER.113
Figure 5.
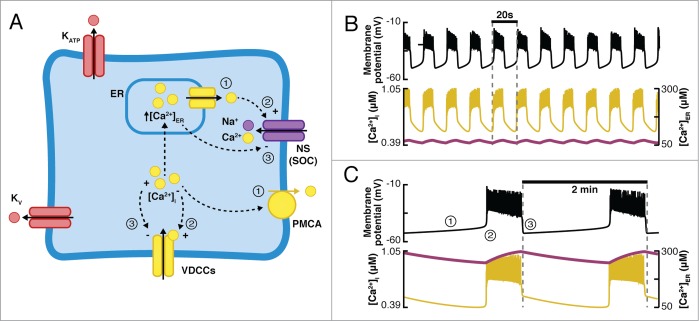
(A) Diagram of the models including ER as a second Ca2+ compartment and a non-specific calcium release-activated current (CRAC). During the silent phase (1), Ca2+ is released from the ER to the cytoplasm and is simultaneously extruded from the cell. This results in the activation of the CRAC current and the Ca2+-inactivated Ca2+ current, driving slow depolarization and initiation of a burst of action potentials (2). As [Ca2+]i increases and Ca2+ is captured by the ER during the active phase, both the CRAC and the Ca2+-inactivating Ca2+ currents are inhibited, resulting in membrane repolarization (3). (B and C) Simulations using the model of Chay111 including ER. Fast (B) and slow (C) bursting is produced by modifying the release rate of Ca2+ from the ER. In both cases, Vm (top, black curve), [Ca2+]i, and [Ca2+]ER (bottom, yellow and purple curves, respectively) are shown.
Other authors have proposed alternative mechanisms to explain the differences in the periodicity of bursting. Bertram et al.25 developed a model based on the idea that the periodicity of bursting is determined by the interaction between a fast and a slow oscillatory variables. These models are capable of producing bursting with an intermediate period, distinct from the periods of the fast and slow variables. Because of this behavior, the models based on this principle are called phantom bursters. In addition, models using the phantom bursting mechanism can also produce fast and slow bursting, mediated entirely by fast and slow variables, respectively. Actually, the models described above that included the ER for the first time110,111 are phantom bursters, though they were identified as such later (see ref. 16), after the appearance of phantom bursting proposal.25 The identity of the fast and slow processes has been extensively investigated by means of mathematical models (see below).
With the discovery of a slow KCa current (TEA and charybdotoxine-insensitive) by Gopel et al.,114 the feedback of Ca2+ onto the KCa channels returned as a feasible candidate mechanism responsible for the periodicity of bursting activity. This was explored theoretically by Goforth et al.115 Simulations by Fridlyand et al.26 support the idea that bursting with a periodicity of seconds could be driven by the Ca2+-dependent K+ current. However, this remains to be established experimentally.
Fridlyand et al.106 proposed Na+ concentration ([Na+]) as an alternative slow mechanism (Fig. 6A). This model includes components that regulate the dynamics of Na+ in β-cells, namely the Na+/Ca2+ exchanger (NCX) and the Na+/K+ pump. They suggested that the increase of [Ca2+]i during the active phase drives Na+ influx through the NCX exchanger, provoking a slow increase in [Na+]i (Fig. 6B). This activates the Na+/K+ pump, carrying the net outward current responsible for burst repolarization. In the course of the silent phase, [Na+]i decreases due to a reduction in the activity of the NCX exchanger, leading to the inhibition of the outward current generated by the Na+/K+ pump and membrane depolarization. Eventually, a new burst is initiated and the cycle is repeated. Other slow processes were also considered (i.e., ADP, IP3, [Ca2+]ER). This model was later extended in order to include more detailed models for the interactions between [Ca2+]i, ATP/ADP, conductance of the KATP channels, and consumption of oxygen and glucose.116 It is important to note that in these models [Ca2+]i shows a sawtooth like behavior that is followed by both [Na+]i and the INa+/K+ current (see Fig. 6B). As mentioned before, experiments29,30 have shown a more square shaped time course of [Ca2+]i resembling the behavior of Vm. The model of Fridlyand et al.106,116 is capable of generating square-shaped oscillations in [Ca2+]i by modifying certain parameters (e.g., decreasing the rate of IP3 synthesis, see Fig. 3 in ref. 106) or by fixing other slow variables (e.g., [ATP]i, [Na+]i and [IP3]i) to a constant value (see Fig. 6 in ref. 106). The role of [Na+]i in β-cells has not been sufficiently studied. However, there is evidence of occasional oscillations of [Na+]i in mouse β-cells, which can be associated with Ca2+ influx and the periodic activation of the NCX exchanger.117 To our knowledge, simultaneous measurements of Vm, [Ca2+]i, and [Na+]i in β-cells have not been performed, which could clarify the role of Na+ in GSIS. The framework of the models of Fridlyand et al.106,116 was used by Cha et al.118 to analyze the contribution of the ionic channels involved in the GSIS in the distinct electric behaviors observed at different glucose levels. The authors concluded that the KATP channels mediate bursting at the physiological range of glucose. In addition, their simulations predicted that at higher glucose levels, the role of the KATP channels becomes practically negligible, as the electrogenic transport mechanisms (i.e. PMCA, NCX and Na+/K+ pump), together with a nonselective current, become more important for the regulation of bursting. Cha et al.119 further identified the fast ([ATP]i or the inactivation gate of the Ca2+ current) and slow ([Na+]i or [Ca2+]ER) processes in their model as defined by the phantom bursting mechanism.
Figure 6.
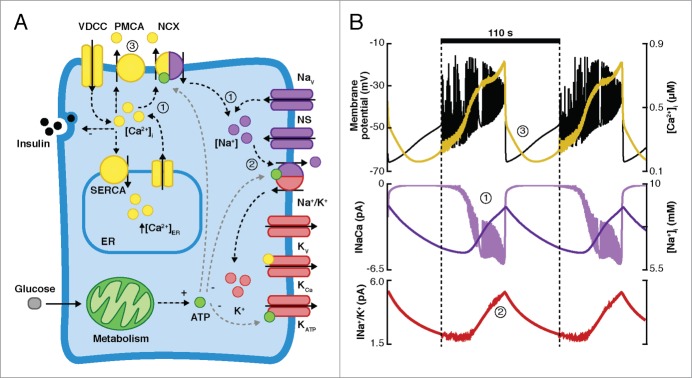
[Na+]i as a pacemaker variable. (A) The model of Fridlyand et al.106 is shown schematically. Entry of Ca2+ during the active phase activates the Na+/Ca2+ exchanger, inducing an increase of [Na+]i (1). This promotes the activity of an outward current through the Na+/K+ pump, eventually repolarizing the membrane (2). In the silent phase, Ca2+ influx is inhibited, resulting in a reduction in both the activity of the NCX exchanger and the Na+/K+ pump, promoting slow depolarization (3). (B) Simulation of slow electrical activity. Top: Vm (black curve) and [Ca2+]i (yellow curve). Middle: Current through the NCX exchanger (INaCa, light purple) and [Na+]i (dark purple). Bottom: Current through the Na+/K+ pump (INa+/K+, red curve).
Bertram and Sherman16 proposed a model using the phantom bursting mechanism with 3 slow processes, [Ca2+]i, [Ca2+]ER, and ATP/ADP. Using a simple representation of these mechanisms, this model was able to reproduce several experimental findings, including the effects of acetylcholine and thapsigargin on electrical activity and the full range of periods of bursting. In a later model, called the Dual Oscillator Model (DOM, Fig. 7A), Bertram et al.120 combined a model of glycolysis,76 a model of mitochondrial metabolism,100,101 and a model of electrical activity.121 The DOM model reproduces the full range of periods observed in bursting activity as well as the compound or mixed oscillations that are often observed (shown in Fig. 7D). In the DOM model, slow bursting is mediated by the glycolytic oscillations driving changes in the production of ATP and the conductance of the KATP channels (Fig. 7B). On the other hand, fast bursting depends entirely on the electrical component (Fig. 7C). Finally, compound bursting is driven by both the electrical and glycolytic components (Fig. 7D). In the DOM model, the glycolytic oscillations are mediated by the feedback of the product FBP onto the PFK reaction. Although this hypothesis has been questioned,107,108 in recent years, some of the predictions of the DOM model have acquired experimental support. For example, oscillations in the membrane conductance of mouse β-cells were associated with changes in the conductance of KATP channels due to intrinsic metabolic oscillations and not because of oscillations produced by the effects of Ca2+ in the production of ATP.122 Moreover, direct experimental evidence of oscillations in the glycolytic pathway123,124 have recently been presented. In addition, it is important to mention that the DOM model is the only model capable of reproducing other recent experimental observations. For instance, Merrins et al.125 showed that in some cells, metabolic oscillations persisted in the absence of Ca2+ oscillations, while in the majority of cells the metabolic oscillations were abolished. In the latter case, it was possible to restore metabolic oscillations by a non-oscillating elevation of [Ca2+]i (i.e by depolarizing with KCl). The DOM model reproduces these observations125 given that Ca2+ oscillations are not needed by the model to produce metabolic oscillations. Moreover, based on their simulations with a reduced version of the DOM model, the authors have proposed that the distinct behaviors mentioned above could be mediated by different rates of the enzyme glucokinase among the cells.126 In contrast, in other models (e.g., the models of Fridlyand et al.,106,116 Keizer and Magnus99 and Diederichs107,108), metabolic oscillations are secondary to Ca2+ oscillations, thus membrane hyperpolarization (i.e., preventing Ca2+ influx) and a fixed [Ca2+]i, mandatorily abolishes metabolic oscillations. It is worth noting that, as in the case of the models based on the cyclical changes in the conductance of the KATP channels as the mechanism underlying bursting electrical activity, the DOM model is not able to explain the origin of the oscillations in Vm and [Ca2+]i observed in β-cells lacking functional KATP channels.104,105
Figure 7.

Intrinsic metabolic oscillations (DOM model). (A) Diagram of the DOM model. The interactions between glycolytic, metabolic, and electrical components drive different electrical behaviors (simulations shown in B–D) depending on the regime of the glycolytic and electrical components. Glucose is metabolized by the glycolytic and metabolic components controlling the production of ATP, which mediate the changes in the conductance of the KATP channels, depolarization, and Ca2+ influx. The 3 compartments (glycolytic, electrical, and metabolic) are affected by the changes in [Ca2+]i. (B) Slow bursting is produced entirely by oscillatory glycolysis. (C) Fast bursting produced by the electrical component. (D) The combination of glycolytic and electrical components produces compound bursting activity. (B–D) Top: Vm (black curve) and the state of glycolysis (represented by F6P, orange curve). Bottom: [Ca2+]i (yellow curve) and [ATP]i (green curve).
Other models of the rodent β-cell127,128 have focused on the role of the ionic channels and transport mechanisms in the glucose induced electrical activity by including a more complete description of the electrophysiological properties of the cell. In fact, recent proposals17,98 of the potential role of the different ionic currents in the electrical activity of the mouse β-cell involves the participation of several ionic transport mechanisms. In order to test the plausibility of this proposal by means of a computational model, an accurate and complete representation of all the mechanisms involved must be included.
Models of human β-cells
All the models described so far have been built based on rodent experimental data, assuming that these are a reasonable model for the human β-cell. However, it has been shown that there are several important differences between species at different levels, including, for example, the proportions and distribution of the different cells in the islets of Langerhans,129,130 the glucose threshold at which insulin starts to be secreted,131,132 the kinetics of insulin exocytosis,133 and the ionic channels expressed and their role in electrical activity and insulin secretion.10,20,22 Human β-cells have ATP-dependent K+ channels; T, L, and P/Q-type Ca2+ channels; voltage-gated Na+ channels; large and small conductance Ca2+-activated K+ channels (SK and BK respectively); inwardly rectifying and delayed rectifier K+ channels; HERG K+ channels; and transient receptor potential (TRP) channels.10,22,24 Interestingly, in contrast to rodent cells, the most frequently observed electrical patterns in human β-cells consist of single action potential firing or fast bursting,10,24,27 although slow bursting has been recently reported.28
Based on these differences, mathematical models of human β-cells have recently been developed. Pedersen27 built the first mathematical model based entirely on electrophysiological data from human β-cells.22,23,134,135 A limitation of this model is the absence of Ca2+ dynamics, metabolism, and SK channels, considering only the interaction between ionic channels. On the other hand, Fridlyand et al.24 also proposed a model based on human data, but in contrast to Pedersen's model, their model included Ca2+ dynamics (although based on mouse experimental data), the SK current, and a minimal model of insulin secretion. Despite their limitations, several experimental observations can be reproduced using these models, like the firing of action potentials, fast bursting, and the effect of channel blockers in electrical activity.
Recently, Riz et al.28 added the SK channels and Ca2+ dynamics to Pedersen's model of the human β-cell (Fig. 8A). Specifically, a cytosolic and a sub-membrane Ca2+ compartment were included. Besides the action potential firing (Fig. 8B) and fast bursting (Fig. 8C) produced by the sub-membrane Ca2+-feedback onto the SK channels (resembling the mechanism of the CK model), this model reproduced slow bursting activity (Fig. 2B and Fig. 8D) due to the addition of a slow glycolytic component that drives changes in ATP and the conductance of the KATP channels.
Figure 8.
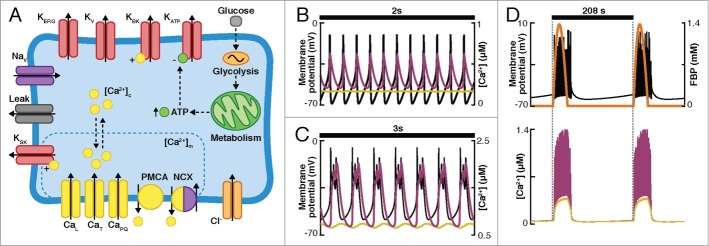
(A) Diagram of the mechanisms included in the model of Riz et al. of human β-cells.28 Channels included in the model: ATP-dependent K+ channels (KATP), big and small conductance Ca2+-dependent K+ channels (KBK and KSK), voltage-dependent K+ channels (Kv), HERG-K+ channels (KERG), voltage-dependent Na+ channels (Nav), L, T and P/Q-type Ca2+ channels (CaL, CaT, CaPQ, respectively), Cl− channels (representing the current mediated by the neurotransmitter γ-aminobutiric acid, GABA). The Ca2+ dynamics included a cytoplasmic and a submembrane compartment and the plasma membrane Ca2+-ATPase (PMCA) and Na+/Ca2+ exchanger (NCX). (B–D) Simulations of Vm (black curve), submembrane Ca2+ (pink curve), intracellular Ca2+ (yellow curve), and glycolysis (FBP, orange curve) are shown. (B) Action potential firing. (C) Fast bursting. (D) Slow bursting.
It is evident that models of human β-cells are in an early stage compared to models of rodent β-cells. However, the former are likely to evolve rapidly and contribute to the understanding of the pathogenesis of T2D and other related diseases.
Discussion
Insulin-secreting β-cells have been intensively studied in the last decades, both experimentally and theoretically. In this review, we have described the main hypotheses behind the mathematical models of β-cells from a physiological viewpoint. It has been shown how models have evolved and grown in complexity as experimental evidence has emerged. Although models have contributed to a better understanding of the GSIS at the cellular level, there are still several open questions. One of the most important is to elucidate the origin of the heterogeneous oscillations observed in β-cells when exposed to stimulatory concentrations of glucose. This has been one of the main objectives of the models of β-cells. In a recent review, Fridlyand et al.26 analyzed both the experiments and the mathematical models in order to identify possible cellular mechanisms behind these different behaviors. They concluded that a single mechanism is not capable of generating all the electrical behaviors, but that each of these behaviors could be driven by a different mechanism. In contrast, Bertram et al.77,136 have proposed that different regimes of a single mechanism composed by the interacting glycolytic, electrical, and mitochondrial components (DOM model) can explain the variety of behaviors observed in β-cells from rodents. The latter proposal has received both indirect and direct evidence (as discussed above). In our opinion, given the experimental support it has acquired, the DOM model is currently the most comprehensive mathematical model in terms of both the experimental observations it can reproduce and the cellular mechanisms that it includes. Merrins et al.123 have developed a technique to measure glycolytic oscillations, which opens the door to the possibility of testing the validity of the assumptions and predictions of the DOM model experimentally. In fact, using this novel technique, the authors presented convincing evidence that glycolytic oscillations are in phase with the mitochondrial redox potential,123 which was also predicted by the DOM model.77
Another important question is how the differences between rodent and human β-cells affect the secretion of insulin. As mentioned above, most of the models are based on experimental data from rodent β-cells, while models for human β-cells were only recently developed. Models of rodent β-cells have achieved a high level of complexity; to such an extent that detailed mathematical descriptions of glucose metabolism and Ca2+-handling have already been incorporated. On the other hand, mathematical models of human β-cells are still incomplete because of the lack of sufficient experimental data. In this regard, detailed measurements of intracellular ionic concentrations and metabolic variables would be extremely helpful to extend the current models and simulate the human β-cell more accurately. In spite of these limitations, significant and substantial progress has been made recently, by identifying the possible role of the ionic channels in the generation of action potentials firing and fast bursting,24,27 and the possible participation of metabolism in slow bursting behavior.28
In the beginning, mathematical models of the electrical activity of pancreatic β-cells were devoted to finding plausible explanations for experimental observations. However, interesting applications have been given to these models in order to use them in more realistic and complex scenarios. Some of the models have been extended to study the dynamics of insulin granule exocytosis. For example, Pedersen et al.137 used a model of the electrical activity of the human β-cell27 along with a compartmental description of Ca2+ dynamics and insulin exocytosis to evaluate the contribution of the different Ca2+ channels during exocytosis.
Models of β-cells have also been useful for investigating the importance of β-cell coupling in the islets of Langerhans, given that it has been proposed that in order to obtain proper insulin secretion in response to a glucose stimulus, the secretion of the β-cells must be synchronized (intra-islet synchronization). This has been tested theoretically, assuming there is electrical coupling between β-cells through gap junctions within the islets of Langerhans.138,139 Similarly, mathematical models have been used to identify possible mechanisms for islet synchronization (inter-islet synchronization).136,138 In a recent review, Han et al.140 described how mathematical models have been used to study the effect of both β-cell interconnection through gap-junctions and paracrine interactions between islet cells.
Another of the aspects recently explored is the inclusion of models of β-cells in multiscale models. For example, Chew et al.141 coupled the Dual Oscillator Model to a model that describes the whole-body glucose regulation system during an oral glucose tolerance test. The aim of this model was to study the changes in the electrical pattern of β-cells due to real changes in blood glucose concentration, as opposed to the models of single β-cells, in which glucose is assumed to be in steady state. It would be interesting to adopt this multiscale approach using a model of human β-cells, such that differences between species are considered.
Given that β-cell dysfunction is implicated in the pathogenesis of T2D, it is likely that mathematical models of human β-cells will evolve rapidly as more experimental data become available. It is also expected that all this progress in the field of mathematical models of β-cells will contribute to the design of new therapies for treating diseases related to the glucose-insulin regulatory system, like T2D. For instance, it has been suggested that mathematical models of β-cells could establish the principles of design for engineered cells capable of sensing glucose and secreting insulin.112,142
Considering the importance of the changes in [Ca2+]i in GSIS, it is surprising that the spatial aspects have not been explicitly considered in the models of β-cells. We think that a necessary extension to the models is the inclusion of a more realistic description of the spatiotemporal distribution of [Ca2+]i, such as its effects on the different cellular processes (e.g., regulation of ionic channels, metabolism, insulin exocytosis) occurring at different locations of the intracellular space are adequately simulated.
For several reasons, mathematical modeling is limited by unavoidable simplifications and assumptions at different levels. For example, when the CK model89 appeared, detailed information about the cellular mechanisms involved in the electrical activity of the β-cell was lacking, which was reflected in the simplicity of the model. The same can be said about the model of the human β-cell of Pedersen,27 given that the number of studies on human β-cells is scarce in comparison to those of rodent cells, perhaps because of the limited availability of human tissue. However, these minimal models have served as a starting point for further development. It is important to note that as more pieces of experimental evidence have emerged, models have been modified consequently. This can be seen for example in the evolution of the models of the different groups (e.g., Chay,89,94,111 Fridlyand et al.26,106,116 and Bertram et al.16,25,77), that have been extended progressively.
Most of the models reviewed in this work have been built in order to reproduce specific experimental observations at the cellular level, aiming to propose plausible hypotheses that explain the origin of the phenomenon under study. This kind of models (often referred to as "whole cell models")143 are constructed by combining individual models of each cellular process considered (e.g., ionic channels, Ca2+ handling, metabolism), hence simplifications and/or assumptions can be made in each of the individual models depending on the objective of the study. It can be said that the majority of the models attempt to capture the qualitative, rather than the quantitative aspects of the functioning of the β-cells. In our opinion, most of the simplifications and assumptions are understandable given the complexity of the system, as long as the implications of the resulting simulations, whether hypotheses or predictions, are bounded accordingly.
Funding
G. Félix-Martínez was supported by a graduate scholarship (No. 228853) from CONACYT (Mexican Council of Science and Technology).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Matthews DR, Lang DA, Burnett MA, Turner RC. Control of pulsatile insulin secretion in man. Diabetologia 1983; 24:231-7; PMID:6345247; http://dx.doi.org/ 10.1007/BF00282705 [DOI] [PubMed] [Google Scholar]
- 2. Hellman B. Pulsatility of insulin release – a clinically important phenomenon. Ups J Med Sci 2009; 114:193-205; PMID:19961265; http://dx.doi.org/ 10.3109/03009730903366075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lang DA, Matthews DR, Burnett M, Turner RC. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 1981; 30:435-9; PMID:7014311; http://dx.doi.org/ 10.2337/diab.30.5.435 [DOI] [PubMed] [Google Scholar]
- 4. Michael DJ, Xiong W, Geng X, Drain P, Chow RH. Human insulin vesicle dynamics during pulsatile secretion. Diabetes 2007; 56:1277-88; PMID:17317765; http://dx.doi.org/ 10.2337/db06-0367 [DOI] [PubMed] [Google Scholar]
- 5. Henquin JC, Ravier MA, Nenquin M, Jonas JC, Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. Eur J Clin Invest 2003; 33:742-50; PMID:12925032; http://dx.doi.org/ 10.1046/j.1365-2362.2003.01207.x [DOI] [PubMed] [Google Scholar]
- 6. Henquin JC. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in β-cells. Diabetes Res Clin PR 2011; 93:S27-31; http://dx.doi.org/ 10.1016/S0168-8227(11)70010-9 [DOI] [PubMed] [Google Scholar]
- 7. Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 2009; 52:739-51; PMID:19288076; http://dx.doi.org/ 10.1007/s00125-009-1314-y [DOI] [PubMed] [Google Scholar]
- 8. Basu A, Pedersen MG, Cobelli C. Prediabetes: Evaluation of β-cell function. Diabetes 2012; 61:270-1; PMID:22275083; http://dx.doi.org/ 10.2337/db11-1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bennett K, James C, Hussain K. Pancreatic β-cell KATP channels: Hypoglycaemia and hyperglycaemia. Rev Endocr Metab Disord 2010; 11:157-63; PMID:20878482; http://dx.doi.org/ 10.1007/s11154-010-9144-2 [DOI] [PubMed] [Google Scholar]
- 10. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 2013; 75:155-79; PMID:22974438; http://dx.doi.org/ 10.1146/annurev-physiol-030212-183754 [DOI] [PubMed] [Google Scholar]
- 11. Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, Zhang E, Almgren P, Ladenvall C, Axelsson AS, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 2012; 61:1726-33; PMID:22492527; http://dx.doi.org/ 10.2337/db11-1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012; 52:28-35; PMID:22502861; http://dx.doi.org/ 10.1016/j.ceca.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanat M, Mari A, Norton L, Winnier D, DeFronzo RA, Jenkinson C, Abdul-Ghani MA. Distinct β-cell defects in impaired fasting glucose and impaired glucose tolerance. Diabetes 2012; 61:447-53; PMID:22275086; http://dx.doi.org/ 10.2337/db11-0995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rinzel J. Bursting oscillations in an excitable membrane model. In: Sleeman BD, Jarvis RJ, ed. Lecture Notes in Mathematics, Vol. 1151 Berlin: Springer Heidelberg; 1985:pp. 304-16 [Google Scholar]
- 15. Bertram R, Sherman A. Negative calcium feedback: the road from Chay-Keizer. In: Combes S, Bressloff PC, editors. Bursting: The Genesis of Rhythm in the Nervous System. Singapore: World Scientific; 2005:pp. 19-48 [Google Scholar]
- 16. Bertram R, Sherman A. A calcium-based phantom bursting model for pancreatic islets. B Math Biol 2004; 66:1313-44; http://dx.doi.org/ 10.1016/j.bulm.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 17. Drews G, Krippeit-Drews P, Düfer M. Electrophysiology of islet cells. Adv Exp Med Biol 2010; 654:115-63; PMID:20217497; http://dx.doi.org/ 10.1007/978-90-481-3271-3_7 [DOI] [PubMed] [Google Scholar]
- 18. Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Prog Biophys Molec Biol 1989; 54:87-143; http://dx.doi.org/ 10.1016/0079-6107(89)90013-8 [DOI] [PubMed] [Google Scholar]
- 19. Meissner H, Schmelz H. Membrane potential of beta-cells in pancreatic islets. Pflugers Arch 1974; 351:195-206; PMID:4608967; http://dx.doi.org/ 10.1007/BF00586918 [DOI] [PubMed] [Google Scholar]
- 20. Hiriart M, Aguilar-Bryan L. Channel regulation of glucose sensing in the pancreatic β-cell. Am J Physiol Endocrinol Metab 2008; 295:E1298-306; PMID:18940941; http://dx.doi.org/ 10.1152/ajpendo.90493.2008 [DOI] [PubMed] [Google Scholar]
- 21. Ashcroft FM, Rorsman P. KATP channels and islet hormone secretion: new insights and controversies. Nat Rev Endocrinol 2013; 9(11):660-69; PMID:24042324; http://dx.doi.org/ 10.1038/nrendo.2013.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic β-cells: electrophysiological characterization and role in insulin secretion. Diabetes 2008; 57:1618-28; PMID:18390794; http://dx.doi.org/ 10.2337/db07-0991 [DOI] [PubMed] [Google Scholar]
- 23. Misler S, Barnett DW. Electrophysiology of stimulus-secretion coupling in human β-cells. Diabetes 1992; 41:1221-8; PMID:1397696; http://dx.doi.org/ 10.2337/diab.41.10.1221 [DOI] [PubMed] [Google Scholar]
- 24. Fridlyand LE, Jacobson DA, Philipson LH. Ion channels and regulation of insulin secretion in human β-cells: a computational systems analysis. Islets 2013; 5:1-15; PMID:23624892; http://dx.doi.org/ 10.4161/isl.24166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertram R, Previte J, Sherman A, Kinard TA, Satin LS. The phantom burster model for pancreatic β-cells. Biophys J 2000; 79:2880-92; PMID:11106596; http://dx.doi.org/ 10.1016/S0006-3495(00)76525-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fridlyand LE, Tamarina N, Philipson LH. Bursting and calcium oscillations in pancreatic β-cells: specific pacemakers for specific mechanisms. Am J Physiol Endocrinol Metab 2010; 299:E517-32; PMID:20628025; http://dx.doi.org/ 10.1152/ajpendo.00177.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedersen MG. A biophysical model of electrical activity in human β-cells. Biophys J 2010; 99:3200-7; PMID:21081067; http://dx.doi.org/ 10.1016/j.bpj.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riz M, Braun M, Pedersen MG. Mathematical modeling of heterogeneous electrophysiological responses in human β-cells. PLoS Comp Biol 2014; 10(1):e1003389; http://dx.doi.org/ 10.1371/journal.pcbi.1003389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Soria B, Valdeolmillos M. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflugers Arch 1991; 418:417-22; PMID:1876486; http://dx.doi.org/ 10.1007/BF00550880 [DOI] [PubMed] [Google Scholar]
- 30. Valdeolmillos M, Santos RM, Contreras D, Soria B, Rosario LM. Glucose-induced oscillations of intracellular Ca2+ concentration resembling bursting electrical activity in single mouse islets of Langerhans. FEBS Lett 1989; 259:19-23; PMID:2689228; http://dx.doi.org/ 10.1016/0014-5793(89)81484-X [DOI] [PubMed] [Google Scholar]
- 31. Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet β-cells: evidence for roles in the generation of action potentials and insulin secretion. Pflug Arch Eur J Phy 1995; 431:272-82; http://dx.doi.org/ 10.1007/BF00410201 [DOI] [PubMed] [Google Scholar]
- 32. Pressel DM, Misler S. Sodium channels contribute to action potential generation in canine and human pancreatic islet B cells. J Membrane Biol 1990; 116:273-80; http://dx.doi.org/ 10.1007/BF01868466 [DOI] [PubMed] [Google Scholar]
- 33. Gilon P, Henquin J. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. J Biol Chem 1992; 267(29), 20713-20; PMID:1400388 [PubMed] [Google Scholar]
- 34. Ammälä C, Eliasson L, Bokvist K, Larsson O, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J Physiol 1993; 472:665-88; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilon P, Shepherd RM, Henquin JC. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J Biol Chem 1993; 22275-68 [PubMed] [Google Scholar]
- 36. Hellman B, Gylfe E, Bergsten P, Grapengiesser E, Lund PE, Berts A, Tengholm A, Pipeleers DG, Ling Z. Glucose induces oscillatory Ca2+ signalling and insulin release in human pancreatic beta cells. Diabetologia 1994; 37:S11-20; PMID:7821725; http://dx.doi.org/ 10.1007/BF00400821 [DOI] [PubMed] [Google Scholar]
- 37. Chen L, Koh D-S, Hille B. Dynamics of calcium clearance in mouse pancreatic β-cells. Diabetes 2003; 52(7), 1723-31; PMID:12829639; http://dx.doi.org/ 10.2337/diabetes.52.7.1723 [DOI] [PubMed] [Google Scholar]
- 38. Herchuelz A, Kamagate A, Ximenes H, van Eylen F. Role of Na/Ca exchange and the plasma membrane Ca2+-ATPase in β-Cell function and death. Ann NY Acad Sci 2007; 1099:456-67; PMID:17446486; http://dx.doi.org/ 10.1196/annals.1387.048 [DOI] [PubMed] [Google Scholar]
- 39. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003; 4 (7):517-29; PMID:12838335; http://dx.doi.org/ 10.1038/nrm1155 [DOI] [PubMed] [Google Scholar]
- 40. Arredouani A, Henquin JC, Gilon P. Contribution of the endoplasmic reticulum to the glucose-induced [Ca2+]c response in mouse pancreatic islets. Am J Physiol Endocrinol Metab 2002; 282:E982-91; PMID:11934662 [DOI] [PubMed] [Google Scholar]
- 41. Gilon P. Uptake and release of Ca2+ by the endoplasmic reticulum contribute to the oscillations of the cytosolic Ca2+ concentration triggered by Ca2+ influx in the electrically excitable pancreatic B-cell. J Biol Chem 1999; 274:20197-205; PMID:10400636; http://dx.doi.org/ 10.1074/jbc.274.29.20197 [DOI] [PubMed] [Google Scholar]
- 42. Tamarina NA, Kuznetsov A, Rhodes CJ, Bindokas VP, Philipson LH. Inositol (1,4,5)-trisphosphate dynamics and intracellular calcium oscillations in pancreatic β-cells. Diabetes 2005; 54:3073-81; PMID:16249428; http://dx.doi.org/ 10.2337/diabetes.54.11.3073 [DOI] [PubMed] [Google Scholar]
- 43. Islam MS. The ryanodine receptor calcium channel of β-cells: molecular regulation and physiological significance. Diabetes 2002; 51:1299-309; PMID:11978625; http://dx.doi.org/ 10.2337/diabetes.51.5.1299 [DOI] [PubMed] [Google Scholar]
- 44. Hellman B, Gylfe E. Mobilization of different intracellular calcium pools after activation of muscarinic receptors in pancreatic beta-cells. Pharmacology 1986; 32:257-67; PMID:3086908; http://dx.doi.org/ 10.1159/000138178 [DOI] [PubMed] [Google Scholar]
- 45. Beauvois MC, Merezak C, Jonas JC. Glucose-induced mixed [Ca2+]c oscillations in mouse β-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am J Cell Physiol 2006; 290:C1503-11; http://dx.doi.org/ 10.1152/ajpcell.00400.2005 [DOI] [PubMed] [Google Scholar]
- 46. Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett 1980; 119:1-8; PMID:7000543; http://dx.doi.org/ 10.1016/0014-5793(80)80986-0 [DOI] [PubMed] [Google Scholar]
- 47. Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab 2006; 290:E143-48; PMID:16144817; http://dx.doi.org/ 10.1152/ajpendo.00216.2005 [DOI] [PubMed] [Google Scholar]
- 48. Magnus G, Keizer J. Minimal model of β-cell mitochondrial Ca2+ handling. Am J Physiol 1997; 273:C717-33; PMID:9277370 [DOI] [PubMed] [Google Scholar]
- 49. Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PloS ONE 2012; 7:e39722; PMID:22829870; http://dx.doi.org/ 10.1371/journal.pone.0039722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Nat Acad Sci U S A 2010; 107:436-41; PMID:20018762; http://dx.doi.org/ 10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maechler P. Mitochondrial function and insulin secretion. Mol Cell Endocrinol 2013; 379:12-8; PMID:23792187; http://dx.doi.org/ 10.1016/j.mce.2013.06.019 [DOI] [PubMed] [Google Scholar]
- 52. Wiederkehr A, Wollheim CB. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic β-cell. Cell Calcium 2008; 44:64-76; PMID:18191448; http://dx.doi.org/ 10.1016/j.ceca.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 53. Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Mol Cell Endocrinol 2012; 353:128-37; PMID:21784130; http://dx.doi.org/ 10.1016/j.mce.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 54. Prentki M, Matschinsky FM, Madiraju SRM. Metabolic signaling in fuel-induced insulin secretion. Cell Metab 2013; 18:162-85; PMID:23791483; http://dx.doi.org/ 10.1016/j.cmet.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 55. Li J, Shuai HY, Gylfe E, Tengholm A. Oscillations of sub-membrane ATP in glucose-stimulated beta cells depend on negative feedback from Ca2+. Diabetologia 2013; 56:1577-86; PMID:23536115; http://dx.doi.org/ 10.1007/s00125-013-2894-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GC, Miyashita Y, Kasai H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc Natl Acad Sci U S A 1999; 96:760-5; PMID:9892707; http://dx.doi.org/ 10.1073/pnas.96.2.760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wiederkehr A, Szanda G, Akhmedov D, Mataki C, Heizmann CW, Schoonjans K, Pozzan T, Spät A, Wollheim CB. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab 2011; 13:601-11; PMID:21531342; http://dx.doi.org/ 10.1016/j.cmet.2011.03.015 [DOI] [PubMed] [Google Scholar]
- 58. Civelek VN, Deeney JT, Shalosky NJ, Tornheim K, Hansford RJ, Prentki M Corkey BE. Regulation of pancreatic β-cell mitochondrial metabolism: influence of Ca2+, substrate and ADP. Biochem J 1996; 318:615-21; PMID:8809055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCormack JG, Longo EA, Corkey BE. Glucose-induced activation of pyruvate dehydrogenase in isolated rat pancreatic islets. Biochem J 1990; 267:527-30; PMID:2185742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 2009; 1787:1309-16; PMID:19413950; http://dx.doi.org/ 10.1016/j.bbabio.2009.01.005 [DOI] [PubMed] [Google Scholar]
- 61. Krippeit-Drews P, Düfer M, Drews G. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem Biophys Res Commun 2000; 267:179-83; PMID:10623595; http://dx.doi.org/ 10.1006/bbrc.1999.1921 [DOI] [PubMed] [Google Scholar]
- 62. Kindmark H, Köhler M, Brown G, Bränström R, Larsson O, Berggren PO. Glucose-induced oscillations in cytoplasmic free Ca2+ concentration precede oscillations in mitochondrial membrane potential in the pancreatic β-cell. J Biol Chem 2001; 276:34530-6; PMID:11445566; http://dx.doi.org/ 10.1074/jbc.M102492200 [DOI] [PubMed] [Google Scholar]
- 63. Brookes PS, Yoon Y, Robotham JL. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004; 287(4):C817-33; PMID:15355853 [DOI] [PubMed] [Google Scholar]
- 64. Pralong WF, Spät A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. J Biol Chem 1994; 269:27310-4; PMID:7961642 [PubMed] [Google Scholar]
- 65. Hellman B, Idahl L-A, Lernmark A, Sehlin J, Taljedal I-B. The pancreatic β-cell recognition of insulin secretagogues. Effects of calcium and sodium on glucose metabolism and insulin release. Biochem J 1974; 138:33-45; PMID:4601168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fridlyand LE, Philipson LH. Glucose sensing in the pancreatic beta cell: a computational systems analysis. Theor Biol Med Model 2010; 7:15; PMID:20497556; http://dx.doi.org/ 10.1186/1742-4682-7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fridlyand LE, Phillipson LH. Mechanisms of glucose sensing in the pancreatic β-cell: A computational systems-based analysis. Islets 2011; 3:224-30; PMID:21814042; http://dx.doi.org/ 10.4161/isl.3.5.16409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maechler P, Kennedy ED, Pozzan T, Wollheim CB. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β-cells. EMBO J 1997; 16:3833-41; PMID:9233793; http://dx.doi.org/ 10.1093/emboj/16.13.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nilsson T, Schultz V, Berggren PO, Corkey BE, Tornheim K. Temporal patterns of changes in ATP/ADP ratio, glucose 6-phosphate and cytoplasmic free Ca2+ in glucose-stimulated pancreatic β-cells. Biochem J 1996; 314:91-4; PMID:8660314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kennedy HJ, Pouli AE, Ainscow EK. Glucose generates sub-plasma membrane ATP microdomains in single islet β-cells. J Biol Chem 1999; 274(19):13281-91; PMID:10224088; http://dx.doi.org/ 10.1074/jbc.274.19.13281 [DOI] [PubMed] [Google Scholar]
- 71. Kennedy RT, Kauri LM, Dahlgren GM, Jung S-K. Metabolic oscillations in β-cells. Diabetes 2002; 51(Suppl 1):S152-61; http://dx.doi.org/ 10.2337/diabetes.51.2007.S152 [DOI] [PubMed] [Google Scholar]
- 72. Ainscow EK, Rutter GA. Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet β-cells: evidence for a Ca2+-dependent mechanism. Diabetes 2002; 51(Suppl 1):S162-70; PMID:11815476; http://dx.doi.org/ 10.2337/diabetes.51.2007.S162 [DOI] [PubMed] [Google Scholar]
- 73. Luciani DS, Misler S, Polonsky KS. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J Physiol 2006; 572:379-92; PMID:16455690; http://dx.doi.org/ 10.1113/jphysiol.2005.101766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jung SK, Aspinwall CA, Kennedy RT. Detection of multiple patterns of oscillatory oxygen consumption in single mouse islets of Langerhans. Biochem Biophys Res Commun 1999; 259:331-5; PMID:10362508; http://dx.doi.org/ 10.1006/bbrc.1999.0784 [DOI] [PubMed] [Google Scholar]
- 75. Tengholm A, Gylfe E. Oscillatory control of insulin secretion. Mol Cell Endocrinol 2009; 297:58-72; PMID:18706473; http://dx.doi.org/ 10.1016/j.mce.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 76. Smolen P. A model for glycolytic oscillations based on skeletal muscle phosphofructokinase kinetics. J Theor Biol 1995; 174:137-48; PMID:7643610; http://dx.doi.org/ 10.1006/jtbi.1995.0087 [DOI] [PubMed] [Google Scholar]
- 77. Bertram R, Satin LS, Pedersen MG, Luciani DS, Sherman A. Interaction of glycolysis and mitochondrial respiration in metabolic oscillations of pancreatic islets. Biophys J 2007; 92:1544-55; PMID:17172305; http://dx.doi.org/ 10.1529/biophysj.106.097154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Detimary P, Gilon P, Henquin J. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem J 1998; 333:269-74; PMID:9657965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tanaka T, Nagashima K, Inagaki N, Kioka H, Takashima S, Fukuoka H, Noji H, Kakizuka A, Imamura H. Glucose-stimulated single pancreatic islets sustain increased cytosolic ATP levels during initial Ca2+ influx and subsequent Ca2+ oscillations. J Biol Chem 2014; 289:2205-16; PMID:24302735; http://dx.doi.org/ 10.1074/jbc.M113.499111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maechler P, Carobbio S, Rubi B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int J Biochem Cell B 2006; 38(5-6), 696-709; http://dx.doi.org/ 10.1016/j.biocel.2005.12.006 [DOI] [PubMed] [Google Scholar]
- 81. Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta 2014; 1840:1404-12; PMID:24246956; http://dx.doi.org/ 10.1016/j.bbagen.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 82. Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Sign 2010; 12:537-77; http://dx.doi.org/ 10.1089/ars.2009.2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, Del Prato S, Rabuazzo AM, Purrello F, Marchetti P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005; 48:282-9; PMID:15654602; http://dx.doi.org/ 10.1007/s00125-004-1627-9 [DOI] [PubMed] [Google Scholar]
- 84. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005; 54(3):727-35; PMID:15734849; / 10.2337/diabetes.54.3.727 [DOI] [PubMed] [Google Scholar]
- 85. Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature 1968; 219:389-90; PMID:4873864; http://dx.doi.org/ 10.1038/219389a0 [DOI] [PubMed] [Google Scholar]
- 86. Dean PM, Matthews EK. Glucose-induced electrical activity in pancreatic islet cells. J Physiol 1970; 210:255-64; PMID:5501259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kinard TA, de Vries G, Sherman A, Satin LS. Modulation of the bursting properties of single mouse pancreatic β-cells by artificial conductances. Biophys J 1999; 76:1423-35; PMID:10049324; http://dx.doi.org/ 10.1016/S0006-3495(99)77303-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gilon P, Ravier MA, Jonas JC, Henquin JC. Control mechanisms of the oscillations of insulin secretion in vitro and in vivo. Diabetes 2002; 51(Suppl 1):S144-51; PMID:11815474; http://dx.doi.org/ 10.2337/diabetes.51.2007.S144 [DOI] [PubMed] [Google Scholar]
- 89. Chay TR, Keizer J. Minimal model for membrane oscillations in the pancreatic β-cell. Biophys J 1983; 42:181-9; PMID:6305437; http://dx.doi.org/ 10.1016/S0006-3495(83)84384-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Atwater I, Rosario L, Rojas E. Properties of the Ca-activated K+ channel in pancreatic β-cells. Cell Calcium 1983; 4:451-61; PMID:6323007; http://dx.doi.org/ 10.1016/0143-4160(83)90021-0 [DOI] [PubMed] [Google Scholar]
- 91. Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive K(Ca) channel is not involved in glucose-induced electrical activity in pancreatic β-cells. J Membrane Biol 1991; 119:187-95; http://dx.doi.org/ 10.1007/BF01871418 [DOI] [PubMed] [Google Scholar]
- 92. Houamed KM, Sweet IR, Satin LS. BK channels mediate a novel ionic mechanism that regulates glucose-dependent electrical activity and insulin secretion in mouse pancreatic β-cells. J Physiol 2010; 588:3511-23; PMID:20643769; http://dx.doi.org/ 10.1113/jphysiol.2009.184341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic β-cells under voltage-clamp conditions. J Physiol 1986; 374:531-50; PMID:2427706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chay TR. The effect of inactivation of calcium channels by intracellular Ca2+ ions in the bursting pancreatic β-cells. Cell Biophys 1987; 11(1):77-90; PMID:2450671; http://dx.doi.org/ 10.1007/BF02797114 [DOI] [PubMed] [Google Scholar]
- 95. Plant TD. Properties and calcium-dependent inactivation of calcium currents in cultured mouse pancreatic B-cells. J Physiol 1988; 404:731-47; PMID:2855352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cook D. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984; 311:271-3; PMID:6090930; http://dx.doi.org/ 10.1038/311271a0 [DOI] [PubMed] [Google Scholar]
- 97. Ashcroft FM, Harrison DE, Ashcroft SJH. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 1984; 312:446-8; PMID:6095103; http://dx.doi.org/ 10.1038/312446a0 [DOI] [PubMed] [Google Scholar]
- 98. Rorsman P, Eliasson L, Kanno T, Zhang Q, Göpel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 2011; 107:224-35; PMID:21762719; http://dx.doi.org/ 10.1016/j.pbiomolbio.2011.06.009 [DOI] [PubMed] [Google Scholar]
- 99. Keizer J, Magnus G. ATP-sensitive potassium channel and bursting in the pancreatic beta cell. A theoretical study. Biophys J 1989; 56:229-42; PMID:2673420; http://dx.doi.org/ 10.1016/S0006-3495(89)82669-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Magnus G, Keizer J. Model of β-cell mitochondrial calcium handling and electrical activity. II. Mitochondrial variables. Am J Physiol Cell Physiol 1998; 274:1158-73; [DOI] [PubMed] [Google Scholar]
- 101. Magnus G, Keizer J. Model of β-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am J Physiol Cell Physiol 1998; 274:1174-84. [DOI] [PubMed] [Google Scholar]
- 102. Smolen P, Keizer J. Slow voltage inactivation of Ca2+ currents and bursting mechanisms for the mouse pancreatic beta-cell. J Membrane Biol 1992; 127:9-19; http://dx.doi.org/ 10.1007/BF00232754 [DOI] [PubMed] [Google Scholar]
- 103. Larsson O, Kindmark H, Brandstrom R, Fredholm B, Berggren PO. Oscillations in KATP channel activity promote oscillations in cytoplasmic free Ca2+ concentration in the pancreatic β cell. Proc Natl Acad Sci U S A 1996; 93:5161-5; PMID:8643546; http://dx.doi.org/ 10.1073/pnas.93.10.5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Düfer M, Haspel D, Krippeit-Drews P, Aguilar-Bryan L, Bryan J, Drews G. Oscillations of membrane potential and cytosolic Ca2+ concentration in SUR1-/- beta cells. Diabetologia 2004; 47:488-98; PMID:14872319; http://dx.doi.org/ 10.1007/s00125-004-1348-0 [DOI] [PubMed] [Google Scholar]
- 105. Ravier MA, Nenquin M, Miki T, Seino S, Henquin JC. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6.2. Endocrinology 2009; 150:33-45; PMID:18787024; http://dx.doi.org/ 10.1210/en.2008-0617 [DOI] [PubMed] [Google Scholar]
- 106. Fridlyand LE, Tamarina N, Philipson LH. Modeling of Ca2+ flux in pancreatic β-cells: role of the plasma membrane and intracellular stores. Am J Physiol Endocrinol Metab 2003; 285:138-54 [DOI] [PubMed] [Google Scholar]
- 107. Diederichs F. Mathematical simulation of membrane processes and metabolic fluxes of the pancreatic β-cell. B Math Biol 2006; 68:1779-818; http://dx.doi.org/ 10.1007/s11538-005-9053-9 [DOI] [PubMed] [Google Scholar]
- 108. Diederichs F. Ion homeostasis and the functional roles of SERCA reactions in stimulus-secretion coupling of the pancreatic β-cell: A mathematical simulation. Biophys Chem 2008; 134:119-43; PMID:18321634; http://dx.doi.org/ 10.1016/j.bpc.2008.02.001 [DOI] [PubMed] [Google Scholar]
- 109. Smith PA, Ashcroft FM, Rorsman P. Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K+-currents in isolated mouse pancreatic β-cells. FEBS Lett 1990; 261:187-90; PMID:2407553; http://dx.doi.org/ 10.1016/0014-5793(90)80667-8 [DOI] [PubMed] [Google Scholar]
- 110. Bertram R, Smolen P, Sherman A, Mears D, Atwater I, Martin F, Soria B. A role for calcium release-activated current (CRAC) in cholinergic modulation of electrical activity in pancreatic β-cells. Biophys J 1995; 2323-32; PMID:7647236; http://dx.doi.org/ 10.1016/S0006-3495(95)80414-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chay TR. Electrical bursting and luminal calcium oscillation in excitable cell models. Biol Cybern 1996; 75:419-31; PMID:8983163; http://dx.doi.org/ 10.1007/s004220050307 [DOI] [PubMed] [Google Scholar]
- 112. Sherman A. Lessons from models of pancreatic beta cells for engineering glucose-sensing cells. Math Biosci 2010; 227:12-9; PMID:20580727; http://dx.doi.org/ 10.1016/j.mbs.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gilon P, Henquin J-C. Mechanisms and physiological significance of the cholinergic control of pancreatic β-Cell function. Endocr Rev 2001; 22(5):565-604; PMID:11588141 [DOI] [PubMed] [Google Scholar]
- 114. Göpel S, Kanno T, Barg S, Eliasson L. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. J Gen Physiol 1999; 114:759-69; PMID:10578013; http://dx.doi.org/ 10.1085/jgp.114.6.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Goforth PB. Calcium-activated K+ Channels of mouse β-cells are controlled by both store and cytoplasmic Ca2+: experimental and theoretical studies. J Gen Physiol 2002; 120:307-22; PMID:12198088; http://dx.doi.org/ 10.1085/jgp.20028581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fridlyand LE, Ma L, Philipson LH. Adenine nucleotide regulation in pancreatic β-cells: modeling of ATP/ADP-Ca2+ interactions. Am J Physiol Endocrinol Metab 2005; 289:E839-48; PMID:15985450; http://dx.doi.org/ 10.1152/ajpendo.00595.2004 [DOI] [PubMed] [Google Scholar]
- 117. Grapengiesser E. Glucose induces cytoplasmic Na+ oscillations in pancreatic β-cells. Biochem Biophys Res Commun 1996; 226:830-5; PMID:8831697; http://dx.doi.org/ 10.1006/bbrc.1996.1436 [DOI] [PubMed] [Google Scholar]
- 118. Cha CY, Nakamura Y, Himeno Y, Wang J, Fujimoto S, Inagaki N, Earm YE, Noma A. Ionic mechanisms and Ca2+ dynamics underlying the glucose response of pancreatic β cells: a simulation study. J Gen Physiol 2011; 138:21-37; PMID:21708953; http://dx.doi.org/ 10.1085/jgp.201110611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cha CY, Santos E, Amano A, Shimayoshi T, Noma A. Time-dependent changes in membrane excitability during glucose-induced bursting activity in pancreatic β cells. J Gen Physiol 2011; 138:39-47; PMID:21708954; http://dx.doi.org/ 10.1085/jgp.201110612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bertram R, Sherman A, Satin LS. Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am J Physiol Endocrinol Metab 2007; 293:E890-E900; PMID:17666486; http://dx.doi.org/ 10.1152/ajpendo.00359.2007 [DOI] [PubMed] [Google Scholar]
- 121. Bertram R, Satin L, Zhang M, Smolen P, Sherman A. Calcium and glycolysis mediate multiple bursting modes in pancreatic islets. Biophys J 2004; 87:3074-87; PMID:15347584; http://dx.doi.org/ 10.1529/biophysj.104.049262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ren J, Sherman A, Bertram R, Goforth PB, Nunemaker CS, Waters CD, Satin LS. Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am J Physiol Endocrinol Metab 2013; 305:E805-17; PMID:23921138; http://dx.doi.org/ 10.1152/ajpendo.00046.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA, Satin LS. Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer(FRET) biosensors for pyruvate kinase M2 activity. J Biol Chem 2013; 288(46):33312-22; PMID:24100037; http://dx.doi.org/ 10.1074/jbc.M113.508127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Merrins MJ, Bertram R, Sherman A, Satin LS. Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase modulates oscillations of pancreatic islet metabolism. PloS ONE 2012; 7:e34036; PMID:22532827; http://dx.doi.org/ 10.1371/journal.pone.0034036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Merrins M, Fendler B, Sherman A. Metabolic oscillations in pancreatic islets depend on the intracellular Ca2+ level but not Ca2+ Oscillations. Biophys J 2010; 99:76-84; PMID:20655835; http://dx.doi.org/ 10.1016/j.bpj.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Watts M, Fendler B, Merrins MJ, Satin LS. Calcium and Metabolic Oscillations in Pancreatic Islets: Who's Driving the Bus?. SIAM J Appl Dyn Syst 2014; 13(2):683-703; http://dx.doi.org/ 10.1137/130920198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Meyer-Hermann ME. The electrophysiology of the β-Cell based on single transmembrane protein characteristics. Biophys J 2007; 93:2952-68; PMID:17573431; http://dx.doi.org/ 10.1529/biophysj.107.106096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fridlyand LE, Jacobson DA, Kuznetsov A, Philipson LH. A model of action potentials and fast Ca2+ dynamics in pancreatic β-cells. Biophys J 2009; 96:3126-39; PMID:19383458; http://dx.doi.org/ 10.1016/j.bpj.2009.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005; 53:1087-97; PMID:15923354; http://dx.doi.org/ 10.1369/jhc.5C6684.2005 [DOI] [PubMed] [Google Scholar]
- 130. Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M. Islet architecture: A comparative study. Islets 2009; 1:129-36; PMID:20606719; http://dx.doi.org/ 10.4161/isl.1.2.9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Harrison DE, Christie MR. Properties of isolated human islets of Langerhans: insulin secretion, glucose oxidation and protein phosphorylation. Diabetologia 1985; 28:99-103; PMID:3884420 [DOI] [PubMed] [Google Scholar]
- 132. Henquin JC, Dufrane D, Nenquin M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes 2006; 55:3470-7; PMID:17130494; http://dx.doi.org/ 10.2337/db06-0868 [DOI] [PubMed] [Google Scholar]
- 133. Braun M, Ramracheya R, Johnson PR, Rorsman P. Exocytotic properties of human pancreatic β-cells. Ann NY Acad Sci 2009; 1152:187-93; PMID:19161389; http://dx.doi.org/ 10.1111/j.1749-6632.2008.03992.x [DOI] [PubMed] [Google Scholar]
- 134. Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, Olivotto M, Wanke E. Glucose-and arginine-induced insulin secretion by human pancreatic β-cells: the role of HERG K+ channels in firing and release. FASEB J 2000; 14:2601; PMID:11099479; http://dx.doi.org/ 10.1096/fj.00-0077com [DOI] [PubMed] [Google Scholar]
- 135. Herrington J, Sanchez M, Wunderler D, Yan L, Bugianesi RM, Dick IE, Clark SA, Brocho RM, Priest BT, Kohler MG, et al. Biophysical and pharmacological properties of the voltage-gated potassium current of human pancreatic β-cells. J Physiol 2005; 567:159-75; PMID:15932888; http://dx.doi.org/ 10.1113/jphysiol.2005.089375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bertram R, Sherman A, Satin LS. Electrical bursting, calcium oscillations, and synchronization of pancreatic islets. Adv Exp Med Biol 2010; 654:261-79; PMID:20217502; http://dx.doi.org/ 10.1007/978-90-481-3271-3_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Pedersen MG, Cortese G, Eliasson L. Mathematical modeling and statistical analysis of calcium-regulated insulin granule exocytosis in β-cells from mice and humans. Prog Biophys Mol Biol 2011; 107:257-64; PMID:21839108; http://dx.doi.org/ 10.1016/j.pbiomolbio.2011.07.012 [DOI] [PubMed] [Google Scholar]
- 138. Pedersen MG, Bertram R, Sherman A. Intra-and inter-islet synchronization of metabolically driven insulin secretion. Biophys J 2005; 89:107-19; PMID:15834002; http://dx.doi.org/ 10.1529/biophysj.104.055681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sherman A, Rinzel J. Model for synchronization of pancreatic β-cells by gap junction coupling. Biophys J 1991; 59:547-59; PMID:1646657; http://dx.doi.org/ 10.1016/S0006-3495(91)82271-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Han K, Kang H, Kim J, Choi M. Mathematical models for insulin secretion in pancreatic β-cells. Islets 2012; 4:94-107; PMID:22627505; http://dx.doi.org/ 10.4161/isl.19569 [DOI] [PubMed] [Google Scholar]
- 141. Chew YH, Shia YL, Lee CT, Majid FAA, Chua LS, Sarmidi MR, Aziz RA. Modeling of oscillatory bursting activity of pancreatic beta-cells under regulated glucose stimulation. Mol Cell Endocrinol 2009; 307:57-67; PMID:19524127; http://dx.doi.org/ 10.1016/j.mce.2009.03.005 [DOI] [PubMed] [Google Scholar]
- 142. Georgiou P, Toumazou C. A silicon pancreatic beta cell for diabetes. IEEE Trans Biomed Circuits Syst 2007; 1:39-49; PMID:23851519; http://dx.doi.org/ 10.1109/TBCAS.2007.893178 [DOI] [PubMed] [Google Scholar]
- 143. Sherman AS, Li YX, Keizer JE. Whole-cell models. In Fall CP, Marland ES, Wagner JM, Tyson JJ, editors. Computational Cell Biology. New York: Springer; 2002. pp. 101-139 [Google Scholar]