

Abstract
We developed a pharmacophore model for type II inhibitors that was used to guide the construction of a library of kinase inhibitors. Kinome-wide selectivity profiling of the library resulted in the identification of a series of 4-substituted 1H-pyrrolo[2,3-b]pyridines that exhibited potent inhibitory activity against two mitogen-activated protein kinases (MAPKs), TAK1 (MAP3K7) and MAP4K2, as well as pharmacologically well interrogated kinases such as p38α (MAPK14) and ABL. Further investigation of the structure–activity relationship (SAR) resulted in the identification of potent dual TAK1 and MAP4K2 inhibitors such as 1 (NG25) and 2 as well as MAP4K2 selective inhibitors such as 16 and 17. Some of these inhibitors possess good pharmacokinetic properties that will enable their use in pharmacological studies in vivo. A 2.4 Å cocrystal structure of TAK1 in complex with 1 confirms that the activation loop of TAK1 assumes the DFG-out conformation characteristic of type II inhibitors.
Introduction
There are approximately 518 kinases encoded in the human genome, and they have been demonstrated to play pivotal roles in virtually all aspects of cellular physiology. Dysregulation of kinase activity through numerous mechanisms results in many pathologies, but therapeutically relevant kinases have been extensively investigated as anticancer agents. Clinically the most successful kinase inhibitors directly target mutationally activated kinases such as BCR-ABL, EML4-ALK, mutant EGFR, B-RAF, and c-KIT that are bonafide oncogenes in the context of tumors that harbor these lesions. Unfortunately, although genome-wide cancer sequencing efforts have uncovered a large number of kinases that are mutated, very few have been biologically validated as being singularly important to tumor function. One challenge to efficient pharmacological validation of new potential kinase targets is the absence of selective inhibitors for the majority of kinases. We sought to develop efficient kinase inhibitor discovery strategies that will enable the identification of inhibitors with sufficient potency and selectivity that they can be used to interrogate the functional and therapeutic consequences of inhibiting less well characterized kinases.
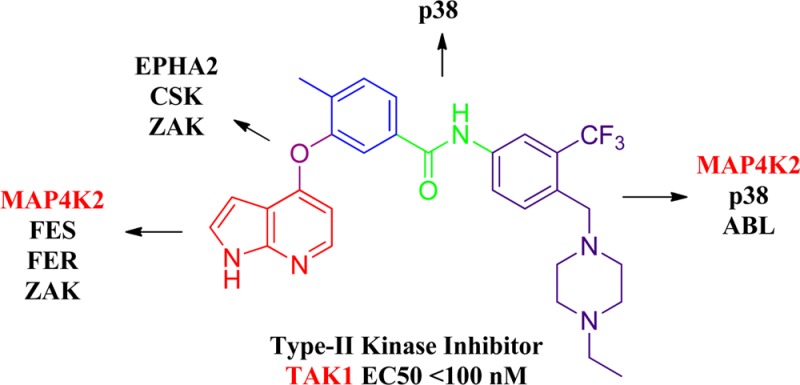
A large majority of known small molecule kinase inhibitors target the ATP binding site with the kinase assuming a conformation otherwise conducive to catalysis (type I inhibitors). A second, broad class of kinase inhibitors (type II) binds to the ATP binding pocket in addition to an adjacent hydrophobic pocket that is created when the activation loop, which contains the conserved DFG motif, is in an “out” conformation. A number of clinically approved inhibitors such as imatinib, nilotinib, and sorafinib have been crystallographically proven to be type II inhibitors of kinases such as ABL, c-KIT, B-RAF, and p38 kinases.1−4 Upon the basis of these crystal structures, we developed a pharmacophore model that defined the structural features needed to access this type II binding conformation (Figure 1b).5 The model posits the need for a “head” heterocyclic motif that occupies the adenosine binding pocket typically making one to three hydrogen bonds to the kinase hinge segment, a linker moiety that traverses the region occupied by the “gatekeeper” residue, a hydrogen bond donor/acceptor motif and a hydrophobic “tail” that occupies the pocket created by the flip of the “DFG” motif of the kinase activation loop (Figure 1b). We constructed a library of potential type II inhibitors based upon this model and performed kinome-wide selectivity profiling in an effort to identify new inhibitors and the kinases that might be susceptible to inhibition by type II inhibitors.6,7 A library of approximately 100 potential type II inhibitors was screened against a panel of over 420 kinases using the KinomeScan approach.3 Two compounds to emerge from this effort are 1 (reported as “NG25” in refs (8) and (9)) and 2 which show binding affinity for TAK1 and MAP4K2 (also known as GCK) (Figure 2). These compounds possess a 4-substituted 1H-pyrrolo[2,3-b]pyridine as the hinge-interacting “head” motif, a 1,3-benzoic acid linker motif inspired by imatinib and a 3-trifluormethylbenzamide “tail” motif inspired by sorafinib/nilotinib.
Figure 1.

General pharmacophore model for the rational design of type II inhibitors. (a) Examples of known type II inhibitors, which can be divided into a “type I” head (black) attached to a “type II” tail (blue). (b) Schematic representation of the rational design of new type II kinase inhibitors: A, hydrogen bond acceptor; D, hydrogen bond donor; HRB, hinge-region binding; HM, hydrophobic motif.
Figure 2.

Chemical structures of “lead” compounds.
Herein, we report the syntheses and target identification of two novel type II kinase inhibitors 1 and 2 of two mitogen activated protein kinases: TAK1 (MAP3K7) and MAP4K2 (GCK). While there have been extensive efforts to target a limited number of MAPKs such as A,B,C-RAF (MAP3Ks), MEK1 (MAP2K1), ERK2 (MAPK1), p38α,β,δ,γ (MAPK14,11,13,12), and JNK1,2,3 (MAPK8,9,10), a majority of other MAPKs have not been subjected to significant inhibitor development efforts. The mitogen activated kinases are typically classified as MAP4Ks (6 members), MAP3Ks (21 members), MAP2Ks (7 members), and MAPKs (14 members) for a total of 49 kinases. TAK1 mediates signaling downstream of multiple cytokine receptors and is functionally important in mitogen, immune, and inflammatory signaling pathways.10,11 Recently several inhibitors of TAK1 have been reported and characterized with respect to their anti-inflammatory and anticancer activity.12−15 The biological functions of MAP4K2 are less well elucidated, but it may be a regulator of NF-κB signaling contributing to cancer development for a subset of malignancies,16,17 and its pharmacological inhibition has been reported to reduce the viability of colon cancer cells.17 MAP4K2 has also been reported to be required to transduce signals from TGFβ receptor to p38.18 Clearly more selective inhibitors of MAP4K2 would be very useful to further elucidate the functions of this kinase. We demonstrate that 1 and 2 and other lead compounds (Figure 2) can inhibit phosphorylation of proteins predicted to be downstream of TAK1 and MAP4K2 such as IKK, p38, and JNK at concentrations of less than 100 nM. Further modification of 1 and 2 resulted in the identification of compounds such as 16 and 17 that exhibit selectivity for MAP4K2 relative to TAK1. Broad kinase selectivity profiling using KiNativ, revealed that a subset of the inhibitors could also target additional kinases such as ABL, ARG, p38, SRC, CSK, FER, FES, and EPH-family kinases, suggesting that these kinases are also susceptible to type II inhibition providing a wealth of potential starting points for further elaborating inhibitors of these kinases.
Results
The synthesis of lead compound 1 is outlined in Scheme 1. First, pyrrolopyridine 27 was coupled with 3-hydroxy-4-methylbenzoic acid using K2CO3 as base. The resulting intermediate was subjected to hydrogenolysis using Pd/C to afford the dechlorinated acid 28 which was then reacted with aniline 29 using HATU/DIEA to provide the desired amide 30. The SEM group of 30 was deprotected under sequential acid and basic conditions to furnish compound 1. This four-step sequence proceeded with high overall yield.
Scheme 1. Synthesis of 3-((1H-Pyrrolo[2,3-b]pyridin-4-yl)oxy)-N-(4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-4-methylbenzamide 1.

Reagents and conditions: (a) 3-hydroxy-4-methylbenzoic acid, K2CO3, DMSO, 100 °C; (b) Pd/C, MeOH; (c) 29, HATU, DMAP, DIEA, CH2Cl2; (d) (i) TFA, CH2Cl2, (ii) LiOH, H2O/THF.
The preparation of compound 2 is outlined in Scheme 2. The iodide intermediate 32 was prepared by reacting aniline 29 and acid 31. Meanwhile, a methoxy group was introduced into aldehyde 33 using sodium methoxide followed by Wittig olefination to afford the terminal olefin 34. The SEM protecting group was replaced with a Boc protecting group to provide compound 35 which was coupled with iodide 32 through Heck reaction to furnish compound 36 in good yield and high trans/cis ratio (>20:1). Deprotection of the Boc protecting group using mildly acidic conditions provided compound 2 with retention of the high E/Z ratio in good overall yield.
Scheme 2. Synthesis of (E)-N-(4-((4-Ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-(2-(4-methoxy-1H-pyrrolo[2,3-b]pyridin-5-yl)vinyl)-4-methylbenzamide 2.

Reagents and conditions: (a) 29, HATU, DMAP, DIEA, CH2Cl2; (b) NaOMe, MeOH, 60 °C; (c) MePPh3I, n-BuLi, THF, −78 °C to rt; (d) (i) TFA, DCM, (ii) LiOH, H2O/THF; (e) Boc2O, DMAP, CH2Cl2; (f) 32, Pd2(dba)3, P(t-Bu)3, DIEA, 80 °C; (g) TFA/CH2Cl2, 0 °C.
Compounds 1 and 2 were screened against a diverse panel of over 420 kinases (DiscoveRX, KinomeScan) using an in vitro ATP-site competition binding assay at a concentration of 10 or 1 μM.6 These two compounds exhibited relatively selective profiles, and for some kinases known to accommodate a type II binding mode, e.g., p38α and ABL, both compounds exhibited potent inhibition. In addition, they also exhibited promising inhibition against several other kinases known to play important roles in cancer-related signaling pathways but whose functions remain uncharacterized because of a lack of highly selective inhibitors, e.g., TAK1, MAP4K2, and EPHA2. To complement the KinomeScan profiling, both compounds were further profiled at 5 and 0.5 μM concentrations on A375 melanoma cells utilizing the KiNativ technology (ActiveX Biosciences).19,20 This live-cell-treatment approach measures the ability of the compounds to protect a subset of kinases in lysates from labeling with a lysine reactive ATP or ADP-biotin probe. Kinases that exhibited moderate or high inhibition of labeling by compounds 1 and 2 are listed in Table 1 (see the Supporting Information for full profiling data). Consistent with the KinomeScan data, at a concentration of 0.5 μM, both compounds inhibited TAK1, MAP4K2, ZAK, p38α, SRC, and LYN while compound 1 also inhibited FER and compound 2 inhibited CSK and EPH-family kinases, respectively. To confirm the observed binding of 1 and 2 to TAK1 and MAP4K2 by the KiNativ approach, we determined the IC50 values using biochemical enzyme assays (Invitrogen, SelectScreen).21 Compounds 1 and 2 inhibited TAK1 with IC50 values of 149 and 41 nM, respectively, and inhibited MAP4K2 with IC50 values of 22 and 98 nM respectively. The in vitro IC50 values of most other targets from KiNativ profiling were also determined and some of them were further confirmed while others were not (Table 1). Compound 1 was also screened at 0.1 μM against another panel of over 100 protein kinases (International Centre for Kinase Profiling, http://www.kinase-screen.mrc.ac.uk/) and gave similar results (Figure S1 in Supporting Information); the IC50 values against TAK1 and MAP4K2 were 15 and 17 nM, respectively, in that assay.
Table 1. Kinase Hits for 1 and 2a.
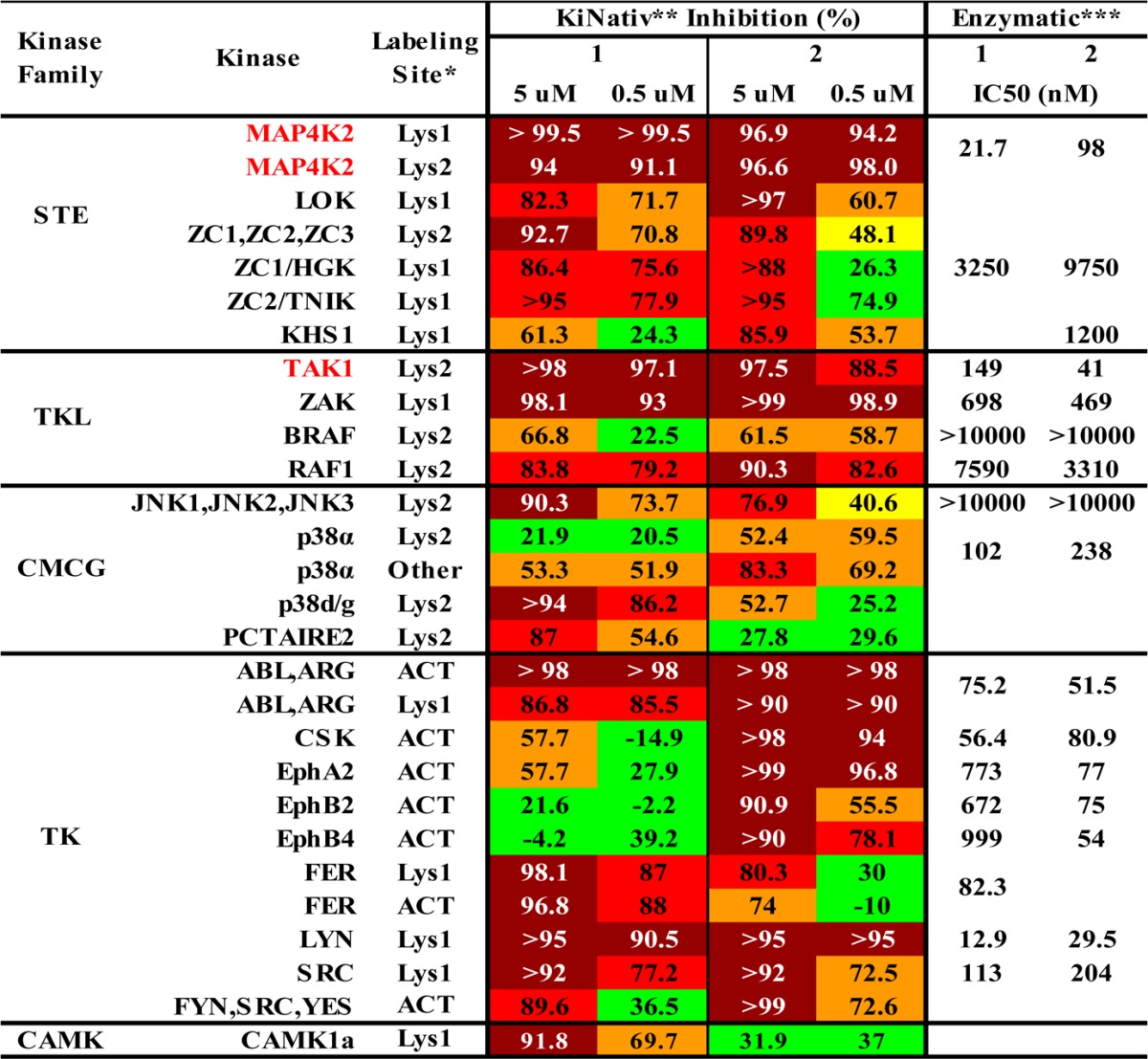
Footnotes: *ACT = activation loop; Lys1 = conserved Lysine 1; Lys2 = conserved lysine 2; other = labeling of residue outside the protein kinase domain, possibly not in ATP binding site. **A375 live cell were treated with 1 and 2 in 5 and 0.5 μM, lysed, and probe-labeled. ***The enzymatic assays were using the SelectScreen kinase profiling service.
To further explore the potential of 1 and 2 to be optimized for potency and selectivity against these kinases, we synthesized 24 diverse analogues (Table 2). To approach this optimization in a systematic fashion, this chemotype was divided into three sections: head, linker, and tail. Each of these units was varied sequentially. The head unit was altered from pyrrolopyridin-4-yl in 1 to pyrrolopyrimidin-4-yl (e.g., 3, 13) and substituted pyrimidin-4-yls (e.g., 6, 7); however, analogues of lead compound 2 contained the original head unit. The linker unit was varied through deletion of the methyl group (e.g., 10, 24), reversing the amide orientation (11, 22) and altering the alkene in compound 2 via saturation (14) or replacement with an ether linkage (15). The tail unit contained various hydrophilic groups instead of the 4-ethylpiperazin-1-yl moiety, along with variations of the substitution site from the 4-position to the 3-position. These changes resulted in minor variation to the syntheses with most analogues being prepared via similar routes and in high yields.
Table 2. Structures of All Compounds.
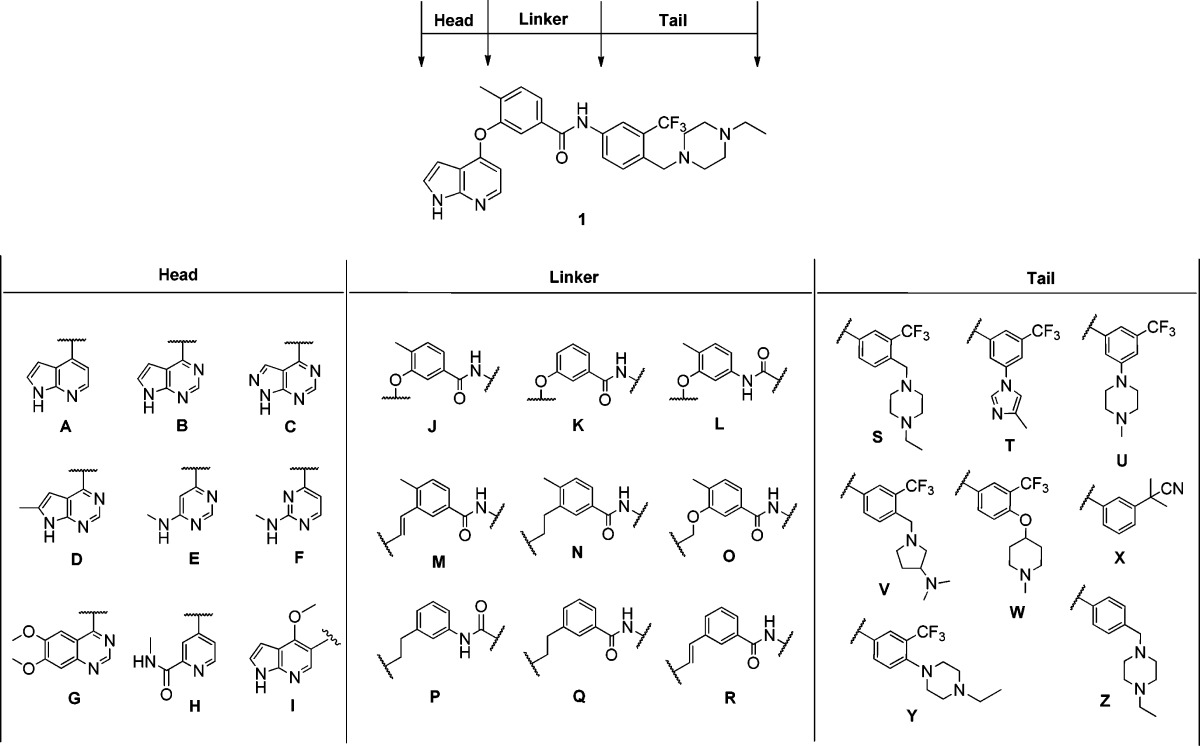
| compd | structure | compd | structure | compd | structure |
|---|---|---|---|---|---|
| 1 | A-J-S | 10 | A-K-S | 19 | I-M-W |
| 2 | I-M-S | 11 | A-L-S | 20 | I-M-X |
| 3 | B-J-S | 12 | A-J-T | 21 | I-N-T |
| 4 | C-J-S | 13 | B-J-U | 22 | I-P-S |
| 5 | D-J-S | 14 | I-N-S | 23 | I-Q-S |
| 6 | E-J-S | 15 | I-O-S | 24 | I-R-S |
| 7 | F-J-S | 16 | E-J-T | 25 | I-N-Y |
| 8 | G-J-S | 17 | E-J-U | 26 | I-M-Z |
| 9 | H-J-S | 18 | I-M-V |
All of the prepared analogues were subjected to KiNativ profiling using HUH7 cell lysates which typically detects approximately 150 protein and lipid kinases of interest. This profiling effort revealed that this class of compounds in general provide good multitargeted scaffolds that can bind to TAK1, MAP4K2, p38α, Abl, ZAK, CSK, FER, FES, and EPHA2 with varying degrees of potency and selectivity. Herein we primarily focus our discussion on how the structural changes affected the ability of the compounds to inhibit TAK1 and MAP4K2 kinases (Table 3). With compound 1, we first changed the head from 1H-pyrrolo[2,3-b]pyridine-4-yl to 7H-pyrrolo[2,3-d]pyrimidin-4-yl (3) and 1H-pyrazolo[3,4-d]pyrimidin-yl (4) and found that the activities against MAP4K2 were maintained while those against TAK1 were clearly improved. However, the addition of a methyl group at the 6-position of the 7H-pyrrolo[2,3-d]pyrimidin-4-yl moiety (5) led to more potent inhibition of MAP4K2 and a decreased potency toward TAK1. We next introduced the monocyclic head units 6-(methylamino)pyrimidin-4-yl (6) and 2-(methylamino)pyrimidin-4-yl (7); 6 exhibited more potent inhibition of MAP4K2 and decreased potency against TAK1, and 7 showed slightly decreased activities. To our surprise, the introduction of a 6,7-dimethoxyquinazolin-4-yl head unit (8) yielded potent activity against both kinases while introduction of 4-substituted N-methylpicolinamide (9) eliminated both TAK1 and MAP4K2 potency. We then varied the linker region and found that deletion of the methyl group (10) had little impact on activity while other modifications only led to loss of activity (11). We next investigated the effect of a variety of tail units and found that all changes led to a decrease in activity against both kinases. However, substitution at the 3-position of the aromatic ring in the tail unit with 4-methyl-1H-imidazol-1-yl (12) and 4-methylpiperazin-1-yl (13) led to only a slight decrease in activity against MAP4K2 but almost complete elimination of TAK1 activity. All analogues of compound 2 generally showed reduced activity against both kinases, with the exception of 14, which showed similar activity, and 15, which exhibited a slight decrease in activity against TAK1.
Table 3. SAR for TAK1 and MAP4K2a.
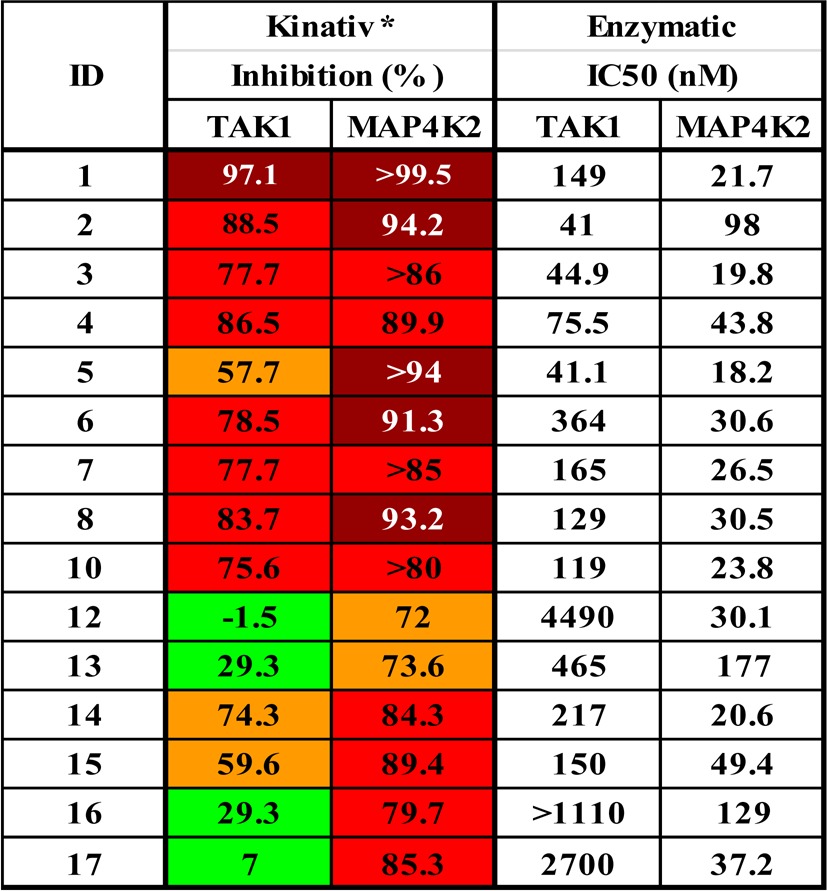
Footnote: *1 and 2 were profiled at 0.5 μM on A375 live cell, while all others were at 1.0 μM on HUH7 lysate.
These results indicated that modification of the head unit can improve activity against both kinases, although in most cases more dramatically for MAP4K2 with the exception of 8. Alteration of the linker unit had little or even negative effect against MAP4K2 and TAK1, while changes in the tail unit always decreased activity against both kinases, although this reduction was small for MAP4K2 but significant for TAK1. These data prompted us to prepare compound 16, which combined the head of 6 and the tail of 12 and maintained the original linker of 1 and exhibited similar inhibitory activity for MAP4K2 compared to 12. Further modification of the tail of 16 led to 17 which exhibited good biochemical selectivity for inhibiting MAP4K2 relative to TAK1 (37 and 2700 nM IC50 values), along with best KiNativ score for MAP4K2 (85% at 1 μM). The results of KiNativ profiling using HUH7 cell lysates showed that only MAP4K2 and ABL among 220 protein and lipid kinases of interest were inhibited by 17 (Supporting Information Table 3), and the inhibition of ABL was rather weak compared with MAP4K2 (49% compared with 85% at 1 μM). Furthermore, the weak inhibition of p38α and ZAK of 16 was eliminated with 17 (Table 4). 17 was also subjected to KiNativ live-cell profiling using HUH7 cells, and strong inhibition of MAP4K2 (88% at 1 μM) labeling was observed suggesting that 17 is a potent and relatively selective cellular MAP4K2 inhibitor (Supporting Information Table 4).
Table 4. Kinase Selectivity of 16 and 17a.
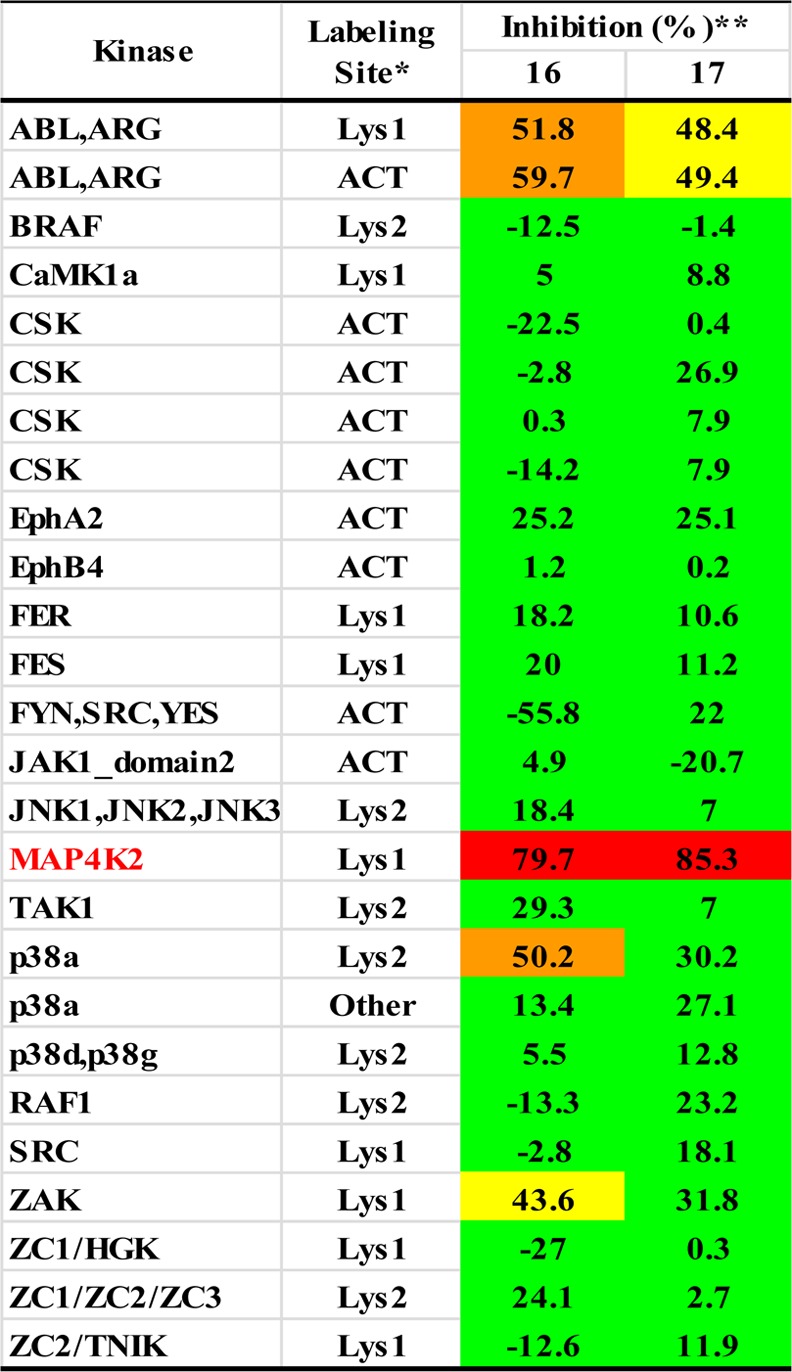
Footnotes: *ACT = activation loop; Lys1 = conserved lysine 1; Lys2 = conserved lysine 2; other = labeling of residue outside the protein kinase domain, possibly not in ATP binding site. **16 and 17 were profiled at 1.0 μM on HUH7 lysate.
To further understand the SAR of TAK1/MAP4K2 inhibitors, we solved cocrystal X-ray structures of TAK1 in complex with 1 (PDB code 4O91). X-ray diffraction extended to 2.4 Å, and the structure was solved using molecular replacement with PDB compound 2YIY as the initial search model. Examination of the active site revealed strong additional electron density into which 1 was easily modeled (Figure S2A). Final statistics for diffraction and the model are given in Table S1. Interactions with the active site residues are schematically depicted in Figure S2B. In summary, the nitrogens in the pyrrolopyridine moiety form hydrogen bonds with Ala107 in the hinge binding region. The linker amide is stabilized inside the deeper binding pocket by hydrogen-bonding with Glu77 and the backbone amide of Asp175. These observations further explain why the reversal of the amide (11) resulted in a loss of activity. The nitrogen of the tertiary amine in the piperazinyl tail interacts with carbonyls of Ile153 and His154 on the activation loop (Figure 3c). Compared with the structure of adenosine bound to the active site of TAK1 (PDB code 2EVA) which shows DFG in the active, flipped-in conformation (Figure 3b), binding of 1 with TAK1 results in an inactive, DFG-out conformation characteristic of type II inhibitors (Figure 3a).
Figure 3.

Compound 1 is a type II inhibitor. (a) Binding of 1 to the active site of TAK1–TAB1 results in the DFG-out conformation characterized by type II inhibitors. (b) The structure of adenosine bound to the active site of TAK1–TAB1 (PDBID 2EVA) is provided for comparison and shows the DFG-in conformation. The DFG motif is highlighted in red. (c) Key interactions of 1 with TAK1. (d) Molecular model of the binding mode of MAP4K2 with 1.
Simulated docking studies using MAP4K2 as the receptor resulted in a similar overall inhibitor pose relative to the TAK1/1 cocomplex structure. However, the long side chain of Gln80 in the C-helix of TAK1 is located very close to the tail of 1 with only 3.6 Å distance between the carbonyl of Gln80 side chain and the 5-position of 3-trifluormethylphenyl group, whereas the shorter side chain of Ile64 in MAP4K2 is not so proximate in the MAP4K2/1 modeling. Substitutions at the 5-position of 3-trifluormethylphenyl group may be too bulky for the side chain of Gln80 of TAK1, whereas they could possibly interact with the side chains of Ile64 and Gln60 of MAP4K2. This potential interaction might compensate for the loss of interactions with the two carbonyls of Ile133 and His134, providing a rationale for why 12, 16, and 17 exhibit a preference for binding to MAP4K2 (Figure 3c,d).
Many of our compounds also exhibited potent inhibition of p38α; the data from KiNativ and enzymatic assays are shown in Table S2. Analogues of 1 that contained the same linker and tail as 1 were all active against p38α (3–9), with 4 being the most potent and 9 being inactive against TAK1 and MAP4K2. These results indicated that modification of the head unit was tolerated on this scaffold. We were also surprised to find that reversing the amide connectivity of 1 afforded a very selective p38α inhibitor (11). To prepare analogues of compound 2, we first explored different tail units and found several compounds with potent activity against p38α (18–20), with 19 being the most selective. Modifications of the linker unit, as in compounds 14 and 15, were also tolerated, and these changes sometimes led to potent activity in compounds with tail units that had previously demonstrated poor activity (i.e., 21, IC50 < 10 nM). Similarly, the reversed amide connectivity (22) retained potent activity when the methyl of the linker was deleted. In sharp contrast, its analogues with the original amide connectivity (23, 24) were inactive against p38α.
Many of the compounds exhibited potent binding activity against ABL kinase which is known to be potently inhibited by type II inhibitors such as imatinib and nilotinib. To evaluate whether these new compounds could inhibit ABL activity in a cellular context, they were evaluated for their antiproliferative activity against BCR-ABL-dependent Ba/F3 cells (Table S3). To evaluate their potential for nonspecific cytotoxicity, they were also evaluated against parental Ba/F3 cells grown in the presence of interleukin 3. The results demonstrated that many of our compounds selectively inhibited the proliferation of BCR-ABL-dependent Ba/F3 cells with EC50 values of less than 400 nM. Several compounds (e.g., 2, 10) exhibited more than 50% cell growth inhibition at 10 nM, with analogues of 2 generally being the most active. These compounds exhibited comparable cellular potency to clinically approved second-generation BCR-ABL inhibitors such as nilotinib and dasatinib. Replacement of the (4-ethylpiperazin-1-yl)methyl tail with a 4-ethylpiperazin-1-yl tail (25) provided very selective inhibition against ABL (both enzymatic and cellular); more surprising was that elimination of the trifluoromethyl group afforded a very strong inhibitor with high selectivity (26).
Some of our compounds exhibited potent inhibition of a number of additional kinases (Table S4). In general, analogues of compound 2 were more likely to inhibit ZAK, with 14 being the most active with >96% inhibition at 1 μM (KiNativ) and an IC50 of 72 nM (SelectScreen). Additionally, compound 9, an analogue of 1, was very selective against ZAK. Only analogues of 2 were found to inhibit CSK, with 14 being the most potent. Compounds 2 and 14 exhibited potent activity against EPHA2, although further modification of their scaffolds led to a severe loss of activity. Surprisingly, we found that only analogues of compound 1 exhibited potency against FES and FER, with the activity of 8 being very strong (93% and 95% inhibition at 1 μM and IC50 values of 51 and 36 nM, respectively).
To further validate inhibitory activity against TAK1 and MAP4K2 by these inhibitors, we next measured their ability to block the downstream signals induced by different cytokines. First we checked their effect on NF-κB signaling in mouse L929 cells following TNFα stimulation, where we found that most inhibitors with strong TAK1 inhibition in previous assays effectively reduced the phosphorylation level of IKKα/β at 100 nM concentrations. The degradation of IκB-α was also effectively inhibited. The reported irreversible TAK1 inhibitor 37 (5Z-7-oxozeaenol)22 was used as positive control, and our lead compounds 1 and 2 exhibited comparable cellular potency (Figure 4a). The selective MAP4K2 inhibitors (12, 16, and 17) and p38α inhibitors 11, 38 (SB203580),23 and 39 (PH-797804)24 were also tested and demonstrated no effect at the same concentrations (Figure 4b, Figure S3B). The most selective MAP4K2 inhibitor 17 was not very effective even at concentrations up to 1 μM (Figure S3A), which suggested that MAP4K2 does not contribute much to this pathway under these conditions.
Figure 4.

Evaluation of the ability of compounds to block TNFα-induced phosphorylation of IKKα/β and stabilize IκB-α in L929 cells. L929 cells were pretreated with indicated (a) TAK1/MAP4K2 inhibitors (1, 2, 3, 4, 5, 7, 10) and covalent TAK1 inhibitor (37), (b) more selective MAP4K2 inhibitors (12, 16, 17, 11) and p38 inhibitor (38) at a concentration of 100 nM for 30 min, and then TNFα was added and incubated for 5 min. Samples were collected and subjected to Western blot. Tubulin was used as a loading control.
Next we checked the effect of 1 in MEFs and human 293 IL-1R cells stimulated with IL-1α and found that the phosphorylation levels of downstream p105, p38, and JNK1/2 were all inhibited in a dose-dependent manner (Figure 5a,b). Similar results were obtained in mouse RAW cells following LPS stimulation (Figure 5c). We also discovered that compound 1 (1 μM) blocked signaling following stimulation of human Gen2.2 cells with the TLR7 agonist CL097; analogue S1 (Figure S4) which possessed moderate TAK1 and MAP4K2 inhibitory activity (IC50 values of 292 and 296 nM, respectively) only partially inhibited signaling. The selective MAP4K2 inhibitor 16 was not able to block CL097 induced signaling (Figure 5d).
Figure 5.

Evaluation of the ability of compounds to inhibit IL-1α, LPS, or CL097-induced phosphorylation of p105, p38, and JNK in several cell types. (a–c) 293 IL-1R, MEFs, and RAW cells were pretreated with 1 at different concentrations for 1 h and then stimulated with IL-1α or LPS for 30 min. Samples were collected and subjected to Western blot for phosphorylation of p105, p38, JNK1/2, and total p38. (d) Gen2.2 cells were pretreated with 1, S1, and 16 at 1 μM for 1 h and then stimulated with CL097 for 30 min. Samples were collected and subjected to Western blot.
It was more challenging to validate the functional cellular consequences of MAP4K2 inhibition, since the effector pathways of this kinase are less well characterized. TAK1 mediates much of the IL-1-induced phosphorylation of p38,10,25 although recent reports suggest that TAK1 does not mediate TGFβ-induced phosphorylation of p38 in TAK1-null MEFs and HaCaT cells.18 A siRNA-based screen targeting all MAP3Ks individually in HaCaT cells suggested roles for MAP3K4 and MAP3K10 in mediating TGFβ-induced phosphorylation of p38.18 A similar siRNA screen targeting MAP4Ks in HaCaT cells demonstrated that depletion of MAP4K2 but not MAP4Ks 1, 3, and 5 resulted in substantial inhibition of both IL-1 and TGFβ-induced phosphorylation of p38, while TGFβ-induced phosphorylation of SMAD2 was unaffected (Figure 6a). Cumulatively these studies suggest a possible role for MAP4K2 in IL-1 and TGFβ-induced phosphorylation of p38. We therefore tested the ability of our selective MAP4K2 inhibitors 16 and 17 to inhibit IL-1 and TGFβ induced phosphorylation of p38 MAPK in WT and TAK1-deficient MEFs. Both 16 and 17 caused a dose-dependent inhibition of IL-1 and TGFβ-induced p38 MAPK phosphorylation but did not inhibit the TGFβ-induced phosphorylation of SMAD2 (Figure 6b). Consistent with the primary role of TAK1 in mediating IL-1 signaling, in WT MEFs, 16 and 17 caused ∼50% inhibition in IL-1-induced phosphorylation of p38 at 500 nM (Figure 6b). However, in TAK1-deficient MEFs, both 16 and 17 resulted in complete inhibition of IL-1-induced phosphorylation of p38 at 500 nM, suggesting that MAP4K2 may mediate the residual IL-1-dependent but TAK1-independent phosphorylation of p38 (Figure 6b). The dose-dependent inhibition of TGFβ-induced phosphorylation of p38 by 16 and 17 was similar in both WT and TAK1-deficient MEFs, consistent with the notion that TAK1 does not mediate TGFβ-induced phosphorylation of p38 in these cells (Figure 6b). Compound 1, a dual inhibitor of TAK1 and MAP4K2, exhibited similar potency to 16 and 17 while the covalent natural product TAK1 inhibitor 37 inhibited phosphorylation of both p38 and SMAD2 at very low concentrations regardless of presence of TAK1, consistent with the observation that TAK1 mediates much of the IL-1-induced phosphorylation of p38 and that 37 has several additional kinase targets including MEK1 (Figure S5).
Figure 6.

Evaluation of signaling following depletion of MAP4Ks by siRNA and evaluation of the ability of selective MAP4K2 inhibitors 16 and 17 to inhibit signaling in wild-type and TAK1-null MEF cells. (a) HaCaT cells were transfected with pools of four different siRNAs targeted against the indicated MAP4Ks. At 48 h after transfection cells were left untreated or treated with TGFβ or IL-1β prior to lysis and Western blot. (b) Wild type and TAK1-null MEFs cells were pretreated with selective MAP4K2 inhibitor compounds 16 and 17 for 1 h, followed by stimulation with TGFβ or IL-1α. Samples were collected and subjected to Western blot.
The pharmacokinetic properties of 1–5 were also evaluated following intravenous and oral delivery in mice, respectively. Compounds 1 and 5 demonstrated favorable pharmacokinetic properties, with T1/2 values of 2.0 and 2.9 h, AUC values of 1369 and 2136 following a 10 mg/kg oral dose, and F values of 70.2% and 38.6%, respectively (Table 5). This suggests that these compounds should be useful for murine efficacy studies.
Table 5. Pharmacokinetic Parameters of 1, 2, and Analogues.
| compd | route | dose (mg/kg) | Tmax (h) | Cmax (ng/mL) | AUClast (h·ng/mL) | T1/2 (h) | CL (mL min–1 kg–1) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | iv | 1 | 396 | 195 | 2.03 | 80.8 | 11.9 | ||
| po | 10 | 0.5 | 316 | 1369 | 70.2 | ||||
| 2 | iv | 1 | 47.1 | 403 | 4.66 | 55.4 | 14.2 | ||
| po | 10 | 4.0 | 163 | 295 | 13.7 | ||||
| 3 | iv | 1 | 230.9 | 236.6 | 2.33 | 65.8 | 9.5 | ||
| po | 10 | 2.0 | 133.6 | 516.2 | 21.8 | ||||
| 4 | iv | 1 | 280.3 | 167.9 | 0.87 | 99.2 | 6.1 | ||
| po | 10 | 0.5 | 61.3 | 186.2 | 11.1 | ||||
| 5 | iv | 1 | 317 | 554 | 2.94 | 26.5 | 5.42 | ||
| po | 10 | 4.0 | 233 | 2136 | 38.6 |
Discussion
In conclusion, compounds 1 and 2, along with their analogues, represent two novel “type II” kinase inhibitor scaffolds discovered using a kinome-wide profiling approach and exhibit diverse activities against a limited set of kinase targets. Among these compounds, 1 was especially interesting as a very potent TAK1 inhibitor and has already been used in some studies,8,9 and 2 is also being utilized in cancer studies because of its potent antiproliferative effects against particular tumor types. Profiling of these type II inhibitors reveals that a subset of the compounds also are capable of potently inhibiting LCK, ABL, p38α, etc., and this needs to be considered when using them to interrogate TAK1 and MAP4K2 dependent effects. As for application of TAK1 inhibitors, the most commonly used probe nowadays is 37, which is not a selective inhibitor. 37 strongly inhibits MAP2Ks and MAPKs such as MEK1 (MAP2K1) and ERK1 (MAPK1) and many other targets such as KDR, PDGFR, ZAK, etc. Moreover, the binding to several of these targets is covalent, and it is likely that other reactive cysteines can be targeted by this compound intracellularly.26 Compound 1 provides a complementary pharmacological probe of TAK1 relative to 37 due to its distinct chemical structure, nonoverlapping off-target pharmacology, and reversible mode of inhibition. The cocrystal structure of TAK1/1 has helped in rationalizing the SAR and will be used to design type II TAK1 inhibitors in the future. Meanwhile 16 and 17 represent relatively selective MAP4K2 inhibitors; compound 17 especially exhibits impressive selectivity and excellent potency in cellular assays. 11 is a selective p38α inhibitor; both 25 and 26 are selective ABL inhibitors. By modification of the tail moiety, these inhibitors (11, 17, and 26, etc.) can specifically interact with the allosteric DFG-pockets, which may provide a means of achieving selectivity among otherwise highly homologous kinases. The cocrystal structure of TAK1/1 reveals that Gln80 is proximal (3.6 Å) from the phenyl ring of the benzamide “tail” moiety, which suggests that a unique H-bond may be able to be introduced in this region to gain selectivity for TAK1. Finally, given their wide diversity with respective kinase selectivity, enzymatic and cellular potency, and favorable pharmacokinetic parameters, 4-substituted 1H-pyrrolo[2,3-b]pyridines may represent privileged scaffolds for the development of therapeutic agents targeting various kinases.
Experimental Section
Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on a 400 MHz (Varian 7600 AS) or 600 MHz (Varian Inova) instrument, and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), brs (broad singlet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Symmetry C18 column (19 mm × 50 mm, 5 μm) using a gradient of 5–95% acetonitrile in water containing 0.05% trifluoroacetic acid (TFA) over 8 min (10 min run time) at a flow rate of 30 mL/min. Purification of compounds was performed with either a Teledyne ISCO CombiFlash Rf system or a Waters Micromass ZQ preparative system. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
4-Methyl-3-((1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)oxy)benzoic Acid (28)
To a stirred solution of 27 (570 mg, 2 mmol) and 3-hydroxy-4-methylbenzoic acid (340 mg, 2.2 mmol) in 13 mL of DMSO was added K2CO3 (830 mg, 6 mmol). The reaction mixture was allowed to stand for 5 h at 100 °C and then cooled to room temperature. The mixture was acidified with 1 N HCl solution and extracted with ethyl acetate. The organic phase was washed with brine and dried over Na2SO4, then filtered and evaporated to give a pale yellow liquid (690 mg, 80%). To a stirred solution of this gained liquid (690 mg, 1.6 mmol) in 10 mL of methanol was added Pd/C (10 wt % loading, 120 mg). The reaction mixture was stirred under hydrogen atmosphere for 2 or 3 days. The reaction was monitored by TLC, and more Pd/C might be needed for complete conversion during the period. Then the reaction solution was filtered, concentrated, and purified with column chromatography (hexane/ethyl acetate = 1:1) to give the title compound as a white solid (510 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 5.6 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.90 (s, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 3.6 Hz, 1H), 6.52 (d, J = 3.6, 1H), 6.44 (d, J = 5.6, 1H), 5.78 (s, 2H), 3.65 (t, J = 8.0 Hz, 2H), 2.37 (s, 3H), 0.99 (t, J = 8.0 Hz, 2H), 0.00 (s, 9H). MS (ESI) m/z 399 (M + H)+.
N-(4-((4-Ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-4-methyl-3-((1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)oxy)benzamide (30)
To a stirred solution of 28 (400 mg, 1 mmol) and aniline 29 (430 mg, 1.5 mmol) in 10 mL of dichloromethane were added HATU (570 mg, 1.5 mmol), DMAP (185 mg, 1.5 mmol), and DIEA (520 μL, 3 mmol). The reaction mixture was allowed to stand for 24 h at room temperature, then diluted with ethyl acetate and washed with water and brine. The organic phase was dried over Na2SO4, filtered, then concentrated and purified with column chromatography (dichloromethane/methanol = 20:1) to give the title compound as a colorless oil (600 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.12 (d, J = 5.6 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.74–7.68 (m, 2H), 7.69 (s, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 3.6, 1H), 6.37 (d, J = 3.6, 1H), 6.33 (d, J = 5.6, 1H), 5.64 (s, 2H), 3.61 (s, 2H), 3.55 (t, J = 8.0 Hz, 2H), 2.65–2.40 (m, 8H), 2.50 (q, J = 6.8 Hz, 2H), 2.28 (s, 3H), 1.24 (t, J = 6.8 Hz, 3H), 0.90 (t, J = 8.0 Hz, 2H), −0.08 (s, 9H). MS (ESI) m/z 668 (M + H)+.
3-((1H-Pyrrolo[2,3-b]pyridin-4-yl)oxy)-N-(4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-4-methylbenzamide (1)
To a stirred ice-cooled solution of 30 (330 mg, 0.5 mmol) in 5 mL of dichloromethane was added 1 mL of TFA. The reaction mixture was stirred for 30 min at 0 °C and then warmed to room temperature. After 5 h the reaction mixture was concentrated and dried under vacuum, then dissolved in 5 mL of THF, and 5 mL of 1 N NaOH water solution was added. The reaction mixture was stirred for 24 h and extracted with ethyl acetate. The combined organic phase was washed with brine and dried with Na2SO4, then filtered and concentrated and purified with column chromatography (dichloromethane/methanol = 10:1) to give the title compound as a white solid (188 mg, 70%). 1H NMR (400 MHz, DMSO) δ 11.78 (bs, 1H), 10.44 (s, 1H), 8.16 (d, J = 2.4 Hz, 1H), 8.09 (d, J = 5.6 Hz, 1H), 8.02 (dd, J = 8.4, 1.6 Hz, 1H), 7.88 (dd, J = 8.0, 2.0 Hz, 1H), 7.78 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.38 (dd, J = 3.2, 2.8, 1H), 6.32 (d, J = 5.6, 1H), 6.21 (dd, J = 3.2, 2.0 Hz, 1H), 3.56 (s, 2H), 2.52–2.30 (m, 8H), 2.50 (q, J = 7.2 Hz, 2H), 2.24 (s, 3H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO) δ 164.83, 157.26, 152.90, 151.59, 144.78, 138.50, 134.90, 134.11, 132.48, 132.21, 131.61, 125.31, 125.25, 124.04, 120.62, 117.80, 117.74, 110.13, 101.78, 97.24, 57.81, 52.97, 52.66, 51.94, 16.17, 12.18. MS (ESI) m/z 538 (M + H)+.
N-(4-((4-Ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-iodo-4-methylbenzamide (32)
To a stirred solution of 3-iodo-4-methylbenzoic acid 31 (262 mg, 1 mmol) and aniline 29 (430 mg, 1.5 mmol) in 10 mL of dichloromethane were added HATU (570 mg, 1.5 mmol), DMAP (185 mg, 1.5 mmol), and DIEA (520 μL, 3 mmol). The reaction mixture was allowed to stand for 24 h at room temperature, then diluted with ethyl acetate and washed with water and brine. The organic phase was dried over Na2SO4, filtered, then concentrated and purified with column chromatography (dichloromethane/methanol 20:1) to give the title compound as a yellow solid (480 mg, 90%). 1H NMR (600 MHz, CDCl3) δ 8.35 (br, 1H), 8.33 (s, 1H), 7.91 (m, 2H), 7.80 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 9.0, 1H), 7.32 (d, J = 8.4 Hz, 1H), 3.66 (s, 2H), 3.00–2.58 (m, 8H), 2.71 (m, 2H), 2.48 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H). MS (ESI) m/z 532 (M + H)+.
4-Methoxy-1-((2-(trimethylsilyl)ethoxy)methyl)-5-vinyl-1H-pyrrolo[2,3-b]pyridine (34)
To a stirred solution of 33 (400 mg, 1.20 mmol) in MeOH (4 mL) was added sodium methoxide (348 mg, 6.43 mmol). The mixture was stirred at 70 °C for 5 h before being cooled to room temperature and filtered through a Celite pad. The filtrate was concentrated under reduced pressure and partitioned between ethyl acetate and water. The aqueous layer was extracted with ethyl acetate three times. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was used for the next step without purification. To a stirred solution of methyltriphenylphosphonium iodide (923 mg, 2.28 mmol) in THF (10 mL) was added n-butyllithium (2.5 M in hexane, 0.822 mL, 2.056 mmol) at −78 °C. After 30 min, the reaction mixture was slowly treated with the obtained residue in THF (5 mL) at −78 °C and stirred for 2 h at −78 °C before being quenched with saturated ammonium chloride solution. The reaction mixture was extracted with ethyl acetate three times, and the combined organic layer was washed with brine, dried over Na2SO4, filtered, then concentrated and purified with column chromatography (hexane/ethyl acetate = 3:1) to give title compound as a colorless oil (208 mg, 57% in two steps). 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 7.22 (m, 1H), 6.97 (m, 1H), 6.74 (m, 1H), 5.77–5.50 (m, 1H), 5.25 (m, 1H), 4.33 (s, 3H), 3.55 (m, 2H), 0.90 (m, 2H), −0.06 (s, 9H). MS (ESI) m/z 305 (M + H)+.
tert-Butyl 4-Methoxy-5-vinyl-1H-pyrrolo[2,3-b]pyridine-1-carboxylate (35)
To 34 (140 mg, 0.46 mmol) in CH2Cl2 (5 mL) was added TFA (0.5 mL) at 0 °C. The reaction mixture was stirred for 30 min at 0 °C and then warmed to room temperature. After 5 h, the reaction mixture was concentrated in vacuo, then dissolved in 5 mL of THF and treated with 5 mL of 1 N NaOH. The reaction mixture was stirred for 24 h and extracted with ethyl acetate. The combined organic phase was washed with brine and dried with Na2SO4, then filtered and concentrated in vacuo. The residue was then dissolved in 5 mL of CH2Cl2 and treated with Boc2O (200 mg, 0.92 mmol) and DIEA (240 μL, 1.38 mmol). The reaction mixture was stirred for 24 h and extracted with ethyl acetate, and the combined organic layer was washed with brine, dried over Na2SO4, filtered, then concentrated and purified with column chromatography (hexane/ethyl acetate = 2:1) to give the title compound as a white solid (100 mg, 80% in two steps). 1H NMR (600 MHz, CDCl3) δ 8.63 (s, 1H), 7.54 (m, 1H), 6.94 (m, 1H), 6.76 (m, 1H), 5.81 (m, 2H), 5.36 (m, 1H), 4.31 (s, 3H), 1.69 (s, 9H). MS (ESI) m/z 275 (M + H)+.
(E)-tert-Butyl 5-(5-((4-((4-Ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)carbamoyl)-2-methylstyryl)-4-methoxy-1H-pyrrolo[2,3-b]pyridine-1-carboxylate (36)
To the mixture of 32 (58 mg, 0.110 mmol), 35 (32 mg, 0.116 mmol), DIEA (480 μL, 0.275 mmol), and tri-tert-butylphosphine (1.0 M in toluene, 11 μL, 0.022 mmol) was added Pd2(dba)3 (10 mg, 0.011 mmol) at room temperature. The reaction mixture was degassed and stirred under Ar atmosphere at 80 °C for 15 h before being cooled to room temperature, then filtered and concentrated. The residue was purified with column chromatography (dichloromethane/methanol 20:1) to give the titled compound as a white solid (60 mg, 80%, E/Z > 20:1). 1H NMR (600 MHz, DMSO) δ 8.50 (s, 1H), 8.49 (s, 1H), 8.13 (s, 1H), 7.97 (s, 1H), 7.90 (d, J = 8.4, 1H), 7.71 (d, J = 7.8, 1H), 7.64 (d, (d, J = 8.4, 1H), 7.50 (d, (d, J = 4.2, 1H), 7.30 (d, J = 16.8, 1H), 7.26 (d, J = 16.8, 1H), 7.20 (d, J = 7.8, 1H), 6.71 (d, J = 4.2, 1H), 4.24 (s, 3H), 3.58 (s, 2H), 2.55 (m, 4H), 2.49 (q, J = 7.2, 2H), 2.41 (s, 3H), 2.16 (m, 2H), 1.89 (m, 2H), 1.63 (s, 9H), 1.11 (t, J = 7.2, 3H). MS (ESI) m/z 678 (M + H)+.
(E)-N-(4-((4-Ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-(2-(4-methoxy-1H-pyrrolo[2,3-b]pyridin-5-yl)vinyl)-4-methylbenzamide (2)
To a solution of 36 (60 mg, 0.088 mmol) in CH2Cl2 (2 mL) was added TFA (0.2 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h, then concentrated and purified with column chromatography (dichloromethane/methanol = 10:1) to give the title compound as a white solid (45 mg, 90%, E/Z > 20:1). 1H NMR (600 MHz, DMSO) δ 11.73 (s, 1H), 10.52 (s, 1H), 8.50 (s, 1H), 8.23 (d, J = 14.4, 1H), 8.06 (d, J = 8.4, 1H), 7.78 (d, J = 8.4, 1H), 7.72 (d, J = 8.4, 1H), 7.44 (s, 1H), 7.43 (s, 1H), 7.37 (d, J = 13.8, 1H), 7.36 (d, J = 7.2, 1H), 6.82 (m, 1H), 4.35 (s, 3H), 3.56 (s, 2H), 2.60–2.20 (m, 10H), 2.48 (s, 3H), 0.99 (t, J = 7.2, 3H). 13C NMR (100 MHz, DMSO) δ 166.14, 157.01, 151.66, 143.64, 139.80, 137.41, 132.78, 132.40, 131.64, 130.89, 127.65, 126.59, 125.35, 124.334, 124.22, 124.07, 117.73, 114.16, 108.40, 100.31, 59.52, 57.91, 53.21, 52.80, 52.01, 20.09, 12.38. MS (ESI) m/z 578 (M + H)+.
Compounds 3–26 were synthesized with same procedures as 1 and 2. 37–39 were commercial from Selleckchem.com.
TAK1–TAB1 Expression and Purification
DNA encoding the TAK1–TAB1 fusion protein (kinase domain residues 31–303 and c-terminal domain residues 468–497) was obtained from GeneScript (GenScript USA Inc., 860 Centennial Avenue, Piscataway, NJ 08854, U.S.). This was cloned into the pFastBac His6 TEV LIC cloning vector (4B) (plasmid 30115). TAK1–TAB1 fusion protein was expressed in Hi5 insect cells and purified as described previously.27,28
TAK1–TAB1/1 Crystallization and Structure Determination
TAK1–TAB1 was concentrated to 10 mg/mL and crystallized as reported previously28 with minor modifications. Briefly, the crystals were obtained using the hanging-drop method at 20 °C in 4 μL drops by mixing protein with equal volumes of reservoir solution [0.65–0.75 M sodium citrate, 0.2 M NaCl, 0.1 M Tris, pH 7.0, and 5 mM adenosine]. The crystals were washed three times in reservoir solution without adenosine. A 10 mM solution of 1 was prepared, and crystals were back-soaked for ∼8–12 h. Crystals were frozen for data collection using 20% ethylene glycol as cryoprotectant. Diffraction data were collected at Argonne Advanced Photon Source (beamline 19-D) and processed with HKL3000.29 The structure was solved by molecular replacement using Phaser,30 with inactive TAK1–TAB1 structures (PDB code 2YIY) as search model. Coot was used for model building,31 and refinement was carried out using both Phenix, version 1.8.4,32 and Refmac, version 5.8.0049.18,21 Figures were generated by PyMol (The PyMOL Molecular Graphics System, version 1.6.0.0) and Meastro (version 1.5.014) from Schrödinger, LLC.
Ba/F3 Cell Proliferation Assay
Compound efficacy against cell proliferation was conducted in 96-well plates. Compounds were added in serial dilutions to cell culture. After 48 h cocultured with compounds, cell viability was measured using CellTiter-Glo (Promega, Wisconsin), and IC50 values were determined by XLfit4.0 (IDBS).
Cell Culture and Stimulations
TAK1-deficient (TAK1–/−) and corresponding wild type (WT) mouse embryonic fibroblasts (MEFs) were a generous gift from S. Akira (Osaka University, Japan).25 Human keratinocyte (HaCaT) cells and WT and TAK1–/– MEFs were cultured in 10 cm diameter dishes in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin mix, and 2 mM l-glutamine under a humidified atmosphere with 5% CO2 at 37 °C. In order to knock down the indicated panels of human MAP4Ks, a pool of four siRNA duplexes designed against each target were purchased from Dharmacon (sold as SMARTpool siRNA) and transfected onto HaCaT cells as described previously.18 Prior to stimulation with appropriate ligands, cells were cultured in DMEM containing 0.1% FBS for 16 h. Inhibitors were added 1 h prior to stimulation with TGFβ (50 pM, 45 min), human IL-1β (1 μg/mL, 10 min), or mouse IL-1α (5 ng/mL, 10 min). Cells were lysed in 0.5 mL of ice-cold complete lysis buffer (50 mM Tris-HCl, pH7.5, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 1 mM sodium orthovanadate, 50 mM sodium flouride, 5 mM sodium pyrophosphate, 0.27 M sucrose, 5 mM β-glycerophosphate, 0.1% (v/v) 2-mercaptoethanol, 0.5 μM microcystin-LR, 1 tablet per 25 mL of complete protease inhibitor cocktail). The extracts were cleared by centrifuging at 16000g at 4 °C for 10 min and processed for Western blot analysis as described previously.18
Western Blot
Cells were harvested in lysis buffer consisting of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, and cocktails of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Cell lysates were clarified by centrifugation for 5 min, and the protein concentration of the supernatants was determined using a modified Bradford assay (Bio-Rad, Hercules, CA). For immunoblotting, 20 μg of protein was loaded in each lane and was separated by SDS–PAGE on 4–12% gradient gels (Invitrogen, Carlsbad, CA), transferred to PVDF membranes and detected by immunoblotting with the following primary antibodies. Goat anti-mouse and anti-rabbit secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) conjugated to horseradish peroxidase were used at a 1:3000 dilution, and immunoreactive bands were detected by chemiluminescence (SuperSignal, Pierce, Rockford, IL) and film (Denville Scientific, South Plainfield, NJ).
In Vivo Pharmacokinetic Studies
Male Swiss albino mice were dosed via tail vein (intravenous, solution in 20% w/v hydroxypropyl β-cyclodextrin in 25 mM sodium phosphate buffer, dose 1 mg/kg) or via oral gavage (suspensions in 0.5% w/v Na CMC with 0.1% v/v Tween-80 in water). Blood samples were collected at 0, 0.083 (for iv only), 0.25, 0.5, 1, 2, 4, 6 (for po only), 8, 12, and 24 h for the iv and po groups. The blood samples were collected from sets of three mice at each time point in labeled microcentrifuge tubes containing K2EDTA as an anticoagulant. Plasma samples were separated by centrifugation and stored below −70 °C until bioanalysis. All samples were processed for analysis by precipitation using acetonitrile and analyzed with a partially validated LC/MS/MS method (LLOQ, 1.138 ng/mL). Pharmacokinetic parameters were calculated using the noncompartmental analysis tool of WinNonlin Enterprise software (version 5.2).
Acknowledgments
This study was supported by research funding from NIH Grants CA130876-05 (N.S.G.), HG006097-03 (N.S.G), and CPRIT R1207 (K.D.W.). We thank Diana Tomchick, Zhe Chen, and the staff at the structural biology laboratory at UT Southwestern Medical Center and beamline 19ID for assistance with X-ray data collection and processing. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357. The Gen2.2 cells used in this study were generously provided by Dr. Joel Plumas and Dr. Laurence Chaperot. We thank Dr. Ying Li for her suggestions and help for this study.
Glossary
Abbreviations Used
- TAK1
transforming growth factor β activated kinase 1
- MAP4K2
mitogen-activated protein kinase kinase kinase kinase 2
- GCK
germinal center kinase
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- TGFβ
transforming growth factor β
- IKK
kinase of IκB
- IκB-α
nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, α
- JNK
c-Jun N-terminal kinase
- ABL
Abelson murine leukemia viral oncogene homologue 1
- ARG
Abelson-related gene
- BCR
breakpoint cluster region protein
- EPHA2
ephrin type A receptor 2
- SRC
sarcoma
- CSK
C-SRC tyrosine kinase
- FES
feline sarcoma oncogene
- FER
feline sarcoma oncogen related tyrosine kinase
- LYN
tyrosine-protein kinase Lyn
- ZAK
sterile α motif and leucine zipper containing kinase AZK
- TNFα
tumor necrosis factors α
- IL-1α
interleukin 1α
- LPS
lipopolysaccharide
- TLR7
Toll-like receptor 7
- siRNA
small interfering RNA
- SMAD2
mothers against decapentaplegic homologue 2
- LIC
ligation independent cloning
- T1/2
half-life period
- AUC
area under concentration–time curve
- F
fraction of bioavailability
- SAR
structure–activity relationship
- TLC
thin-layer chromatography
- rt
room temperature
- TMS
tetramethylsilane
- ppm
parts per million
- HPLC
high performance liquid chromatography
- Boc
N-tert-butoxycarbonyl
- DIEA
N,N-diisopropylethylamine
- DMAP
4-(dimethylamino)pyridine
- DMSO
dimethyl sulfoxide
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- Pd2(dba)3
tris(dibenzylideneacetone)dipalladium(0)
- SEM
2-(trimethylsilyl)ethoxymethyl
- TFA
trifluoroacetic acid
Supporting Information Available
Figures S1–S5; structures of 37–39 (Figure S6); Table S1–S4; structures and spectral data of 3–26; Ambit profiling data of 1 and 2; ActiveX profiling data of 1, 2, and 17; KinomeScan profiling data and KiNativ assay data in xlsx format. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
X-ray coordinates and structure factors have been deposited in the Protein Data Bank, accession code 4O91.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- O’Hare T.; Shakespeare W. C.; Zhu X.; Eide C. A.; Rivera V. M.; Wang F.; Adrian L. T.; Zhou T.; Huang W. S.; Xu Q.; Metcalf C. A. 3rd; Tyner J. W.; Loriaux M. M.; Corbin A. S.; Wardwell S.; Ning Y.; Keats J. A.; Wang Y.; Sundaramoorthi R.; Thomas M.; Zhou D.; Snodgrass J.; Commodore L.; Sawyer T. K.; Dalgarno D. C.; Deininger M. W.; Druker B. J.; Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajiwala K. S.; Wu J. C.; Christensen J.; Deshmukh G. D.; Diehl W.; DiNitto J. P.; English J. M.; Greig M. J.; He Y. A.; Jacques S. L.; Lunney E. A.; McTigue M.; Molina D.; Quenzer T.; Wells P. A.; Yu X.; Zhang Y.; Zou A.; Emmett M. R.; Marshall A. G.; Zhang H. M.; Demetri G. D. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould A. E.; Adams R.; Adhikari S.; Aertgeerts K.; Afroze R.; Blackburn C.; Calderwood E. F.; Chau R.; Chouitar J.; Duffey M. O.; England D. B.; Farrer C.; Forsyth N.; Garcia K.; Gaulin J.; Greenspan P. D.; Guo R.; Harrison S. J.; Huang S. C.; Iartchouk N.; Janowick D.; Kim M. S.; Kulkarni B.; Langston S. P.; Liu J. X.; Ma L. T.; Menon S.; Mizutani H.; Paske E.; Renou C. C.; Rezaei M.; Rowland R. S.; Sintchak M. D.; Smith M. D.; Stroud S. G.; Tregay M.; Tian Y.; Veiby O. P.; Vos T. J.; Vyskocil S.; Williams J.; Xu T.; Yang J. J.; Yano J.; Zeng H.; Zhang D. M.; Zhang Q.; Galvin K. M. Design and optimization of potent and orally bioavailable tetrahydronaphthalene Raf inhibitors. J. Med. Chem. 2011, 54, 1836–1846. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Getlik M.; Grutter C.; Pawar V.; Wulfert S.; Rabiller M.; Rauh D. Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J. Am. Chem. Soc. 2009, 131, 13286–13296. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Miduturu C. V.; Deng X.; Kwiatkowski N.; Yang W.; Brault L.; Filippakopoulos P.; Chung E.; Yang Q.; Schwaller J.; Knapp S.; King R. W.; Lee J. D.; Herrgard S.; Zarrinkar P.; Gray N. S. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem. Biol. 2011, 18, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein D. M.; Gray N. S.; Zarrinkar P. P. High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discovery 2008, 7, 391–397. [DOI] [PubMed] [Google Scholar]
- Pauls E.; Shpiro N.; Peggie M.; Young E. R.; Sorcek R. J.; Tan L.; Choi H. G.; Cohen P. Essential role for IKKbeta in production of type 1 interferons by plasmacytoid dendritic cells. J. Biol. Chem. 2012, 287, 19216–19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N.; Inesta-Vaquera F.; Zhang J.; Xie C.; Cai H.; Arthur S.; Tan L.; Choi H.; Gray N.; Cohen P.; Pedrioli P.; Clark K.; Alessi D. R. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One 2012, 7, e39132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [DOI] [PubMed] [Google Scholar]
- Dai L.; Aye Thu C.; Liu X. Y.; Xi J.; Cheung P. C. TAK1, more than just innate immunity. IUBMB Life 2012, 64, 825–834. [DOI] [PubMed] [Google Scholar]
- Buglio D.; Palakurthi S.; Byth K.; Vega F.; Toader D.; Saeh J.; Neelapu S. S.; Younes A. Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood 2012, 120, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger K. R.; Chen X.; Crew A. P.; Kleinberg A.; Ma L.; Mulvihill M. J.; Wang J.; Wilde V. L.; Albertella M.; Bittner M.; Cooke A.; Kadhim S.; Kahler J.; Maresca P.; May E.; Meyn P.; Romashko D.; Tokar B.; Turton R. Discovery of 7-aminofuro[2,3-c]pyridine inhibitors of TAK1: optimization of kinase selectivity and pharmacokinetics. Bioorg. Med. Chem. Lett. 2013, 23, 4511–4516. [DOI] [PubMed] [Google Scholar]
- Hornberger K. R.; Berger D. M.; Crew A. P.; Dong H.; Kleinberg A.; Li A. H.; Medeiros M. R.; Mulvihill M. J.; Siu K.; Tarrant J.; Wang J.; Weng F.; Wilde V. L.; Albertella M.; Bittner M.; Cooke A.; Gray M. J.; Maresca P.; May E.; Meyn P.; Peick W. Jr.; Romashko D.; Tanowitz M.; Tokar B. Discovery and optimization of 7-aminofuro[2,3-c]pyridine inhibitors of TAK1. Bioorg. Med. Chem. Lett. 2013, 23, 4517–4522. [DOI] [PubMed] [Google Scholar]
- Kilty I.; Green M. P.; Bell A. S.; Brown D. G.; Dodd P. G.; Hewson C.; Hughes S. J.; Phillips C.; Ryckmans T.; Smith R. T.; van Hoorn W. P.; Cohen P.; Jones L. H. TAK 1 Inhibition in the DFG-out conformation. Chem. Biol. Drug Des. 2013, 82, 500–505. [DOI] [PubMed] [Google Scholar]
- Ivanov V. N.; Kehrl J. H.; Ronai Z. Role of TRAF2/GCK in melanoma sensitivity to UV-induced apoptosis. Oncogene 2000, 19, 933–942. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; Laughlin J. D.; Park H.; LoGrasso P. V.; Patricelli M.; Nomanbhoy T. K.; Sorger P. K.; Alessi D. R.; Gray N. S. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota G. P. The TGFbeta-induced phosphorylation and activation of p38 mitogen-activated protein kinase is mediated by MAP3K4 and MAP3K10 but not TAK1. Open Biol. 2013, 3, 130067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Szardenings A. K.; Liyanage M.; Nomanbhoy T. K.; Wu M.; Weissig H.; Aban A.; Chun D.; Tanner S.; Kozarich J. W. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Nomanbhoy T. K.; Wu J.; Brown H.; Zhou D.; Zhang J.; Jagannathan S.; Aban A.; Okerberg E.; Herring C.; Nordin B.; Weissig H.; Yang Q.; Lee J. D.; Gray N. S.; Kozarich J. W. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011, 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebakken C. S.; Riddle S. M.; Singh U.; Frazee W. J.; Eliason H. C.; Gao Y.; Reichling L. J.; Marks B. D.; Vogel K. W. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. J. Biomol. Screening 2009, 14, 924–935. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J.; Kajino T.; Ono K.; Ohtomo T.; Matsumoto M.; Shiina M.; Mihara M.; Tsuchiya M.; Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 2003, 278, 18485–18490. [DOI] [PubMed] [Google Scholar]
- Arana-Argaez V. E.; Delgado-Rizo V.; Pizano-Martinez O. E.; Martinez-Garcia E. A.; Martin-Marquez B. T.; Munoz-Gomez A.; Petri M. H.; Armendariz-Borunda J.; Espinosa-Ramirez G.; Zuniga-Tamayo D. A.; Herrera-Esparza R.; Vazquez-Del Mercado M. Inhibitors of MAPK pathway ERK1/2 or p38 prevent the IL-1{beta}-induced up-regulation of SRP72 autoantigen in Jurkat cells. J. Biol. Chem. 2010, 285, 32824–32833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L.; Igea A.; Canovas B.; Dolado I.; Nebreda A. R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 2013, 5, 1759–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S.; Sanjo H.; Takeda K.; Ninomiya-Tsuji J.; Yamamoto M.; Kawai T.; Matsumoto K.; Takeuchi O.; Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol 2005, 6, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Schirmer A.; Kennedy J.; Murli S.; Reid R.; Santi D. V. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.; Vial S. C.; Dedi N.; Long J. M.; Dunster N. J.; Cheetham G. M. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J. Mol. Biol. 2005, 354, 1013–1020. [DOI] [PubMed] [Google Scholar]
- Wu J.; Powell F.; Larsen N. A.; Lai Z.; Byth K. F.; Read J.; Gu R. F.; Roth M.; Toader D.; Saeh J. C.; Chen H. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. [DOI] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.