

Abstract
Several studies have shown that type X collagen (COL X), a marker of late-stage chondrocyte hypertrophy, is expressed in mesenchymal stem cells (MSCs) from osteoarthritis (OA) patients. We recently found that Naproxen, but not other nonsteroidal anti-inflammatory drugs (NSAIDs) (Ibuprofen, Celebrex, Diclofenac), can induce type X collagen gene (COL10A1) expression in bone-marrow-derived MSCs from healthy and OA donors. In this study we determined the effect of Naproxen on COL X protein expression and investigated the intracellular signaling pathways that mediate Naproxen-induced COL10A1 expression in normal and OA hMSCs. MSCs of OA patients were isolated from aspirates from the intramedullary canal of donors (50–80 years of age) undergoing hip replacement surgery for OA and were treated with or without Naproxen (100 μg/mL). Protein expression and phosphorylation were determined by immunoblotting using specific antibodies (COL X, p38 mitogen-activated protein kinase [p38], phosphorylated-p38, c-Jun N-terminal kinase [JNK], phosphorylated-JNK, extracellular signal-regulated kinase [ERK], and phosphorylated-ERK). Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed to determine the expression of COL10A1 and Runt-related transcription factor 2 gene (Runx2). Our results show that Naproxen significantly stimulated COL X protein expression after 72 h of exposure both in normal and OA hMSCs. The basal phosphorylation of mitogen-activated protein kinases (MAPKs) (ERK, JNK, and p38) in OA hMSCs was significantly higher than in normal. Naproxen significantly increased the MAPK phosphorylation in normal and OA hMSCs. NSAID cellular effects include cyclooxygenase, 5-lipoxygenase, and p38 MAPK signaling pathways. To investigate the involvement of these pathways in the Naproxen-induced COL10A1 expression, we incubated normal and OA hMSCs with Naproxen with and without inhibitors of ERK (U0126), JNK (BI-78D3), p38 (SB203580), and 5-lipoxygenase (MK-886). Our results showed that increased basal COL10A1 expression in OA hMSCs was significantly suppressed in the presence of JNK and p38 inhibitors, whereas Naproxen-induced COL10A1 expression was suppressed by 5-lipoxygenase inhibitor. This study shows that Naproxen induces COL X both at transcriptional and translational levels in normal and OA hMSCs. Elevated basal COL10A1 expression in OA hMSCs is probably through the activation of MAPK pathway and Naproxen-induced COL10A1 expression is through the increased 5-lipoxygenase signaling.
Introduction
Osteoarthritis (OA) is a common disorder of articular joints characterized by slow progressive damage and loss of the articular cartilage.1–3 Besides the functional and psychological impact of the disease on the affected individuals, OA is associated with a significant socioeconomic burden.2 All the currently approved treatments for OA, including nonsurgical and surgical approaches, are focused on relieving the symptoms of pain and stiffness as well as improving the function of the joint.4 However, an effective treatment for the regeneration of the damaged articular cartilage is still lacking.
Adult mesenchymal stem cells (MSCs) are multipotent and capable of self-renewal and able to differentiate into chondrocytes, osteoblasts, and other tissues of mesenchymal origin.5 Alternative treatment efforts for cartilage degeneration include tissue engineering of cartilage, using MSCs induced to differentiate into chondrocytes.6,7 Autologous MSCs taken from OA patients are being currently considered in the biological repair of cartilage or disc lesions. However, expression of type X collagen (COL X), which is a marker of late-stage chondrocyte hypertrophic differentiation and endochondral ossification,8,9 in MSCs from OA patients10–12 is an undesirable degenerative phenotype. COL X is a nonfibrillar collagen, primarily a specific marker for terminally differentiated chondrocytes in the hypertrophic zone of growth plates in the growing bones.13,14 Although it is well established that there is constitutive expression of COL X in chondrocytes and MSCs from OA patients,8,11 the factors and intracellular mechanisms that regulate this phenomenon are not well understood.
We have recently observed among the nonsteroidal anti-inflammatory drugs (NSAIDs), commonly prescribed to OA patients, tested in our study, that specifically Naproxen but not Ibuprofen, Diclofenac, or Celebrex induces COL10A1 gene expression as well as osteogenic marker genes, like alkaline phosphatase, osteocalcin, and bone sialoprotein, in normal human MSCs.15 Similarly, several studies have revealed the negative impact of NSAIDs on articular cartilage.16,17 The most important drawback of certain NSAIDs is to inhibit matrix production. NSAIDs can be classified into three categories on the basis of their different effects on chondrocyte GAG synthesis, that is, inhibitory (e.g., Indomethacin and Naproxen), stimulatory (e.g., Aceclofenac, Tenidap, and Tolmetin), or having no effect (e.g., Diclofenac, Aspirin, and Piroxicam). These differential effects may stem from their action on the function of IL-1β and growth factors in chondrocytes.
Since cytokines and growth factors are involved in the regulation of balance between anabolic and catabolic processes, which is disturbed in OA, it is important that the pharmacological agents should act to maintain this balance and preserve and protect the cartilage. Thus, inhibition of cytokine action and stimulation of matrix synthesis as promoted by aceclofenac is favorable for cartilage protection. It has also been shown that licofelone—a potent, competitive inhibitor of 5-LOX and COX pathways—possesses antiplatelet, analgesic, anti-inflammatory, antipyretic, and antibronchoconstrictory properties and also to be safe for the gastrointestinal tract at effective doses.
Preclinical studies indicated that licofenolone is effective in protecting the articular cartilage and the synovial space in degenerative joint disease and also displays antithrombotic activity.18 These drugs, by inducing the expression of markers related to hypertrophy and osteogenesis, might accelerate the OA-related changes. It is necessary to identify the underlying mechanisms in order to devise steps for modulating the expression of hypertrophy- and osteogenesis-related markers in MSCs from OA patients. NSAIDs are known to exert anti-inflammatory effects mainly through cyclooxygenase inhibition, p38 MAPK, and 5-lipoxygenase pathways.19 Previously it was shown that Ibuprofen, Diclofenac, and Celebrex inhibit 5-lipoxygenase,20–24 while Naproxen increases 5-lipoxygenase expression.25 We have previously shown that mitogen-activated protein kinases (MAPKs) regulate the expression of COL X in parathyroid-hormone-treated MSCs.26 In the present study we examined whether Naproxen-induced COL X expression in normal and OA hMSCs also involves 5-lipoxygenase and/or MAPK signaling.
Materials and Methods
Source and isolation of MSCs
Normal human MSCs were purchased from Lonza. Osteoarthritic human mesenchymal stem cells (OA hMSCs) were isolated from bone marrow aspirates of the intramedullary canal of femur from donors (four women of age 56, 62, 72, and 79 years and three men of age 53, 59, and 78 years) undergoing total hip replacement for OA, following a protocol approved by the research ethics committee of the Jewish General Hospital (Montreal, Quebec, Canada).11,12,27 Isolation of MSCs from bone marrow aspirates was carried out using previously described methods.12,28
Briefly, each aspirate was diluted 1:1 (v/v) with Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Wisent, Inc.) and then layered over Ficoll (Ficoll-Paque Plus; GE Healthcare Bio-Sciences). The mononuclear cell layer, after being centrifuged at 900 g for 30 min, was removed from the interface, washed with DMEM, and resuspended in DMEM supplemented with 10% fetal bovine serum (FBS; Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured in 20 mL of medium in 176-cm2 culture dishes and incubated at 37°C in a 5% CO2 humidified atmosphere. After 72 h, nonattached cells were washed off and the adherent cells were thoroughly washed twice with DMEM. Cells were cultured and expanded in complete medium (DMEM high-glucose [4.5 g/L] supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin) and were used within three to four passages.
Treatment of normal and OA hMSCs with Naproxen for determining COL X protein expression
Normal and OA hMSCs were expanded in 100×20-mm2 culture dishes in 10 mL of complete medium to 80–90% confluence. Then, the MSCs were incubated overnight in DMEM containing 1% FBS and then treated without (control) or with Naproxen sodium (Sigma-Aldrich) dissolved in DMEM containing 1% FBS for 72 h at 37°C in a 5% CO2 humidified atmosphere. Cell lysates are prepared in RIPA buffer (Sigma-Aldrich) containing 1% protease inhibitor cocktail (Sigma-Aldrich).
Since the bone marrow is well vascularized, the concentration of Naproxen in our experiments was similar to the blood-circulating levels in patients taking Naproxen. The blood concentration of Naproxen in patients is around 50–75 μg/mL,29 at 220 mg Naproxen dosage.30 Naproxen has an average pharmacokinetic half-life of>12 h when administered at high doses (500 mg b.i.d.) and it has been demonstrated that effective inhibition of COX1 can only be achieved for longer periods (up to 24 h) only when Naproxen is given at 440 mg b.i.d. dose in patients.30
Blood concentrations of Naproxen at this dosage were reported to be about 120 μg/mL, as compared with 220 mg that achieves blood levels of ∼75 μg/mL. In fact, in vitro, Naproxen exerts near complete inhibition of both COX1 and COX2 at ∼100 μg/mL concentration. For these reasons, we chose to employ Naproxen at 100 μg/mL in this study, which corresponds to intermediate blood concentration between low and high dosage of Naproxen.
Treatment of normal and OA hMSCs with Naproxen for examining MAPK phosphorylation
For MAPK phosphorylation studies, normal and OA hMSCs were expanded to 80–90% confluence and were serum deprived overnight and were treated without (basal phosphorylation) or with Naproxen sodium (100 μg/mL) dissolved in serum-free medium, for different time points (0.5, 1, and 6 hours at 37°C in a 5% CO2 humidified atmosphere). Cell lysates are prepared in RIPA buffer containing 1% protease inhibitor and 1% phosphatase inhibitor cocktails (Sigma-Aldrich).
Treatment of normal and OA hMSCs with MAPK and 5-lipoxygenase inhibitors
For the studies with inhibitors, MSCs were cultured in complete DMEM to near confluence and were serum deprived for 24 h followed by treatment with MEK inhibitor (10 μM; 1, 4-diamino-2, 3-dicyano-1, 4-bis [2-aminophenylthio] butadiene, U0126; Sigma-Aldrich), c-Jun N-terminal kinase (JNK) inhibitor (1 μM, BI-78D3; Sigma-Aldrich), p38 inhibitor (20 μM, SB203580; Cell Signaling), and 5-lipoxygenase inhibitor (10 μM, MK-886; Sigma-Aldrich) for 30 min prior to stimulation with Naproxen (100 μg/mL).
Measurement of gene expression by real-time polymerase chain reaction
At the end of incubations total RNA from MSCs was isolated using Trizol reagent (Invitrogen). The resulting RNA pellet was washed with 75% ethanol, and then centrifuged and air-dried. Then, the pellets were suspended in 40 μL of diethylpyrocarbamate-treated (DEPC) water and RNA concentration was determined using Nanodrop 3000 fluorospectrometer. Reverse transcription was performed using the RNA (1 μg) with random primers (final concentration 0.15 μg/μL), dNTP mixture (final concentration 0.5 mM), and DEPC water in a final volume of 12 μL. After the solution was incubated at 65°C for 5 min, it was mixed with first-strand buffer, DTT, RNaseOUT, and Superscript II reverse transcriptase in a final volume of 20 μL. Then, the solution was incubated at 45°C for 50 min and then at 70°C for 15 min to inactivate the reverse transcriptase. For LightCycler real-time polymerase chain reaction (PCR), a master mix of the following reaction components was prepared with final concentrations: 10 μL SYBER PCR master mix (1×) (Qiagen), 8 μL water, 0.5 μL forward primer (0.25 μM), and 0.5 μL reverse primer (0.25 μM) (Table 1). To this mix, 1 μL of cDNA was added as PCR template.
Table 1.
Primer Sequences
| Gene | Sequence | Product size (bp) |
|---|---|---|
| ALOX5 | Forward: TGG AGA GCA AAG AAG ACA TCC | 123 |
| Reverse: GCC GTA CAC GTA GAC ATC GTT | ||
| RUNX2 | Forward: CAG ACC AGC AGC ACT CCA TA | 178 |
| Reverse: CAG CGT CAA CAC CAT CAT TC | ||
| COL10A1 | Forward: AAT GCC TGT GTC TGC TTT TAC | 130 |
| Reverse: ACA AGT AAA GAT TCC AGT CCT | ||
| GAPDH | Forward: TGA AGG TCG GAG TCA ACG GAT | 181 |
| Reverse: TTC TCA GCC TTG ACG GTG CCA |
The reaction conditions included one cycle of 95°C for 15 min, 45 cycles of amplification and quantification (94°C for 15 s, 57°C for 30 s, and 72°C for 30 s), one cycle of melting curve (65–95°C with heating rate of 0.1°C per second with a continuous fluorescence measurement), and a final cooling step to 4°C. The crossing points (CPs) were determined by the Light Cycler software 3.3 (Roche Diagnostics) and were measured at constant fluorescence level. Every sample was run in duplicates and the average value was used in the calculation. The relative gene transcription was determined by the following equation and GAPDH was employed as the reference gene:
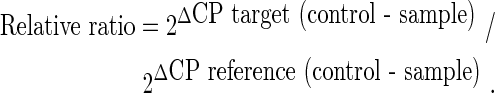 |
Protein expression
Total protein content in the cell lysates was measured by Bradford assay and equal amounts of protein from each sample were resolved on 10% acrylamide gel and, after SDS-PAGE, the separated proteins were transferred to nitrocellulose membranes by semidry electrophoretic transfer using Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad). Protein expression was measured by western blot using specific antibodies directed against COL X (1:1000; Sigma-Aldrich), extracellular signal-regulated kinase (ERK), phosphorylated-ERK (Erk1/2 and pErk1/2; 1:5000), JNK, phosphorylated-JNK (JNK and pJNK; 1:1000), and p38, phosphorylated-p38 (p38, 1:1000 and p-p38, 1:500; all from Cell Signaling). GAPDH (1:10,000; Sigma-Aldrich) was used as a housekeeping gene and utilized to normalize for the expression of COL X. The phosphorylation of ERK, p38, and JNK was normalized to the corresponding nonphosphorylated total forms. Protein bands on the immunoblot were visualized using the chemiluminescent reagents (Bio-Rad Clarity™ Western ECL Substrate) followed by exposure to the photographic film or by Bio-Rad VersaDoc (Bio-Rad Canada). Western blot photographic films were scanned and pixel intensity was quantified by densitometry using ImageJ (NIH) software.
Statistical analysis
Experiments were carried out using the cells from three donors for normal MSCs and from four to seven donors for OA hMSCs. Statistical analysis was performed using analysis of variance followed by Fisher's protected least significant difference post-hoc test using Prism statistical program. Results are presented as the mean±SE. Differences were considered statistically significant with p<0.05.
Results
Effect of Naproxen on COL X protein expression in normal and OA hMSCs
Treatment of normal MSCs with Naproxen (100 μg/mL) for up to 72 h significantly increased COL X protein expression (170%±20%). MSCs isolated from OA patients showed elevated basal COL X expression as compared with normal MSCs (285%±14%) and the expression was further augmented significantly by treatment with Naproxen for 72 h (459%±16%) (Fig. 1).
FIG. 1.

Effect of Naproxen on type X collagen (COL X) expression in normal and osteoarthritis (OA) hMSCs. Normal hMSCs (Lonza) and the MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in DMEM (1% FBS and 1% PS) without or with Naproxen (100 μg/mL) for up to 72 h. COL X protein expression was determined by immunoblotting. GAPDH was used as a housekeeping gene and utilized to normalize the results. Values represent the mean±SE of four experiments (*p<0.05; ***p<0.001 compared with untreated normal hMSCs; ###p<0.001 compared with untreated OA hMSCs). DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; hMSCs, human mesenchymal stem cells.
Basal phosphorylation levels of MAPKs in normal versus OA hMSCs
MAPKs are important mediators of inflammation and modulate intracellular signal transductions involved in expression of osteogenesis markers like Runt-related transcription factor 2 (Runx2), alkaline phosphatase (ALP), and osteocalcin (OC).31 Runx2 is known to regulate transcription of COL10A1.32 Since MSCs from OA patients show elevated COL10A1 expression, we first examined whether there is any relationship between MAPK signaling and the expression of COL X in MSCs. Basal phosphorylation levels of ERK, JNK, and p38 in MSCs from both normal and OA patients were measured and the results showed that the phosphorylation of all these three MAPKs in MSCs from OA patients was significantly higher (pERK1/2 [307%±69%] [Fig. 2A]; pJNK [943%±92%] [Fig. 2B]; and p-p38 [132%±19%] [Fig. 2C]) than in normal MSCs.
FIG. 2.

Increased mitogen-activated protein kinase (MAPK) activation in OA hMSCs as compared with normal hMSCs. MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty and normal human MSCs (Lonza) were cultured in complete DMEM to near confluence. Then, the cells were serum starved for 24 h. The protein expression and phosphorylation of MAPKs (ERK, JNK, and p38) in MSC lysates were determined by immunoblotting. Values represent the mean±SE of seven donors (A: pERK1/2, 307±69, ***p<0.001; B: pJNK, 943±92, ****p<0.0001; and C: p-p38, 132±19, *p<0.05) as percentage of normal human MSCs (100%). ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase.
Effect of Naproxen on MAPK phosphorylation in normal and OA hMSCs
We investigated whether Naproxen has any effect on MAPK signaling in normal MSCs and OA hMSCs. Normal and OA hMSCs were incubated with Naproxen (100 μg/mL) for 0.5 to 6 h. In normal MSCs, phosphorylation of ERK significantly increased by 1 h (153%±12%) following Naproxen treatment and by 6 h it reached below control levels (50%±26%) (Fig. 3A). Similarly Naproxen significantly increased the phosphorylation of JNK by 0.5 h (119%±3%), which also decreased to below control levels by 6 h (Fig. 3B). Naproxen treatment increased phosphorylation of p38 by 0.5 h and reached significant levels by 6 h (282%±106%) (Fig. 3C). In OA hMSCs, phosphorylation of ERK significantly increased by 0.5 h (294±24) following Naproxen treatment and after 6 h reached to nonsignificant levels (Fig. 4A). Naproxen treatment had no significant effect on the phosphorylation of JNK (Fig. 4B) but increased the phosphorylation of p38 by 0.5 h and this increase was statistically significant at 1 h (170±32) and continued to be significant even after 6 h (195±39) (Fig. 4C).
FIG. 3.

Effect of Naproxen on phosphorylation of MAPKs in normal hMSCs. Normal hMSCs were cultured to near confluence in complete DMEM. Then, the cells were serum starved for 24 h followed by treatment with Naproxen (100 μg/mL) or without (CTL) for up to 6 h. Protein expression and phosphorylation of ERK 1/2, JNK, and p38 were determined by immunoblotting and protein bands were quantified using Image J (NIH) software. Total ERK 1/2, JNK, and p38 were used to normalize corresponding phosphorylated forms. Values represent the mean±SE of three experiments. (A) Effect of Naproxen on phosphorylation of extracellular signal-regulated kinase (ERK). Naproxen significantly increased the phosphorylation of ERK at 1 h (153±12, *p<0.05), which reached to below control levels after 6 h (50±26). (B) Effects of Naproxen on phosphorylation of c-Jun N-terminal kinase (JNK). Naproxen significantly increased the phosphorylation of JNK in 0.5 h (119±3, *p<0.05), which reached to control levels after 6 h. (C) Effects of Naproxen on phosphorylation of p38. The phosphorylation of p38 increased by 30 min and this increase was statistically significant at 6 h (282±106, **p<0.01).
FIG. 4.

Effect of Naproxen on phosphorylation of MAPKs in OA hMSCs. OA hMSCs were cultured to near confluence in complete DMEM. Then, the cells were serum starved for 24 h and were treated without (CTL) or with Naproxen (100 μg/mL) for up to 6 h. Protein expression and phosphorylation of ERK 1/2, JNK, and p38 were determined by immunoblotting and protein bands were quantified using Image J (NIH) software. Total ERK 1/2, JNK, and p38 were used to normalize corresponding phosphorylated forms. Values represent the mean±SE of four donors. (A) Effect of Naproxen on phosphorylation of ERK. Naproxen had significantly increased the phosphorylation of ERK by 0.5 h (294±24, ***p<0.001) and reached to nonsignificant levels only after 6 h. (B) Effects of Naproxen on phosphorylation of JNK. Naproxen did not change the phosphorylation of JNK as compared with control. (C) Effects of Naproxen on phosphorylation of p38. The phosphorylation of p38 increased by 0.5 h and this increase was statistically significant at 1 h (170±32, *p<0.05) and 6 h (195±39, *p<0.05).
Effect of MAPK inhibitors on Naproxen-induced COL10A1 expression in normal and OA hMSCs
To investigate whether the MAPKs are implicated in the Naproxen-induced COL10AI expression, normal and OA hMSCs were treated with Naproxen (100 μg/mL) in the presence of MAPK inhibitors. In normal MSCs, MAPK inhibitors did not show any significant effect on Naproxen-induced COL10A1 expression (Fig. 5A) even though COL10AI expression seems to show a downward trend in presence of JNK (BI-78D3) and p38 (SB203580) inhibitors. In OA hMSCs, basal COL10A1 expression, which was significantly higher than in normal MSCs, was attenuated by JNK inhibitor (BI-78D3) and p38 inhibitor (SB203580) by 0.69-fold and 0.43-fold, respectively, while ERK inhibitor (U0126) had no effect. Naproxen-induced COL10A1 expression in OA hMSCs was not altered by ERK and JNK inhibitors; on the other hand, p38 inhibitors appeared to increase its expression further by approximately threefold over Naproxen alone (Fig. 5B).
FIG. 5.

(A) Effect of MAPK inhibitors on Naproxen-induced COL10A1 expression in normal MSCs. Normal MSCs (Lonza) were cultured in complete DMEM to near confluence. Then, the cells were serum deprived for 24 h and were treated with or without Naproxen (100 μg/mL) in the absence or presence of MAPK inhibitors (ERK inhibitor: U0126 [10 μM], JNK inhibitor: BI-78D3 [1 μM], and p38 inhibitor: SB203580 [20 μM]). Gene expression was measured by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Values represent the mean±SE of three experiments (**p<0.01; compared with untreated control [0.1% DMSO]). (B) Effect of MAPK inhibitors on Naproxen-induced COL10A1 expression in OA hMSCs. MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence. Then, the cells were serum deprived for 24 h followed by treatment without or with Naproxen (100 μg/mL) in presence or absence of MAPK inhibitors (ERK inhibitor: U0126 [10 μM], JNK inhibitor: BI-78D3 [1 μM], and p38 inhibitor: SB203580 [20 μM]). Gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Values represent the mean±SE of six donors (***p<0.001, compared with untreated control [0.1% DMSO]; ###p<0.001, compared with Naproxen [100 μg/mL] and 0.1% DMSO treated).
Effect of MAPK inhibitors on Naproxen-induced Runx2 expression in normal and OA hMSCs
In normal MSCs, ERK inhibitor (U0126) did not show any effect on Naproxen-induced Runx2 expression while it was suppressed by JNK inhibitor (BI-78D3) (∼1-fold) and by p38 inhibitor (SB203580) (1.7-fold), which was statistically significant (Fig. 6A). In OA hMSCs, similar to COL10A1, Runx2 expression was significantly attenuated by JNK inhibitor (BI-78D3) and p38 inhibitor (SB203580) by 0.7- and 0.5-fold, respectively, while ERK inhibitor (U0126) had no effect. Naproxen-induced Runx2 expression in OA hMSCs was not altered significantly by any of the MAPK inhibitors (Fig. 6B), but similar to COL10A1, there was a downward trend in expression with JNK and p38 inhibitors.
FIG. 6.

(A) Effect of MAPK inhibitors on Naproxen-induced Runx2 expression in normal MSCs. Normal MSCs (Lonza) were cultured in complete DMEM to near confluence. Then, the cells were serum deprived for 24 h followed by treatment with or without Naproxen (100 μg/mL) in absence or presence of MAPK inhibitors (ERK inhibitor: U0126 [10 μM], JNK inhibitor: BI-78D3 [1 μM], and p38 inhibitor: SB203580 [20 μM]). Gene expression was measured by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Values represent the mean±SE of three experiments (*p<0.05, compared with untreated control [0.1% DMSO]; ##p<0.01, compared with Naproxen [100 μg/mL] and 0.1% DMSO treated). (B) Effect of MAPK inhibitors on Naproxen-induced Runx2 expression in OA hMSCs. MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence and were serum deprived for 24 h. Then, the cells were treated without or with Naproxen (100 μg/mL) in presence or absence of MAPK inhibitors (ERK inhibitor: U0126 [10 μM], JNK inhibitor: BI-78D3 [1 μM], and p38 inhibitor: SB203580 [20 μM]). Gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Values represent the mean±SE of six donors (*p<0.05; ***p<0.001, compared with untreated control [0.1% DMSO]).
Effect of Naproxen on 5-lipoxygenase (ALOX5) and COL10A1 gene expression in normal and OA hMSCs
Naproxen treatment for 24 h significantly increased ALOX5 gene expression (3.29-fold) in normal MSCs. Basal ALOX5 gene expression on OA hMSCs is significantly higher (8-fold) compared with normal hMSCs. OA hMSCs treated with Naproxen had significantly higher ALOX5 gene expression (13-fold over untreated normal hMSCs and 5-fold over untreated OA hMSCs) (Fig. 7A). Similarly Naproxen treatment augmented the COL10A1 expression in both normal (6-fold) and OA hMSCs (18-fold). Basal COL10A1 expression in OA hMSCs was significantly higher (sevenfold) as compared with normal hMSCs (Fig. 7B). These results indicate that there was a clear correlation between the expression of ALOX5 and COL10A1.
FIG. 7.

Effect of Naproxen on 5-lipoxygenase (ALOX5) and type X collagen (COL10A1) gene expression in normal and OA hMSCs. Normal hMSCs (Lonza) and the MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence and were serum deprived for 24 h followed by treatment without or with Naproxen (100 μg/mL). ALOX5 (A) and COL10A1 (B) gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Normal hMSC values represent the mean±SE of three experiments (*p<0.05; **p<0.01; compared with untreated normal hMSC control). OA hMSC values represent the mean±SE of six donors (*p<0.05; **p<0.01; ***p<0.001; compared with untreated normal hMSC control).
Effect of p38 inhibitor on 5-lipoxygenase (ALOX5) gene expression in normal and OA hMSCs
Treatment with p38 inhibitor (SB203580) alone significantly suppressed ALOX5 gene expression in normal (0.78-fold) and OA hMSCs (0.45-fold) while it further upregulated ALOX5 gene expression in the presence of Naproxen both in normal (2.06-fold) and OA hMSCs (2.69-fold) when compared with corresponding p38-inhibitor untreated controls (Fig. 8).
FIG. 8.

Effect of p38 inhibitor on 5-lipoxygenase (ALOX5) gene expression in normal and OA hMSCs with and without Naproxen treatment. Normal hMSCs (Lonza) and the MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence. Then, the cells were serum deprived for 24 h followed by treatment without or with p38 inhibitor (SB203580, 20 μM) and Naproxen (100 μg/mL). ALOX5 gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Normal hMSC values represent the mean±SE of three experiments (***p<0.001, compared with untreated normal hMSC control; #p<0.05, compared with Naproxen-treated normal hMSCs without p38 inhibitor). OA hMSC values represent the mean±SE of six donors (§§p<0.01, compared with untreated OA hMSC control; $p<0.05, compared with Naproxen-treated OA hMSCs without p38 inhibitor).
Effect of 5-lipoxygenase inhibitor (MK-886) on COL10A1 gene expression in normal and OA hMSCs
Naproxen-induced COL10A1 expression was significantly attenuated by 5-lipoxygenase inhibitor in normal (threefold) and OA hMSCs (twofold) (Fig. 9).
FIG. 9.

Effect of 5-lipoxygenase inhibitor (MK-886) on Naproxen-induced type X collagen (COL10A1) gene expression in normal and OA hMSCs. Normal hMSCs (Lonza) and the MSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence and were serum deprived for 24 h followed by treatment without or with 5-LOX inhibitor (MK-886, 10 μM) and Naproxen (100 μg/mL). COL10A1 gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Normal hMSC values represent the mean±SE of three experiments (**p<0.01, compared with untreated normal hMSC control; #p<0.05, compared with Naproxen-treated normal MSCs without 5-LOX inhibitor). OA hMSC values represent the mean±SE of six donors (###p<0.001, compared with Naproxen-treated OA hMSCs without 5-LOX inhibitor).
Effect of 5-lipoxygenase inhibitor (MK-886) on Runx2 gene expression in normal and OA hMSCs
Naproxen-induced Runx2 expression was suppressed by the 5-lipoxygenase inhibitor in normal and OA hMSCs and was statistically significant in OA hMSCs (1.7-fold) (Fig. 10).
FIG. 10.

Effect of 5-lipoxygenase inhibitor (MK-886) on Naproxen-induced RUNX2 gene expression in normal and OA hMSCs. Normal hMSCs (Lonza) and the hMSCs isolated from bone marrow aspirates of OA human donors undergoing total hip anthroplasty were cultured in complete DMEM to near confluence and were serum deprived for 24 h followed by treatment without or with 5-LOX inhibitor (MK-886, 10 μM) and Naproxen (100 μg/mL). RUNX2 gene expression was measured after 24 h of treatment by real-time RT-PCR. GAPDH was used as the housekeeping gene to normalize the results. Normal hMSC values represent the mean±SE of three experiments (*p<0.05, compared with Naproxen-treated normal hMSCs without 5-LOX inhibitor). OA hMSC values represent the mean±SE of six donors (###p<0.001, compared with Naproxen-treated OA hMSCs without 5-LOX inhibitor).
Discussion
The purpose of this study was to investigate the mechanism underlying the induction of COL10A1 by Naproxen in human MSCs and the signal transduction pathways involved. Human MSCs from OA patients, which are used as a potential cell source for cartilage tissue engineering, have elevated expression of COL X, a marker of late-stage chondrocyte hypertrophy and associated with endochondral ossification.15 Other osteogenesis-related markers are also present in increased levels in MSCs from OA patients.8,10–12 We have recently investigated the effect of commonly prescribed drugs to OA patients, including NSAIDs and acetaminophen, on the expression of markers of chondrocyte hypertrophy and osteogenesis in normal and OA hMSCs and our results showed that Naproxen, an NSAID and a nonspecific inhibitor of cyclooxygenase, can significantly increase the expression levels of COL10A1 in human MSCs from normal and OA subjects.15
In the present study, using normal and OA hMSCs, we further show that the effect of Naproxen on COL X expression is not only at the transcriptional level, but also at the protein level. Similar detrimental effects of Naproxen on matrix synthesis both in normal and OA articular cartilage were reported earlier.17 The nonselective NSAIDs like Naproxen, by inhibiting cyclooxygenases 1 and 2, can reduce production of prostaglandins and downstream activation of p38 but this can lead to accumulation of arachidonic acid and its diversion to the formation of leukotrienes via lipoxygenase pathway. Leukotrienes are known to enhance the phosphorylation/activation of p38 MAPK, which in turn stabilizes the cyclooxygenase-2 mRNA.33,34 The COL10A1 transcription factor Runx2 is known to be activated/phosphorylated by p-p38 and thus increases the expression of COL10A1.31,32
Since NSAIDs can alter MAPK signaling, in particular, p38 MAPK, in this study we investigated whether altered MAPK signaling pathways play a role in Naproxen-induced increase in the expression of COL10A1 in human MSCs. Our results indicate MAPK involvement in the elevated COL10A1 expression in OA hMSCs as the increased basal COL10A1 was attenuated with p38 and JNK inhibitors. On the other hand, Naproxen-induced COL10A1 expression in both normal and OA hMSCs was unchanged by MAPK inhibitors, indicating involvement of a different signaling mechanism.
Previously it was shown in human OA chondrocytes that Naproxen increased 5-lipoxygenase mRNA by 330% after 24 h of incubation.25 In our current study we observed a significant increase in 5-lipoxygenase mRNA levels with Naproxen treatment for 24 h, by 3.29-fold in normal MSCs and by 13-fold in OA hMSCs as compared with untreated normal MSCs, indicating a possible diversion of accumulated arachidonic acid toward lipoxygenase pathway. 5-Lipoxygenase catalyzes oxidation of arachidonic acid to leukotrienes (LTB4) through several intermediary steps.35 These leukotrienes are known to activate MAPKs (p38 and ERK),36,37 which are instrumental in activation/phosphorylation of COL10A1 transcription factor Runx2 and thus increasing its expression.31 In the present study, we found that Naproxen-induced COL10A1 and Runx2 expression was significantly reduced both in normal and OA hMSCs by 5-lipoxygenase inhibitor.
Identifying the signaling mechanisms that regulate the expression of markers of chondrocyte hypertrophy in human MSCs is necessary in order to help preventing hypertrophic differentiation and ossification during cartilage tissue engineering.
MAPK signaling has been proposed to be involved in the different stages of chondrogenic differentiation.38 There is growing evidence that MAPK signaling pathways probably play a critical role in OA-related changes of chondrocytes.39–42 Further, it has been shown that inhibition of one or more MAP kinases in chondrocytes both in vitro and in vivo results in slowing down of the progression of OA-related changes.41,43,44 The possible involvement of MAPK in the upregulation of markers of chondrocyte hypertrophy and ossification became evident as all the MAPKs (p38, JNK, and ERK) are activated even under basal conditions in MSCs from OA patients as compared with normal MSCs. Thus, the elevated expression of COL10A1 seen in MSCs from OA patients in earlier studies15 and in the present studies is associated with MAPK signaling. Besides p38 MAPK, ERK1/2 activation may also contribute to the upregulation of COL10A1 gene expression by inactivating the inhibitory transcription factor Foxo3a (Fig. 11). NSAIDs that increase the flux through 5-lipoxygenase pathways and/or increase the expression of 5-lipoxygenase can potentially lead to further elevation in the expression of COL X and aggravate the pathogenesis of OA.
FIG. 11.

Arachidonic acid is liberated from the cell membrane phospholipids by cytoplasmic phospholipase A2 (PLA2). Free arachidonic acid can be metabolized through cyclooxygenase (COX) and the lipoxygenase (LOX) pathways. In the COX pathway, arachidonic acid is enzymatically converted to biological mediators called prostanoids, including prostaglandins (PGs) by specific prostaglandin synthases through several intermediary steps. COX-1 is the constitutive cyclooxygenase and maintains basal levels of prostaglandins, whereas COX-2 is inducible and produces prostaglandins under inflammatory conditions. In the 5-LOX pathway, arachidonic acid is converted into biologically active metabolites, such as leukotrienes (LTB4), which also trigger inflammatory response. Prostaglandin-mediated activation of p38 leads to the stabilization of COX-2 mRNA, which further enhances the prostaglandin production and inflammation through COX-2 pathway. Activated p38-MAPK phosphorylates and activates Runx-2 transcription factor, which triggers the expression of COL10A1 gene. Leukotrienes (LTB4) derived from 5-LOX pathway also activate p38-MAPK as well as ERK1/2 by phosphorylation. Phospho-ERK1/2 can also lead to the activation of Runx-2. Besides activating Runx-2, phospho-ERK1/2 is also known to inactivate and alleviate the inhibitory action of FOXO3a, thus further increasing the COL10A1 gene expression. Naproxen, by inhibiting both the COX-1 and COX-2, causes diversion of arachidonic acid to 5-LOX-pathway-mediated leukotriene production. These leukotrienes (LTB4) in turn upregulate COL10A1 gene expression.
Considering the importance of NSAIDs as the most prescribed antipain medication for patients with OA, the results from the present study point toward caution that needs to be exercised, while suggesting the use of these drugs. This is particularly true for drugs like Naproxen that target only COX and those with similar molecular action. One can speculate that drugs such as licofelone that show dual inhibition of COX and LOX pathways are more likely to have better beneficial effects in preventing inflammatory responses than the COX inhibitors.45 In fact, it has recently been reported in a 2-year clinical trial with 161 knee OA patients that the total knee replacements were much higher in patients receiving Naproxen than in patients given licofelone,46,47 indicating the beneficial significance of choosing a COX/LOX inhibitor over COX-only inhibitor in OA patients. Interestingly, licofelone also possesses anticancer effects and is currently at advanced stage of clinical development status with completed phase III trials.48
In conclusion, the findings of our study contribute to advancing our understanding of the molecular mechanisms underlying COL10A1 expression in human MSCs, implicating MAPK signaling pathways in the chondrocyte hypertrophic differentiation of OA hMSCs. Our study also explains how NSAIDs like Naproxen aggravate the pathogenesis of OA by increasing COL X expression via 5-lipoxygenase. Further research is required to identify and target other signaling pathways involved in this process, which may provide a basis for finding therapy directed toward reverting the hypertrophic changes associated with OA.
Acknowledgment
This study was supported by the Canadian Institutes of Health Research (CIHR).
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Arden N., and Nevitt M.C.Osteoarthritis: epidemiology. Best Pract Res Clin Rheumatol 20,3, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Litwic A., Edwards M.H., Dennison E.M., and Cooper C.Epidemiology and burden of osteoarthritis. Br Med Bull 105,185, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suri P., Morgenroth D.C., and Hunter D.J.Epidemiology of osteoarthritis and associated comorbidities. PM R 4,S10, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Lutzner J., Kasten P., Gunther K.P., and Kirschner S.Surgical options for patients with osteoarthritis of the knee. Nat Rev Rheumatol 5,309, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Tuan R.S., Boland G., and Tuli R.Adult mesenchymal stem cells and cell-based tissue engineering. Arthritis Res Ther 5,32, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qi Y., Feng G., and Yan W.Mesenchymal stem cell-based treatment for cartilage defects in osteoarthritis. Mol Biol Rep 39,5683, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Mobasheri A., Csaki C., Clutterbuck A.L., Rahmanzadeh M., and Shakibaei M.Mesenchymal stem cells in connective tissue engineering and regenerative medicine: applications in cartilage repair and osteoarthritis therapy. Histol Histopathol 24,347, 2009 [DOI] [PubMed] [Google Scholar]
- 8.von der Mark K., Kirsch T., Nerlich A., Kuss A., Weseloh G., Gluckert K., and Stoss H.Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum 35,806, 1992 [DOI] [PubMed] [Google Scholar]
- 9.Shen G.The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod Craniofac Res 8,11, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Mwale F., Girard-Lauriault P.L., Wang H.T., Lerouge S., Antoniou J., and Wertheimer M.R.Suppression of genes related to hypertrophy and osteogenesis in committed human mesenchymal stem cells cultured on novel nitrogen-rich plasma polymer coatings. Tissue Eng 12,2639, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Mwale F., Stachura D., Roughley P., and Antoniou J.Limitations of using aggrecan and type X collagen as markers of chondrogenesis in mesenchymal stem cell differentiation. J Orthop Res 24,1791, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Nelea V., Luo L., Demers C.N., Antoniou J., Petit A., Lerouge S., R Wertheimer M., and Mwale F.Selective inhibition of type X collagen expression in human mesenchymal stem cell differentiation on polymer substrates surface-modified by glow discharge plasma. J Biomed Mater Res Part A 75,216, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Mackie E.J., Ahmed Y.A., Tatarczuch L., Chen K.S., and Mirams M.Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40,46, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Tchetina E., Mwale F., and Poole A.R.Distinct phases of coordinated early and late gene expression in growth plate chondrocytes in relationship to cell proliferation, matrix assembly, remodeling, and cell differentiation. J Bone Miner Res 18,844, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Almaawi A., Wang H.T., Ciobanu O., Rowas S.A., Rampersad S., Antoniou J., and Mwale F.Effect of acetaminophen and nonsteroidal anti-inflammatory drugs on gene expression of mesenchymal stem cells. Tissue Eng Part A 19,1039, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Pelletier J.P.The influence of tissue cross-talking on OA progression: role of nonsteroidal antiinflammatory drugs. Osteoarthritis Cartilage 7,374, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Dingle J.T.The effects of NSAID on the matrix of human articular cartilages. Z Rheumatol 58,125, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Celotti F., and Durand T.The metabolic effects of inhibitors of 5-lipoxygenase and of cyclooxygenase 1 and 2 are an advancement in the efficacy and safety of anti-inflammatory therapy. Prostaglandins Other Lipid Mediat 71,147, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Rao P., and Knaus E.E.Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci 11,81s, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Markworth J.F., Vella L., Lingard B.S., Tull D.L., Rupasinghe T.W., Sinclair A.J., Maddipati K.R., and Cameron-Smith D.Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. Am J Physiol Regul Integr Comp Physiol 305,R1281, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kudo C., Kori M., Matsuzaki K., Yamai K., Nakajima A., Shibuya A., Niwa H., Kamisaki Y., and Wada K.Diclofenac inhibits proliferation and differentiation of neural stem cells. Biochem Pharmacol 66,289, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Maier T.J., Tausch L., Hoernig M., Coste O., Schmidt R., Angioni C., Metzner J., Groesch S., Pergola C., Steinhilber D., et al. Celecoxib inhibits 5-lipoxygenase. Biochem Pharmacol 76,862, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Konstan M.W., Vargo K.M., and Davis P.B.Ibuprofen attenuates the inflammatory response to Pseudomonas aeruginosa in a rat model of chronic pulmonary infection. Implications for antiinflammatory therapy in cystic fibrosis. Am Rev Respir Dis 141,186, 1990 [DOI] [PubMed] [Google Scholar]
- 24.Vanderhoek J.Y., and Bailey J.M.Activation of a 15-lipoxygenase/leukotriene pathway in human polymorphonuclear leukocytes by the anti-inflammatory agent ibuprofen. J Biol Chem 259,6752, 1984 [PubMed] [Google Scholar]
- 25.Chen S.H., Fahmi H., Shi Q., and Benderdour M.Regulation of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase-activating protein/5-lipoxygenase by 4-hydroxynonenal in human osteoarthritic chondrocytes. Arthritis Res Ther 12,R21, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mwale F., Yao G., Ouellet J.A., Petit A., and Antoniou J.Effect of parathyroid hormone on type X and type II collagen expression in mesenchymal stem cells from osteoarthritic patients. Tissue Eng Part A 16,3449, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Mwale F., Wang H.T., Nelea V., Luo L., Antoniou J., and Wertheimer M.R.The effect of glow discharge plasma surface modification of polymers on the osteogenic differentiation of committed human mesenchymal stem cells. Biomaterials 27,2258, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Colter D.C., Class R., DiGirolamo C.M., and Prockop D.J.Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci U S A 97,3213, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevelius H., Runkel R., Segre E., and Bloomfield S.S.Bioavailability of naproxen sodium and its relationship to clinical analgesic effects. Br J Clin Pharmacol 10,259, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capone M.L., Tacconelli S., Sciulli M.G., Anzellotti P., Di Francesco L., Merciaro G., Di Gregorio P., and Patrignani P.Human pharmacology of naproxen sodium. J Pharmacol Exp Ther 322,453, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Greenblatt M.B., Shim J.H., and Glimcher L.H.Mitogen-activated protein kinase pathways in osteoblasts. Annu Rev Cell Dev Biol 29,63, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Zheng Q., Zhou G., Morello R., Chen Y., Garcia-Rojas X., and Lee B.Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol 162,833, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsatsanis C., Androulidaki A., Venihaki M., and Margioris A.N.Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol 38,1654, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Faour W.H., He Y., He Q.W., de Ladurantaye M., Quintero M., Mancini A., and Di Battista J.A.Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J Biol Chem 276,31720, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Martel-Pelletier J., Mineau F., Fahmi H., Laufer S., Reboul P., Boileau C., Lavigne M., and Pelletier J.P.Regulation of the expression of 5-lipoxygenase-activating protein/5-lipoxygenase and the synthesis of leukotriene B(4) in osteoarthritic chondrocytes: role of transforming growth factor beta and eicosanoids. Arthritis Rheum 50,3925, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Gaudreault E., and Gosselin J.Leukotriene B4 potentiates CpG signaling for enhanced cytokine secretion by human leukocytes. J Immunol 183,2650, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Lee S.J., Kim C.E., Yun M.R., Seo K.W., Park H.M., Yun J.W., Shin H.K., Bae S.S., and Kim C.D.4-Hydroxynonenal enhances MMP-9 production in murine macrophages via 5-lipoxygenase-mediated activation of ERK and p38 MAPK. Toxicol Appl Pharmacol 242,191, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Bobick B.E., and Kulyk W.M.Regulation of cartilage formation and maturation by mitogen-activated protein kinase signaling. Birth Defects Res Part C Embryo Today 84,131, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Ding L., Heying E., Nicholson N., Stroud N.J., Homandberg G.A., Buckwalter J.A., Guo D., and Martin J.A.Mechanical impact induces cartilage degradation via mitogen activated protein kinases. Osteoarthritis Cartilage 18,1509, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malemud C.J.Protein kinases in chondrocyte signaling and osteoarthritis. Clin Orthop Relat Res S145, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Loeser R.F., Erickson E.A., and Long D.L.Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr Opin Rheumatol 20,581, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pelletier J.P., Fernandes J.C., Jovanovic D.V., Reboul P., and Martel-Pelletier J.Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: role of cyclooxygenase-2 and inducible nitric oxide synthase. J Rheumatol 28,2509, 2001 [PubMed] [Google Scholar]
- 43.Brown K.K., Heitmeyer S.A., Hookfin E.B., Hsieh L., Buchalova M., Taiwo Y.O., and Janusz M.J.P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J Inflamm (Lond) 5,22, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelletier J.P., Fernandes J.C., Brunet J., Moldovan F., Schrier D., Flory C., and Martel-Pelletier J.In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum 48,1582, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Ulbrich H., Soehnlein O., Xie X., Eriksson E.E., Lindbom L., Albrecht W., Laufer S., and Dannhardt G.Licofelone, a novel 5-LOX/COX-inhibitor, attenuates leukocyte rolling and adhesion on endothelium under flow. Biochem Pharmacol 70,30, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Raynauld J.P., Martel-Pelletier J., Bias P., Laufer S., Haraoui B., Choquette D., Beaulieu A.D., Abram F., Dorais M., Vignon E., et al. Protective effects of licofelone, a 5-lipoxygenase and cyclo-oxygenase inhibitor, versus naproxen on cartilage loss in knee osteoarthritis: a first multicentre clinical trial using quantitative MRI. Ann Rheum Dis 68,938, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Raynauld J.P., Martel-Pelletier J., Haraoui B., Choquette D., Dorais M., Wildi L.M., Abram F., and Pelletier J.P.Risk factors predictive of joint replacement in a 2-year multicentre clinical trial in knee osteoarthritis using MRI: results from over 6 years of observation. Ann Rheum Dis 70,1382, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Steinhilber D., and Hofmann B.Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin Pharmacol Toxicol 114,70, 2014 [DOI] [PubMed] [Google Scholar]