

Abstract
Aortic atherosclerosis (AoA) defined as intima-media thickening or plaques and aortic stiffness (AoS) also considered an atherosclerotic process and defined as decreased vessel distensibility (higher pulse pressure to achieve similar degree of vessel distension) are common in patients with SLE.
Immune-mediated inflammation, thrombogenesis, traditional atherogenic factors, and therapy-related metabolic abnormalities are the main pathogenic factors of AoA and AoS.
Pathology of AoA and AoS suggests an initial subclinical endothelialitis or vasculitis, which is exacerbated by thrombogenesis and atherogenic factors and ultimately resulting in AoA and AoS.
Computed tomography (CT) for detection of arterial wall calcifications and arterial tonometry for detection of increased arterial pulse wave velocity are the most common diagnostic methods for detecting AoA and AoS, respectively. MRI may become a more applicable and accurate technique than CT. Although transesophageal echocardiography accurately detects earlier and advanced stages of AoA and AoS, it is semi-invasive and cannot be used as a screening method.
Although imaging techniques demonstrate highly variable prevalence rates, on average about one third of adult SLE patients may have AoA or AoS.
Age at SLE diagnosis; SLE duration; activity and damage; corticosteroid therapy; metabolic syndrome; chronic kidney disease; and mitral annular calcification are common independent predictors of AoA and AoS. Also, AoA and AoS are highly associated with carotid and coronary atherosclerosis.
Earlier stages of AoA and AoS are usually subclinical. However, earlier stages of disease may be causally related or contribute to peripheral or cerebral embolism, pre-hypertension and hypertension, and increased left ventricular afterload resulting in left ventricular hypertrophy and diastolic dysfunction. Later stages of disease predisposes to visceral ischemia, aortic aneurysms and aortic dissection.
Even earlier stages of AoA and AoS have been associated with increased cardiovascular and cerebrovascular morbidity and mortality of SLE patients.
Aggressive non-steroidal immunosuppressive therapy and non-pharmacologic and pharmacologic interventions for control of atherogenic risk factors may prevent the development or progression of AoA and AoS and may decrease cardiovascular and cerebrovascular morbidity and mortality in SLE.
Keywords: Aorta, Atherosclerosis, Systemic Lupus Erythematosus, Thrombogenesis, Inflammation, Vasculitis
Background
Atherosclerosis is common in patients with systemic lupus erythematosus (SLE) and is associated with a 2-5 fold increased risk of future cardiac, cerebral or peripheral arterial ischemic events and mortality [1-7]. Patients with SLE as compared to age and sex matched controls and after controlling for traditional atherogenic risk factors, have a higher prevalence of subclinical carotid and aortic atherosclerosis (AoA) manifested as intima-media thickening or plaques and a higher prevalence of subclinical coronary artery disease (CAD) manifested as calcifications on computed tomography (CT) [1-4,8,9].
Unlike for carotid and CAD, the prevalence, characteristics, risk factors or predictors, clinical manifestations, prognosis, and therapy of AoA in SLE patients have not been well defined. This is important because AoA may have different clinical and prognostic implications than those of carotid and coronary atherosclerosis [10-12].
Premature aortic stiffness (AoS), generally considered an atherosclerotic process, is also common in patients with SLE [13]. However, in SLE it is unclear if AoS is a precursor of AoA, is a condition that occurs simultaneously but independently of AoA, or is a consequence of AoA.
The purpose of this article is to comprehensively review the pathogenesis, pathology, diagnostic methods, prevalence, risk factors or predictors, associations, clinical manifestations, prognosis, and therapy of AoA and AoS in SLE patients.
Definition
AoA is defined as intima-media thickening or plaques. Although a standardized definition of intima-media thickening is not available, recent reports have metrically defined it as 1.5-2 standard deviations (SD) above the mean intima-media thickness of healthy controls [9,14]. The overall intima-media thickness of the proximal, mid, and distal thoracic aorta in healthy controls is reported as 0.66 ± 0.15 mm. Therefore, intima-media thickening of >0.86 mm, which corresponds to the mean in normal controls plus 1.5SD, provides a specificity of 91% in a receiver-operating curve to detect intima-media thickening. Intima-media thickening of >0.96 mm, which corresponds to the mean in normal controls plus 2SD, provides an even higher specificity. Plaques are defined as >50% focal and protruding wall thickening as compared with the surrounding wall [1,9,14,15] (Figure 1).
Figure 1.
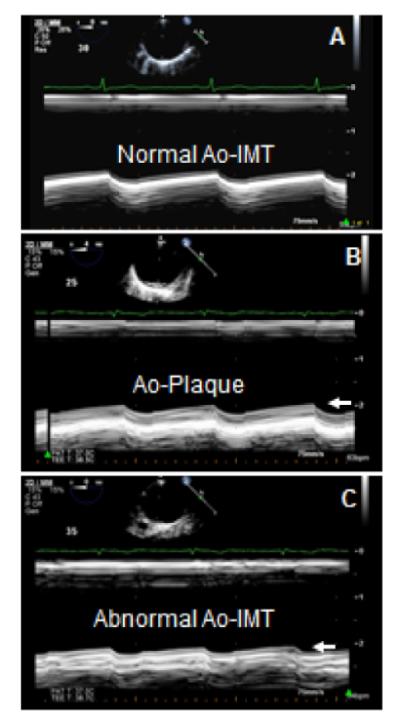
Aortic intima-media thickening and plaques by TEE. A: Short axis 2-dimensional (2-D) guided M-mode image of the anterior wall at the mid-level (30-35 cm) of the descending thoracic aorta demonstrating normal intima media thickness (IMT) of <0.8 mm (arrows) in a patient with SLE. B,C: 2-D guided M-mode image of a 48-year-old woman with SLE demonstrating a well-defined aortic plaque of 3.3 mm at the proximal (B, arrow) and abnormal IMT of 1.2 mm at the distal level of (C, arrow) descending thoracic aorta.
AoS occurs when increasingly higher amounts of force (pulse pressure) are required to achieve the same degree of vessel distension [9,13,16]. Although several parameters have been used to assess AoS, the Pressure–Strain Elastic Modulus is commonly reported and accepted as a standard. The Pressure–Strain Elastic Modulus is defined as [k(sBP-dBP)/(sD-dD/dD)]/10000, where k=133.3 is a conversion factor from mmHg to Nm−2 (Pascal units), sBP=systolic blood pressure, dBP=diastolic blood pressure, sD=systolic diameter, and dD=diastolic diameter. This is a well-established parameter for assessment of static arterial stiffness independently of intima-media thickness or plaques.
Pathogenesis
The highly interrelated pathogenic factors of AoA and AoS in SLE are immune-mediated inflammation, thrombogenesis, traditional atherogenic factors, and therapy-related metabolic abnormalities as delineated below [1,2,8,9,13,17-36]:
Activation of cytotoxic cells leading to production of multiple cytokines (Granulocyte or monocyte colony stimulating factors, vascular endothelial growth factor, interferon-α, β or γ, interleukins, tumor necrosis factor α or β, and macrophage migration inhibition factor): These cytokines are pro-inflammatory, chemotactic, increase proliferation of smooth muscle cells, and activate macrophages causing the release of free radicals, matrix metalloproteinase and elastase (which causes elastin degradation and release of fibroblast growth factors).
Mononuclear and endothelial cell activation with increased production of chemokines (heat shock proteins, CRP, and rheumatoid factor): These chemokines recruit inflammatory cells and up-regulate endothelial production of vascular and intercellular adhesion molecules, which further promotes the adhesion of inflammatory cells, vascular smooth cell proliferation, oxidative stress, endothelial dysfunction and apoptosis, and deposition of extracellular matrix and collagen.
Endothelial dysfunction with increased production of pro-inflammatory HDL and oxidative LDL.
Impairment in phenotype and function of endothelial progenitor cells mediated by interferon-α: Thacker, et al. [27] demonstrated in a mice model that thoracic aorta endothelial and smooth muscle cells exposed to interferon-α reduced the number, increased apoptosis, and impaired the function of endothelial progenitor cells. This correlated with a significant decrease in endothelium-dependent vasodilatory response.
Antiphospholipid antibodies and atherogenesis [28-32]: Antiphospholipid antibodies (aPL) in the form of IgG, IgM, or IgA anti-cardiolipin antibodies, anti-β2-glycoprotein I antibodies (aβ2-GPI), and lupus anticoagulant are common in SLE (with reported frequencies of up to 47%, 33%, and 26%, respectively), and are associated with inflammation and thrombogenesis and probably with atherogenesis. The plasma protein β2-GPI has anticoagulant properties in addition to pleiotropic functions affecting fibrinolysis, angiogenesis, apoptosis, and probably atherogenesis. β2-GPI complexed to anionic phospholipids is the main antigenic target for aPL.
Serum concentration of oxidized LDL is higher in SLE patients than in matched healthy controls. Oxidized LDL is highly proinflammatory and chemotactic for macrophage/monocyte and immune cells. Oxidized LDL interacts with β2GPI to form oxidized LDL/β2GPI complexes, which may generate an immune response producing antibodies against oxidized LDL, β2GPI, or oxidized LDL/β2GPI complexes. These immune-complexes may promote atherogenesis by the following proposed mechanisms: 1) induce subendothelial cell expression of pro-inflammatory genes including interleukin-1β, interleukin-18 receptor 1, and interleukin-6; 2) direct activation of the endothelium or, via cross-reactivity with other antigens interferes with lipoprotein metabolism; and 3) promote binding of oxidized LDL/β2-GPI to macrophages with acceleration of macrophage uptake and intracellular accumulation of lipids within atherosclerotic lesions. However, the association between oxidized LDL/β2-GPI complexes, IgG or IgM anti-oxidized LDL/β2-GPI autoantibodies, and aPL with subclinical atherosclerosis has not been consistently demonstrated in SLE patients. Also, in large inception cohorts of SLE patients, an association of aPL with subclinical and clinical atherosclerosis has been variable.
Inflammatory cytokine-mediated hyperactivity of the reninangiotensin system: It causes an increased production of angiotensin II and endothelin-1 (both potent vasoconstrictors) and increased oxidative stress resulting in AoS. This may lead to pre-hypertension or hypertension, which may perpetuate a vicious cycle that exacerbates AoS and accelerates the development of AoA [33,34].
Active cellular and humoral immunity causing the activation of macrophages, lymphocytes, phagocytes, neutrophils, CD4+CD28 and CD36 T-cells and dendritic cells: These cytotoxic cells (either circulating or adhered to the endovascular epithelium) cause platelets to release platelet-derived growth factors and thromboxane A2 (a vasoconstrictor and platelet activator) and decrease the production of nitric oxide and prostacyclin in endothelial cells. These factors cause vasoconstriction and thrombosis.
Alteration of cholesterol homeostasis: Reiss, et al. [35], demonstrated that incubated human monocytes and aortic endothelial cells in SLE patients’ plasma significantly decreased the cholesterol 27-hydroxylase (an anti-atherogenic cholesterol-degrading enzyme that promotes cellular cholesterol efflux) in monocytes and aortic endothelial cells by about 50% as compared to those cells incubated in healthy controls’ plasma (both p≤0.008). In addition, when macrophages were incubated in 25% SLE plasma and then loaded with acetylated low density lipoprotein, the SLE plasma more than doubled the macrophage foam cell transformation (74 ± 3% vs 35 ± 3% for healthy controls plasma, p<0.001).
Corticosteroid therapy-related metabolic abnormalities including hypertriglyceridemia, hypercholesterolemia, homocysteinemia, and insulin resistance: These metabolic abnormalities may induce AoA or AoS and may also increase perivascular adipose tissue deposition. Shields, et al. [36], using electron beam computed tomography (EBCT) quantified the volume of descending thoracic aortic perivascular adipose tissue, coronary artery calcification, and aortic calcification in 135 SLE patients without clinical cardiovascular disease and in 152 age and race-matched healthy controls. Women with SLE had greater median perivascular adipose tissue deposition (32.2 cm3 as compared to controls 28.6 cm3, p=0.007) and greater median aortic calcification (26.0 vs controls 6.0, p=0.001).
Pathology
The previously described pathogenetic factors of AoA and AoS are supported by autopsy studies. The described pathologic vascular changes in SLE are characterized first by immune complex deposition, subsequent complement activation, swelling, dystrophy and desquamation of endothelial cells, plasmatic impregnation of the walls, fibrinoid necrosis, and infiltrative-proliferative cellular reaction with small vessel luminal thrombosis followed by the development of sclerosis and premature atherosclerosis [37,38]. The aortic wall, including the vasa vasorum, can be affected by these inflammatory processes with immune complex mediated deposition of IgG, C3 and fibrinogen, resulting in either subclinical aortitis or in severe cases in clinically evident aortitis (resembling Takayasu’s arteritis), aortic aneurysms, aortic dissection, and/or thrombosis. With chronicity, all these pathologic changes result in marked atherosclerosis, even in young individuals [39-43]. Thus, autopsy studies indicate that there is first inflammation of the aorta which in combination with superimposed thrombosis and traditional atherogenic risk factors leads to accelerated and premature AoA and AoS in SLE patients.
Diagnostic Methods
Transesophageal echocardiography (TEE)
This diagnostic modality uses a multiplane TEE probe with a 7 MHZ phased array transducer with an axial resolution of 0.1 mm.
At a low depth (3-4 cm) and using a narrow sector scan to improve image lateral resolution, two-dimensional (2-D) guided M-mode images are used to assess intima media thickness (IMT) and plaques of the anterior wall of the aortic arch and proximal (at 25-30 cm from the incisors), mid (at 30-35 cm), and distal (at 35-40 cm) descending thoracic aorta (Figure 1). Also, 2-D TEE images are used to assess aortic IMT and plaques of the medial and lateral walls. Measurements of IMT are performed from the aortic short and long axis views during end-diastole (after the electrocardiographic P wave). At each aortic level, ≥3 measurements (from short and long axis views) of the anterior IMT are recommended to determine the mean ±1 SD, minimum, and maximum aortic IMT values [9,13,14,44].
Semi automated methods with intima-media inner and outer border detection are feasible, practical, and provided similar results in a preliminary study to those of manual measurements by 2-D guided M-mode and 2-D imaging [44] (Figure 2).
Figure 2.
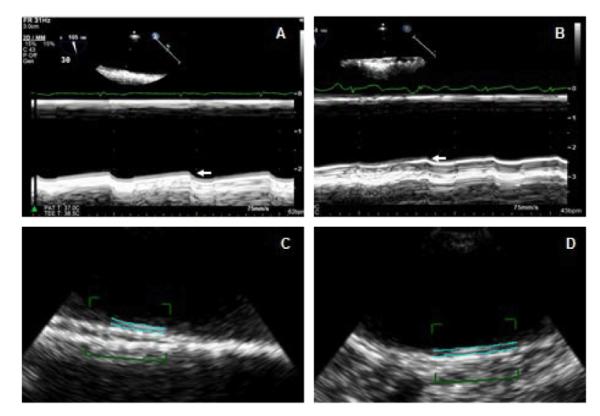
Two-dimensional (2-D) semi-automated imaging of the aortic intima-media thickness. A 41 year old SLE patient with mid (A) and distal (B) aortic IMT of 0.72 mm (arrow) and 0.65 mm (arrow), respectively, by 2-D guided M-mode imaging. Corresponding IMT measurements by 2D-semi automated imaging were 0.75 mm (C) and 0.68 mm (D), respectively.
Recently, three-dimensional TEE (3D-TEE) has emerged as a superior imaging modality as compared to M-mode and 2-D for detection, determination of the extent, and characterization of AoA in non-SLE populations [45]. However, no data is yet available on the diagnostic value of this technique in SLE patients. Preliminary data from our laboratory in a subset of young patients with SLE showed that 3D-TEE detected more often and better defined the extent and characteristics of AoA (unpublished data) (Figure 3).
Figure 3.
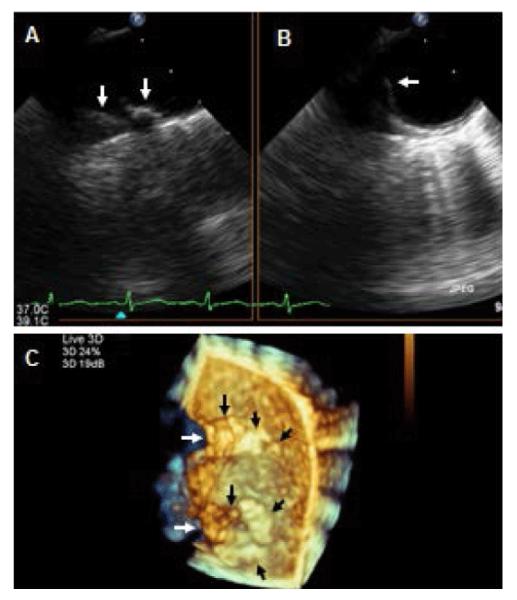
Two-dimensional and three-dimensional TEE for assessment of aortic atherosclerosis. A,B: Biplane 2-D TEE views of the distal arch and proximal descending thoracic aorta in a patient with SLE demonstrating a complex and protruding atherosclerotic disease located on the anterior and medial portions of the aorta (arrows) C: Three-dimensional en-face view of the anterior and in part medial and lateral walls of the distal portion of the aortic arch and proximal descending thoracic aorta demonstrating a tortuous aortic medial wall (white arrows), protruding atherosclerosis on the medial wall of the distal arch and proximal descending thoracic aorta (black arrows), and multiple other early atherosclerotic plaques in both aortic portions (short black arrows).
For assessment of AoS using TEE, the diameters of the descending thoracic aorta are measured from the short and long-axis views during end-systole and end-diastole (peak to end of T wave and after P wave on the electrocardiogram, respectively) (Figure 4). Three or more automatic blood pressure measurements are obtained from the brachial artery and matched in time with measurements of the proximal, mid, and distal aortic diameters.
Figure 4.
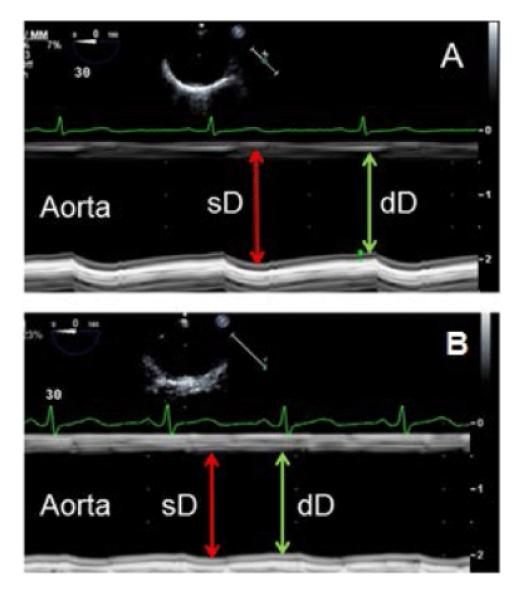
Assessment of aortic stiffness. A: Two-dimensional guided M-mode images of the mid descending thoracic aorta in a 42 year old SLE patient with normal aortic distensibility based on significant increase in aortic diameter during systole (sD) as compared to diastole (dD). B: Two-dimensional guided M-mode images of the mid descending thoracic aorta in a 39 year old SLE patient with aortic stiffness based on minimal change in aortic diameters during systole and diastole.
The following parameters for assessment of AoS have been reported [13,16,46,47]:
1) Pressure-Strain Elastic Modulus: [k(sBP-dBP)/(sD-dD/dD)]/10000, where k=133.3 is a conversion factor from mmHg to Nm-2 (Pascal units), sBP = systolic blood pressure, dBP=diastolic blood pressure, sD=systolic diameter, and dD=diastolic diameter (Figure 4). This is a well-established parameter for assessment of static arterial stiffness independently of IMT or plaques.
2) Strain (%): (sD-dD)/dD, which assesses percent change of vessel deformation independent of blood pressure.
3) Stiffness (Units): (sBP/dBP)/(sD-dD/dD), which as Pressure–Strain Elastic Modulus assesses the amount of pressure required to distend a vessel.
4) Distensibility (Units): (dD/IMT)/(sBP/dBP)/(sD-dD/dD), which assesses changes in vessel diameter as a function of blood pressure and wall thickness.
The limitations of TEE in the assessment of AoA and AoS include: 1) near- field limited resolution precludes accurate assessment of the aortic posterior wall; 2) far-field limited resolution and common off axis views preclude an accurate assessment of IMT of the ascending aorta; 3) 2-D guided M-mode imaging cannot assess the aortic lateral and medial walls; and 4) the study is semi-invasive, not readily available, and requires high expertise.
Transthoracic echocardiography (TTE)
There are no studies using TTE in SLE patients for assessment of AoA and AoS of the ascending aorta, arch or thoracic-abdominal aorta. Lower frequency transducers (2 MHz) with decreased resolution limit the ability of TTE to define the presence of intima-media thickening or early plaques. Limited data in non-SLE populations have demonstrated a low sensitivity and low specificity of TTE as compared to TEE for detection of even advanced AoA in these locations [48].
Computed tomography
Arterial calcium deposition correlates highly with the presence of advanced atherosclerosis. Electron beam CT (EBCT) or 64 slices multi-detector CT (MDCT) are techniques with high sensitivity for detection of calcium deposition (expressed in Agaston units) in the arterial vessels (Figure 5). Few studies using EBCT in SLE patients have demonstrated a higher prevalence of abnormal calcium deposition (defined as calcium score>0) on the aorta, coronary arteries, and carotid arteries as compared to matched controls [2,49-51]. However, the diagnostic value of EBCT and MDCT for detecting earlier stages of intima-media thickening and plaques without calcification may be limited. Also, these techniques are not easily available, are expensive, and require high expertise.
Figure 5.
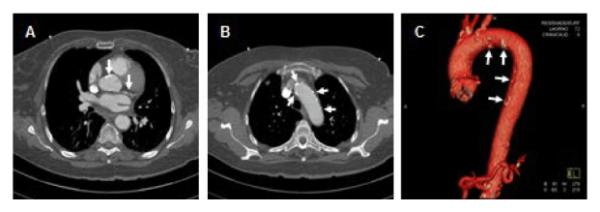
Computed tomography for assessment of aortic atherosclerosis. This computed tomography in an SLE patient demonstrates calcifications of the ascending aorta at the coronary sinuses level (left sided arrow in A), on the coronary arteries (right sided arrow in A), on the distal arch and proximal descending aorta (arrows in B). C. Three-dimensional reconstruction of the entire thoracic aorta demonstrating multiple areas of calcification (arrows in C) predominantly at the aortic arch and descending thoracic aorta.
Magnetic resonance imaging (MRI)
Three-dimensional MRI using T1-weighted imaging with a spatial resolution of 1 mm and its ability to image the entire aorta has been applied in non-SLE populations and has demonstrated superiority over TEE for detection of complex aortic plaques (defined as ≥4 mm intima-media thickness, ulcerated, or containing mobile thrombi) on the ascending aorta, arch, and descending thoracic aorta in patients with cryptogenic stroke [52]. However, limited data are available on the diagnostic value of this technique for detection of AoA in SLE patients. This technique may also be limited for detection of earlier stages of AoA such as intima-media thickening and small or non-complex plaques, is expensive, is not readily available, and requires high expertise.
Arterial tonometry
Arterial tonometry uses simultaneous recordings of Doppler velocimetry and electrocardiography to assess the carotid-to-femoral pulse wave velocity (PWV). An increase in carotid-to-femoral PWV correlates with increased AoS. Arterial flow waves from the right common carotid artery and right femoral artery are measured by a bidirectional transcutaneous velocimeter Doppler device with an 8 MHz transducer. After waveforms collection, the distance between the carotid and femoral arteries sampling sites is measured. PWV is calculated in m/s by dividing the carotid to femoral artery distance by the time difference between the onsets of the 2 arterial velocity waveforms. The stiffer the vessels are, the shorter the time between the 2 arterial velocities is, and therefore the higher the PWV is. Several series using arterial tonometry have demonstrated increased AoS in SLE patients as compared to age and sex matched controls [16,53,54].
QPV interval
The QPV interval is the time interval expressed in milliseconds between the onset of the QRS complex on the ECG and the brachial artery Doppler peak flow velocity (assessed with a 7.5 MHz linear array transducer). The QPV interval assessment is based on the principle that transit time from cardiac ejection to brachial artery peak velocity is shortened in patients with increased arterial stiffness [55]. The true diagnostic accuracy of this method for detecting AoS is undefined because it has not been tested against a gold standard. The reported interobserver and intraobserver variability of this measurement is <10%.
Prevalence
Pathologic or postmortem studies
Although clinical imaging studies indicate that at least one third of patients with SLE have evidence of subclinical carotid, coronary, or AoA disease, the prevalence of AoA in post-mortem studies is not well defined due to a lack of pathologic series specifically assessing the presence and extent of AoA [37-43].
TEE
Roldan, et al. [9], in a controlled study of 47 young SLE patients (44 women, age 38 ± 12 years) and 21 age-and-sex-matched controls (19 women, age 34 ± 12 years) reported higher overall prevalence rates of abnormal aortic IMT (using 1.5 SD above the mean intima-media thickness of controls or >0.86 mm), plaques, or both in SLE patients than in controls (37%, 23%, and 43% versus 14%, 0%, and 14%, respectively, all p≤0.02). The same investigators in a subsequent analysis in 76 SLE patients (69 women; mean age, 37 ± 12 years [range, 18-60 years]) and 26 controls (22 women; mean age, 34 ± 11 years [range, 18-57 years]) reported higher prevalence rates of abnormal IMT (using 2SD above the mean IMT of controls or >0.96 mm), plaques, or both as compared to controls (23%, 22%, and 33% versus 4%, 0%, and 4%, respectively, all p≤0.007) [14].
Computed tomography
Yiu, et al. [49] compared 50 SLE patients to age and sex matched controls without SLE who underwent 64 slices MDCT to evaluate calcium score in the coronary arteries, carotid arteries, and aorta. SLE patients as compared to controls had more often a calcium score>0 in the coronary arteries (42% versus 8%), carotid arteries (24% versus 0%), and aorta (20% versus 4%) (all p≤0.03). The rate of calcification on the aorta was likely underestimated because the mid and distal thoracic aorta was not assessed. In a different report, Yiu, et al. [50], using MDCT demonstrated higher prevalence of aortic and mitral valve calcifications in 52 SLE patients as compared to 60 age and sex-matched healthy controls (19.1% versus 0%, p<0.01) and a higher rate of calcification on the carotid arteries (11.8% versus 1.7%), coronary arteries (32.7% versus 1.7%), ascending aorta (11.8% versus 0%), descending aorta (43.6% versus 6.7%), or any vascular bed (66.4% versus 8.3%) (all p≤0.02).
Lertratanakul, et al. [51], in a cohort of 149 SLE patients and 124 controls (age at first study visit was 43.1 ± 9.9 and 46.3 ± 10.3 years, respectively) who underwent coronary artery and aortic calcium measurements by EBCT or MDCT during an initial and follow-up visits, demonstrated that 32 SLE patients (28.3%) had progression in aortic calcification at follow-up as compared to 22 controls (18.0%). The relative risk for progression of aortic calcification in cases compared to controls was 1.57 (95% CI 0.97-2.53). However, no specific prevalence rates of aortic calcification were provided in this study.
Arterial tonometry
Several series using arterial tonometry have demonstrated either aortic and/or peripheral arterial stiffness manifested by an increased carotid-to-femoral PWV. Yildiz, et al. [53], determined in 24 premenopausal women with SLE and 24 age and sex-matched controls that carotid-femoral PWV was significantly higher in women with SLE (8.98 ± 2.05 vs. 8.05 ± 0.94 m/s, p=0.04). By comparing 27 women with SLE (52-68 years old) and 27 controls, Bjarnegra, et al. [54] demonstrated that carotid-to-femoral PWV was higher in women with SLE after correction for mean arterial pressure and body mass index. Sabio, et al. [56] determined in a study of 128 patients with SLE that those with metabolic syndrome had higher aortic PWV compared to those without metabolic syndrome (9.8 ± 2.4 vs 8.5 ± 1.7 m/s; p=0.002). Finally, Qureshi, et al. [55], prospectively studied 46 female SLE patients and demonstrated that QPV interval correlated inversely with PWV (r=0.39, p=0.007). On multivariate analysis, QPV interval was an independent predictor of PWV after adjusting for age (r=0.26, p<0.001).
However, no cut-off values indicative of abnormal PWV and corresponding prevalence rates of arterial stiffness using this modality have been reported in SLE patients. In normal, non-SLE populations (defined as those with optimal or normal blood pressure and no other atherogenic risk factors) reference PWV values per decade have been reported by the European Society of Cardiology [57] (Table 1). These values will help to define abnormality and prevalence rates of AoS in future non-controlled studies of SLE patients.
Table 1.
Normal Reference Values for Pulse Wave Velocity (m/s) in a Normal Population (n=1455 subjects).
| Age Category | Mean ± 2SD | Median (10-90 percentile) |
|---|---|---|
| <30 | 6.2 (4.7-7.6) | 6.1 (5.3-7.1) |
| 30-39 | 6.5 (3.8-9.2) | 6.4 (5.2-8.0) |
| 40-49 | 7.2 (4.6-9.8) | 6.9 (5.9-8.6) |
| 50-59 | 8.3 (4.5-12.1) | 8.1 (6.3-10.0) |
| 60-69 | 10.3 (5.5-15.0) | 9.7 (7.9-13.1) |
| ≥70 | 10.9 (5.5-16.3) | 10.6 (8.0-14.6) |
Normal population was defined as those subjects with normal or optimal blood pressure without other additional traditional atherogenic risk factors.
SD=standard deviation; 10 percentile=the upper limit of the 10th percentile; 90 percentile=the lower limit of the 90th percentile.
Risk Factors or Predictors
SLE activity, SLE duration, and corticosteroid use have been reported as nontraditional risk factors for development of AoA and AoS in SLE patients [9,13,58-62]. SLE patients also have a higher prevalence of traditional atherogenic risk factors [9,13-15,20,63,64] as compared to controls without SLE.
Antiphospholipid (aPL) antibodies are common in SLE patients and are considered risk factors for atherosclerosis, albeit with mixed results [65]. Other autoantibodies with possible roles in SLE-associated AoA are directed against apolipoprotein A1 and high-density lipoprotein (HDL) [66]. HDL normally serves a protective role in atherosclerosis by mediating reverse cholesterol transport and protecting LDL from oxidation. On the other hand, proinflammatory HDL is associated with increased oxidized LDL formation, carotid plaques, and carotid intima-media thickness [35,66] and is present in as many as 45% of SLE patients.
In the study by Roldan, et al. [9], age at diagnosis of SLE was the only positive independent predictor of AoA [OR 1.12 per year from diagnosis of SLE, 95% confidence interval (CI) 1.0-1.19, p=0.001] and cyclophosphamide therapy was the only negative independent predictor of AoA (OR 0.186, 95% CI 0.153-0.95, p=0.04, equivalent to 5.4 times less likely to develop AoA).
Lertratanakul, et al. [51], in a cohort of 149 SLE patients and 124 controls who underwent coronary artery and aortic calcium measurements by EBCT or MDCT during initial and follow-up visits, demonstrated that lower C3, lower C4, use of corticosteroids, higher corticosteroid dose, higher lipoprotein(a), higher homocysteine levels, and presence of SLE were associated with aortic calcification.
As in general populations, aortic valve and mitral annular calcification have been associated with atherosclerosis in SLE patients. In the series of Yiu, et al. [50] using MDCT, the presence of mitral but not aortic valve calcification was independently associated with coronary or any vascular bed calcification after adjustment for other clinical variables.
Molad, et al. [67], in a study of 107 SLE patients (mean age 45.9 ± 14.7 years) undergoing 2D TTE demonstrated mitral annular calcification in 24 patients (22.6%) and aortic valve calcification in 22 patients (20.1%). Both mitral annular and aortic valve calcification were associated with traditional atherogenic risk factors, renal dysfunction, SLE damage index, IgA anticardiolipin antibodies, and stroke. In addition, mitral annular calcification was associated with coronary artery disease (r=0.2, p=0.05). Both mitral annular and aortic valve calcifications were associated with death during the follow-up period (r=0.20, p=0.05; r=0.20, p=0.03, respectively). Thus, mitral annular and aortic valve calcifications are prevalent among young SLE patients, positively correlate with premature diffuse atherosclerosis, and are risk factors for subsequent all-cause mortality.
Shields, et al. [36], use EBCT in 135 SLE patients without clinical cardiovascular disease and in 152 age and race-matched healthy controls. They quantified coronary artery and aortic calcification using Agatston scores and thoracic aortic perivascular adipose tissue using standard Hounsfield Units. Women with SLE had greater median aortic perivascular adipose tissue and greater median aortic valve calcification than controls (both p≤0.007). In SLE patients, aortic perivascular adipose tissue was independently associated with aortic calcification (odd ratio of 6.78 [95% confidence interval 2.0-23], p=0.002) even after adjusting for circulating inflammatory markers (p=0.007) and coronary artery calcification (p=0.003).
Palmieri, et al. [68], in a study of 45 SLE patients without clinical cardiovascular events (56% with an SLE damage score=0, 44% with a damage score=1-4) determined that the prevalence of preclinical carotid atherosclerosis (28% versus 16%), low global arterial compliance (18% versus 10%), aortic valve fibrosclerosis (10% versus 8%), and flow mediate arterial dilatation <10% were similar in SLE patients with a damage score=0 and >0. These findings suggest that SLE patients have subclinical atherosclerotic disease independently of the degree of SLE damage score.
In the study by Roldan, et al. [13], 50 patients with SLE, 94% women, with a mean age of 38 ± 12 years, and 22 age and gender-matched healthy controls underwent clinical and laboratory evaluations and multiplane TEE to assess AoS, of the proximal, mid, and distal descending thoracic aorta. AoS at each aortic level and overall AoS assessed by the pressure-strain elastic modulus was higher in patients than in controls after adjusting for age (overall, 8.25 ± 4.13 versus 6.1 ± 2.5 Pascal units, p=0.01). Age at SLE diagnosis and nonneurologic damage score were the only independent SLE-specific predictors of AoS (both p≤0.01).
Selzer, et al. [16], in a non-controlled study of 214 SLE women without clinical cardiovascular disease with a mean age of 45.2 ± 10.5 years (including patients >60 years old) demonstrated that AoS determined by PWV was associated with older age, higher systolic blood pressures, higher C3 levels, lower white blood cell count, higher insulin levels, and renal disease.
In the study by Yildiz, et al. [53], AoS was correlated with age, body mass index, waist-to-hip ratio, heart rate, and blood pressure.
In the study by Bjarnegra, et al. [54], aortic pulse wave velocity was positively associated with levels of C-reactive protein and complement factor.
In the study by El Gramal, et al. [69], 16 adolescent patients with active SLE (mean age 15 ± 2.42 years; 16 females), 14 patients with inactive SLE (mean age 15.7 ± 1.89 years; 4 males, 10 females), and 16 age and sex-matched healthy adolescents (15.5 ± 1.71 years; 4 males, 12 females) underwent echocardiography for assessment of proximal aortic PWV [defined as Ao distance/Ao wave transit time in the aortic arch]. Patients with active SLE had significantly higher PWV values than controls (p<0.05), while no difference was found between patients with inactive SLE and controls.
Finally, Sabio, et al. [56], determined in a study of 128 SLE patients that patients with metabolic syndrome as compared to those without had higher aortic PWV (9.8 ± 2.4 vs 8.5 ± 1.7 m/s; p=0.002). Increased PWV was independently associated with age, male sex, metabolic syndrome, duration of SLE, and CRP levels.
Association with Atherosclerosis in other Arterial Beds
Studies in SLE using EBCT for simultaneous imaging of the carotid and coronary arteries and aorta have demonstrated calcium deposition in the 3 arterial beds, with higher prevalence rates in the coronary arteries, followed by the aorta, and then by the carotid arteries [49] (Figure 5). However, calcium deposition implies significant and generalized atherosclerotic disease and therefore it is uncertain if a particular arterial bed is affected earlier and to a greater extent in earlier stages of the disease. In our experience, earlier stages of atherosclerotic disease appear to be more prevalent and occur earlier in the aorta than in the carotid arteries [13,70]. In different study populations, the prevalence rates of AoA using TEE are similar to those reported for coronary artery calcification by EBCT and higher than those of carotid atherosclerosis by ultrasonography [1,2,9,71]. However, larger cross-sectional and longitudinal studies are needed to confirm these findings in the same populations and using the same diagnostic method.
Clinical Manifestations
Commonly silent or subclinical
In cross-sectional controlled studies, at least one third of SLE patients have subclinical premature atherosclerotic disease manifested as increased carotid intima-media thickness or plaques, coronary artery calcification on CT, and intima-media thickening or plaques on the aorta independently of traditional risk factors [1-4,9,13].
Pre-hypertension and hypertension
Preceding or concomitant AoS with subclinical AoA may lead to a decrease in aortic distensibility and higher levels of systolic blood pressure, pre-hypertension or hypertension, which then may self-perpetuate AoS and accelerate AoA [13,33].
Left ventricular hypertrophy and left ventricular diastolic dysfunction
Roldan, et al. [47], demonstrated in a 6-year-duration cross-sectional study comparing 76 SLE patients (69 women; mean age, 37 ± 12 years) to 26 age and sex-matched healthy controls that SLE patients had higher AoS (8.14 ± 4.25 versus 5.97 ± 2.31 U, p<0.001), lower mitral inflow E/A ratio, lower septal and lateral mitral annulus tissue Doppler E’/A’ velocity ratios, longer isovolumic relaxation time, lower septal and lateral mitral annulus E’ velocities, and higher mitral E/septal E’ and mitral E/lateral E’ velocity ratios (all p≤0.03), all indicative of left ventricular diastolic dysfunction. In patients with SLE, AoS was correlated with parameters of left ventricular diastolic dysfunction (all p<0.05), was independently and negatively associated with E/A and E’/A’ ratios and E’ velocities, and was positively associated with E/E’ ratios (P≤0.02 for each parameter and p<0.001 for all parameters as a profile). Also, in SLE patients, AoS was correlated with left ventricular mass and left atrial volume indexes(all p<0.05).These findings suggest that SLE-associated immune-mediated inflammation may cause endothelial dysfunction and apoptosis, increased thrombogenesis, smooth-muscle-cell proliferation, intima-media thickening, vessel stiffness and increased impedance, and consequently prehypertension or hypertension-which may cause further AoS and AoA, increased blood pressure and left ventricular afterload; increase left ventricular mass; and ultimately cause left ventricular diastolic dysfunction.
Cerebrovascular and coronary artery disease
Patients with SLE have a higher prevalence, incidence, and recurrence of acute stroke or TIA and (to a lesser degree) of acute coronary syndromes. Non-obstructive carotid, intracerebral, or coronary artery disease is most commonly found in these patients. Therefore, important pathogenetic or exacerbating factors of cerebrovascular and cardiovascular disease in SLE may include underlying early and mild atherosclerosis associated with endothelial dysfunction, decreased vasodilatory response or increased arterial stiffness in combination with prevalent primary or secondary increased platelet activity and coagulation [1-7,9,14,67].
Intestinal angina
Matsuda, et al. [72], reported a case of a patient with long standing SLE with postprandial abdominal pain as a manifestation of intestinal angina and evidence on CT of severe stenosis of the superior mesenteric artery with complete occlusion of the celiac and inferior mesenteric arteries. Histopathology of the superior mesenteric artery and abdominal aorta showed atherosclerosis with no vasculitis or thrombosis.
Peripheral arterial embolism
Aortic plaques compounded by underlying primary or secondary thrombogenesis may be a common source of subclinical and clinical peripheral embolism in SLE patients [73] (Figure 6).
Figure 6.
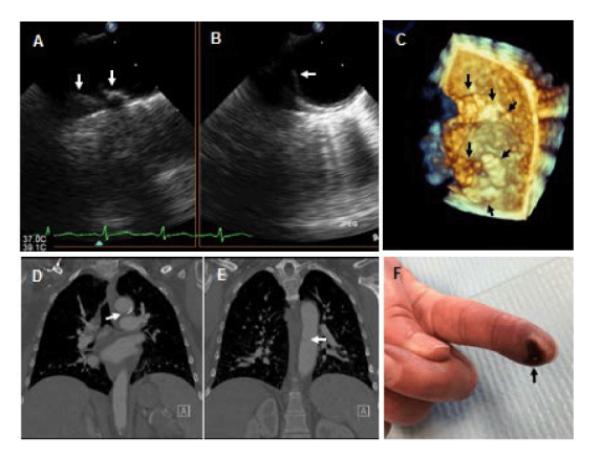
Fifty-two year old woman with SLE and peripheral arterial embolism. A-C: Two-dimensional (A,B) and 3-dimensional (C) TEE views of the distal arch to proximal descending thoracic aorta demonstrating complex and protruding atherosclerotic disease (arrows). Her thoracic computed tomography demonstrated significant calcifications and a filling defect suggestive of an atheroma with thrombus on the anterior wall of the aortic arch (arrow in D) and a second filling defect on the medial wall of the mid descending thoracic aorta (arrow in E). Left index finger tip ulceration (arrow in F) and ischemic discoloration consistent with peripheral atheroembolism
Aortic aneurysms and aortic dissection
Finally, atherosclerosis compounded, preceded, or exacerbated by vasculitis and cystic medial necrosis can lead to aortic aneurysmal formation and aortic dissection. Wang, et al. [74], in a retrospective cohort study of a Taiwanese population database of 15,209 SLE patients (89.9% women, mean age of 38.3 years) reported 20 cases with aortic aneurysm and 13 cases with aortic dissection (overall incidence rate, 4.26 per 10,000 person-years). Compared with control patients, the overall incidence rate ratio for developing aortic aneurysm or aortic dissection was 3.34 (95% confidence intervals, 1.71-6.91; p<0.001). Kurata, et al. [75], in a meta-analysis of relevant studies published over the past 40 years (1969-2008) identified 35 SLE patients with aortic aneurysms. Thoracic aneurysms were associated with atherosclerosis, cystic medial degeneration, and vasculitis, whereas abdominal aneurysms were associated predominantly with atherosclerosis.
Prognosis
In SLE patients, as is the case in non-SLE populations, a strong association exists between AoA, coronary artery disease, and cerebrovascular disease. Therefore, AoA is a marker of generalized atherosclerosis and a predictor of a 2-5 fold increased risk of future new or recurrent acute coronary syndromes, stroke or TIA, and cardiac or cerebrovascular mortality. Also, AoA and its associated AoS may be an important pathogenic factor or a marker of prehypertension, hypertension, increased left ventricular afterload, left ventricular hypertrophy, and consequently subclinical and clinically evident left ventricular diastolic dysfunction with heart failure. In addition, AoA exacerbated by hypercoagulability in SLE patients may be a pathogenic factor for peripheral arterial thrombotic or thromboembolic ischemic events. Furthermore, AoA is a pathogenic factor for the development of aortic aneurysms and aortic dissection with a consequent high mortality in these immunocompromised patients.
Therapy
In our experience and that of others, the later the age at diagnosis of SLE, the higher the likelihood of developing AoA or AoS. Also, SLE-related inflammation requiring steroid therapy may lead to a metabolic syndrome which may lead to functional and structural arterial disease. In contrast, non-steroidal immunosuppressive therapy and disease modifying anti-rheumatic drugs may have a vascular protective effect. These findings emphasize the need for early diagnosis of SLE, aggressive non-steroidal immunosuppressive therapy for control of inflammation, and aggressive non-pharmacologic and pharmacologic interventions for control of traditional atherogenic risk factors for primary prevention or secondary prevention of the progression of AoA and AoS [76]. These interventions have the potential to decrease the prevalence and incidence of cardiac and cerebrovascular morbidity and mortality in SLE patients.
Statins
Mendez-Fernandez, et al. [77], reported the effects of pravastatin in combination with an apolipoprotein-AI (Apo-AI) mimetic peptide in a mouse model of lupus-accelerated atherosclerosis. Although this combined therapy caused an increase in size of aortic root atherosclerotic lesions, it did significantly reduce systemic inflammation and changed the phenotype of the lesion to a more stable plaque. Woo, et al. [78], in a murine lupus model of accelerated atherosclerosis (apoE−/−Fas−/−C57BL/6 mice) demonstrated that after 35 weeks, L-4F-treated mice, in the presence of pravastatin, had significantly lower serum levels of IgG antibodies to dsDNA (p<0.05) and oxidized phospholipids (p<0.005) as compared to controls. Although treatment resulted in larger aortic root lesions as compared to controls, atheromas in the combination treatment mice had less infiltration with CD68+ macrophages, increased mean alpha-actin stained area, and lower levels of VCAM-1 (all p<0.05). Thus, increased smooth muscle content, decreased macrophage infiltration, and decreased pro-atherogenic chemokines in L-4F plus pravastatin treated mice suggest protective mechanisms not only on lupus-like disease, but also on potential plaque remodeling. In another study [79], SLE patients with inactive disease and subclinical atherosclerosis were randomized in a double-blinded manner to receive either rosuvastatin (10 mg/day) or matching placebo (half in each group were also randomly allocated to low-dose aspirin) for 12-24 months. Plasma levels of homocysteine, high-sensitivity C-reactive protein (hsCRP), soluble vascular cell adhesion molecule 1, P-selectin, and thrombomodulin were measured at baseline, 6 months, and 12 months. At 12 months, a significant decrease in the mean low-density lipoprotein cholesterol and median hsCRP levels was seen in the rosuvastatin group (both p≤0.02). A subgroup analysis of patients with a SLE Disease Activity Index score≤2 revealed a significant decrease in hsCRP and thrombomodulin levels (both p≤0.04) with rosuvastatin treatment. At 24 months, IMT of the internal carotid arteries was also decreased in patients treated with rosuvastatin. These animal and human data support a beneficial effect of statins in the treatment of autoimmune-mediated inflammation and atherosclerosis in SLE.
Aspirin
The clinical benefit of anti-platelet therapy in SLE patients with antiphospholipid antibodies have been studied in limited non-randomized and non-controlled clinical studies. Findings from these studies support the prophylactic use of aspirin for all patients with SLE and positive antiphospholipid antibodies to prevent arterial and venous thrombotic manifestations and improve survival [80,81]. However, limited are available on the beneficial effect of aspirin in SLE patients without antiphospholipid antibodies.
Non-steroid immunosuppressive anti-inflammatory therapy
Mycophenolate mofetil: It is a potent immunosuppressive agent that is commonly used for treatment of patients with active SLE. In a study of gld.apoE (−/−) mice on a high cholesterol Western diet with or without mycophenolate mofetil, the mice receiving mycophenolate mofetil showed a decrease in atherosclerotic lesion area compared to the control group, after 12 weeks on the diet [82]. Mycophenolate mofetil treatment also improved the SLE phenotype by significantly decreasing circulating autoantibody levels.
Cyclophosphamide: In the study by Roldan, et al. [9], cyclophosphamide therapy was an independent negative predictor of AoA.
Disease modifying anti-rheumatic drugs
Antimalarial drugs, mainly hydroxychloroquine, are prescribed in most SLE patients to treat constitutional symptoms, rashes, and arthritis, and to prevent flares. The immune-modulatory effect of hydroxychloroquine, including antagonizing Toll-like receptor activation, results in inhibition of interferon-α (IFN-α) expression and activation of multiple IFN-α-mediated pathways. Although processing of low-affinity antigens (e.g., self-antigens) is blocked, the immune response against high-affinity antigens (e.g., bacterial peptides) is not impaired and therefore causes an effective immunomodulation without immunosuppression. The use of hydroxychloroquine is associated with less damage, improvement in disease activity, lipid profile, glucose control, prevention of metabolic syndrome, prevention of thromboembolic events, and ultimately in improved survival [83-87].
Hypoglycemic agents
Juarez-Rojas, et al. [88], in a double-blind trial of 30 premenopausal women with SLE (30 ± 8 years old) randomized to pioglitazone (30 mg/day) or placebo for 3 months demonstrated that pioglitazone significantly increased HDL-cholesterol plasma levels (14.2%), reduced fasting insulin plasma levels (23.6%) and significantly reduced the levels of C-reactive protein (70.9%) and serum amyloid A (34.9%). Thus, pioglitazone may have a beneficial effect for the prevention of the development or progression of AoA and AoS in SLE.
Acknowledgments
Grant Support: Some research content of this manuscript was funded by the grant RO1-HL04722-01-A6 by the National Institutes of Health/National Heart Lung and Blood Institute and in part by the grant 8UL1-TR000041 by the National Center for Research Resources.
References
- 1.Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–2406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- 2.Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–2415. doi: 10.1056/NEJMoa035611. [DOI] [PubMed] [Google Scholar]
- 3.Jiménez S, García-Criado MA, Tàssies D, Reverter JC, Cervera R, et al. Preclinical vascular disease in systemic lupus erythematosus and primary antiphospholipid syndrome. Rheumatology (Oxford) 2005;44:756–761. doi: 10.1093/rheumatology/keh581. [DOI] [PubMed] [Google Scholar]
- 4.Becker-Merok A, Nossent J. Prevalence, predictors and outcome of vascular damage in systemic lupus erythematosus. Lupus. 2009;18:508–515. doi: 10.1177/0961203308099233. [DOI] [PubMed] [Google Scholar]
- 5.Ippolito A, Petri M. An update on mortality in systemic lupus erythematosus. Clin Exp Rheumatol. 2008;26:S72–79. [PubMed] [Google Scholar]
- 6.Nossent J, Cikes N, Kiss E, Marchesoni A, Nassonova V, et al. Current causes of death in systemic lupus erythematosus in Europe, 2000--2004: relation to disease activity and damage accrual. Lupus. 2007;16:309–317. doi: 10.1177/0961203307077987. [DOI] [PubMed] [Google Scholar]
- 7.Cook RJ, Gladman DD, Pericak D, Urowitz MB. Prediction of short term mortality in systemic lupus erythematosus with time dependent measures of disease activity. J Rheumatol. 2000;27:1892–1895. [PubMed] [Google Scholar]
- 8.Roldan CA. Valvular and coronary heart disease in systemic inflammatory diseases: Systemic Disorders in heart disease. Heart. 2008;94:1089–1101. doi: 10.1136/hrt.2007.132787. [DOI] [PubMed] [Google Scholar]
- 9.Roldan CA, Joson J, Sharrar J, Qualls CR, Sibbitt WL., Jr Premature aortic atherosclerosis in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. J Rheumatol. 2010;37:71–78. doi: 10.3899/jrheum.090665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen S, Hinderliter A, Sen PK, Simmons J, Beck J, et al. Aortic arch atheroma progression and recurrent vascular events in patients with stroke or transient ischemic attack. Circulation. 2007;116:928–935. doi: 10.1161/CIRCULATIONAHA.106.671727. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka M, Yasaka M, Nagano K, Otsubo R, Oe H, et al. Moderate atheroma of the aortic arch and the risk of stroke. Cerebrovasc Dis. 2006;21:26–31. doi: 10.1159/000089590. [DOI] [PubMed] [Google Scholar]
- 12.Varga A, Gruber N, Forster T, Piros G, Havasi K, et al. Atherosclerosis of the descending aorta predicts cardiovascular events: a transesophageal echocardiography study. Cardiovasc Ultrasound. 2004;22:2–21. doi: 10.1186/1476-7120-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roldan CA, Joson J, Qualls CR, Sharrar J, Sibbitt WL., Jr Premature aortic stiffness in systemic lupus erythematosus by transesophageal echocardiography. Lupus. 2010;19:1599–1605. doi: 10.1177/0961203310377088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roldan CA, Sibbitt WL, Jr, Qualls CR, Jung RE, Greene ER, et al. Libman-Sacks endocarditis and embolic cerebrovascular disease. JACC Cardiovasc Imaging. 2013;6:973–983. doi: 10.1016/j.jcmg.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manzi S, Selzer F, Sutton-Tyrrell K, Fitzgerald SG, Rairie JE, et al. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum. 1999;42:51–60. doi: 10.1002/1529-0131(199901)42:1<51::AID-ANR7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, et al. Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001;37:1075–1082. doi: 10.1161/01.hyp.37.4.1075. [DOI] [PubMed] [Google Scholar]
- 17.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 18.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valdivielso P, Gómez-Doblas JJ, Macias M, Haro-Liger M, Fernández-Nebro A, et al. Lupus-associated endothelial dysfunction, disease activity and arteriosclerosis. Clin Exp Rheumatol. 2008;26:827–833. [PubMed] [Google Scholar]
- 20.Colombo BM, Cacciapaglia F, Puntoni M, Murdaca G, Rossi E, et al. Traditional and non traditional risk factors in accelerated atherosclerosis in systemic lupus erythematosus: role of vascular endothelial growth factor (VEGATS Study) Autoimmun Rev. 2009;8:309–315. doi: 10.1016/j.autrev.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Reiss AB, Wan DW, Anwar K, Merrill JT, Wirkowski PA, et al. Enhanced CD36 scavenger receptor expression in THP-1 human monocytes in the presence of lupus plasma: linking autoimmunity and atherosclerosis. Exp Biol Med (Maywood) 2009;234:354–360. doi: 10.3181/0806-BC-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Carvalho JF, Bonfá E, Borba EF. Systemic lupus erythematosus and “lupus dyslipoproteinemia”. Autoimmun Rev. 2008;7:246–250. doi: 10.1016/j.autrev.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 23.Roman MJ, Crow MK, Lockshin MD, Devereux RB, Paget SA, et al. Rate and determinants of progression of atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2007;56:3412–3419. doi: 10.1002/art.22924. [DOI] [PubMed] [Google Scholar]
- 24.Tani C, Mosca M, d’Ascanio A, Versari D, Virdis A, et al. Chronic inflammation and endothelial dysfunction: analysis of a cohort of patients with SLE and UCTD. Reumatismo. 2006;58:212–218. doi: 10.4081/reumatismo.2006.212. [DOI] [PubMed] [Google Scholar]
- 25.Santos LL, Morand EF. Macrophage migration inhibitory factor: a key cytokine in RA, SLE and atherosclerosis. Clin Chim Acta. 2009;399:1–7. doi: 10.1016/j.cca.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Frieri M. Accelerated atherosclerosis in systemic lupus erythematosus: role of proinflammatory cytokines and therapeutic approaches. Curr Allergy Asthma Rep. 2012;12:25–32. doi: 10.1007/s11882-011-0236-1. [DOI] [PubMed] [Google Scholar]
- 27.Thacker SG, Duquaine D, Park J, Kaplan MJ. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelialdysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus. 2010;19:288–99. doi: 10.1177/0961203309353773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Artenjak A, Lakota K, Frank M, Nik S, Rozman B, et al. Antiphospholipid antibodies as non-traditional risk factors in atherosclerosis based cardiovascular diseases without overt autoimmunity. A critically updated review. Autoimmun Rev. 2012;11:873–82. doi: 10.1016/j.autrev.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Bassi N, Zampieri S, Ghirardello A, Tonon M, Zen M, et al. oxLDL/beta2GPI complex and anti-oxLDL/beta2GPI in SLE: prevalence and correlates. Autoimmunity. 2009;42:289–291. doi: 10.1080/08916930902828247. [DOI] [PubMed] [Google Scholar]
- 30.Matsuura E, Lopez LR, Shoenfeld Y, Ames PR. Î22-glycoprotein I and oxidative inflammation in early atherogenesis: a progression from innate to adaptive immunity? Autoimmun Rev. 2012;12:241–249. doi: 10.1016/j.autrev.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Narshi CB, Giles IP, Rahman A. The endothelium: an interface between autoimmunity and atherosclerosis in systemic lupus erythematosus? Lupus. 2011;20:5–13. doi: 10.1177/0961203310382429. [DOI] [PubMed] [Google Scholar]
- 32.Petri M. Update on anti-phospholipid antibodies in SLE: the Hopkins’ Lupus Cohort. Lupus. 2010;19:419–423. doi: 10.1177/0961203309360541. [DOI] [PubMed] [Google Scholar]
- 33.Ryan MJ. The pathophysiology of hypertension in systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1258–1267. doi: 10.1152/ajpregu.90864.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Booth AD, Wallace S, McEniery CM, Yasmin, Brown J, et al. Inflammation and arterial stiffness in systemic vasculitis: a model of vascular inflammation. Arthritis Rheum. 2004;50:581–588. doi: 10.1002/art.20002. [DOI] [PubMed] [Google Scholar]
- 35.Reiss AB, Anwar K, Merrill JT, Chan ES, Awadallah NW, et al. Plasma from systemic lupus patients compromises cholesterol homeostasis: a potential mechanism linking autoimmunity to atherosclerotic cardiovascular disease. Rheumatol Int. 2010;30:591–8. doi: 10.1007/s00296-009-1020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shields KJ, Barinas-Mitchell E, Gingo MR, Tepper P, Goodpaster BH, et al. Perivascular adipose tissue of the descending thoracic aorta is associated with systemic lupus erythematosus and vascular calcification in women. Atherosclerosis. 2013;231:129–35. doi: 10.1016/j.atherosclerosis.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ansari A, Larson PH, Bates HD. Vascular manifestations of systemic lupus erythematosus. Angiology. 1986;37:423–432. doi: 10.1177/000331978603700601. [DOI] [PubMed] [Google Scholar]
- 38.Kawai S, Fukuda Y, Okada R. Atherosclerosis of the coronary arteries in collagen disease and allied disorders, with special reference to vasculitis as a preceding lesion of coronary atherosclerosis. Jpn Circ J. 1982;46:1208–1221. doi: 10.1253/jcj.46.1208. [DOI] [PubMed] [Google Scholar]
- 39.Sato J, Kawakami T, Nakabayashi K, Fukuoka K, Hirano K, et al. Multiple aortic aneurysms complicated by a rupture in the systemic lupus erythematosus: a case report. Pathol Res Pract. 2008;204:845–850. doi: 10.1016/j.prp.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Silver AS, Shao CY, Ginzler EM. Aortitis and aortic thrombus in systemic lupus erythematosus. Lupus. 2006;15:541–543. doi: 10.1191/0961203306lu2342cr. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, French SW, Chuang CC, McPhaul L. Pathologic quiz case: an unusual complication of systemic lupus erythematosus. Arch Pathol Lab Med. 2000;124:324–326. doi: 10.5858/2000-124-0324-PQC. [DOI] [PubMed] [Google Scholar]
- 42.Willett WF, Kahn MJ, Gerber MA. Lupus aortitis: a case report and review of the literature. J La State Med Soc. 1996;148:55–59. [PubMed] [Google Scholar]
- 43.Yoshimoto K, Saima S, Nakamura Y, Ishikawa H, Kinoshita M, et al. A case of acute dissecting aneurysm of the aorta in systemic lupus erythematosus. Nihon Jinzo Gakkai Shi. 1989;31:1211–1216. [PubMed] [Google Scholar]
- 44.Macias L, Schevchuck A, Roldan P, Qualls CR, Sibbitt WL, Roldan CA. Aortic Intima-Media Thickness by M-mode or 2-Dimensional Semi-Automated Imaging: Do These Methods Provide Equivalent Results? Arteriosclerosis, Thrombosis and Vascular Biology (ATVB) 2013;33:A238. [Google Scholar]
- 45.Piazzese C, Tsang W2, Sotaquira M3, Kronzon I4, Lang RM5, et al. Semiautomated detection and quantification of aortic plaques from three-dimensional transesophageal echocardiography. J Am Soc Echocardiogr. 2014;27:758–766. doi: 10.1016/j.echo.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Godia EC, Madhok R, Pittman J, Trocio S, Ramas R, et al. Carotid artery distensibility: a reliability study. J Ultrasound Med. 2007;26:1157–1165. doi: 10.7863/jum.2007.26.9.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roldan CA, Alomari IB, Awad K, Boyer NM, Qualls CR, et al. Aortic stiffness is associated with left ventricular diastolic dysfunction in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. Clin Cardiol. 2014;37:83–90. doi: 10.1002/clc.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmed S, Rehan A, Ahmad I, Gardin JM, Nanda NC, Cohen GI. Can Transthoracic Echocardiography with Subcostal View Predict Abdominal Aortic Atherosclerosis? Echocardiography. 2005;22:736–42. doi: 10.1111/j.1540-8175.2005.00077.x. [DOI] [PubMed] [Google Scholar]
- 49.Yiu KH, Wang S, Mok MY, Ooi GC, Khong PL, et al. Pattern of arterial calcification in patients with systemic lupus erythematosus. J Rheumatol. 2009;36:2212–2217. doi: 10.3899/jrheum.090312. [DOI] [PubMed] [Google Scholar]
- 50.Yiu KH, Wang S, Mok MY, Ooi GC, Khong PL, et al. Relationship between cardiac valvular and arterial calcification in patients with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol. 2011;38:621–627. doi: 10.3899/jrheum.100844. [DOI] [PubMed] [Google Scholar]
- 51.Lertratanakul A, Wu P, Dyer AR, Kondos G, Edmundowicz D, et al. Risk factors in the progression of subclinical atherosclerosis in women with systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2014;66:1177–1185. doi: 10.1002/acr.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harloff A, Brendecke SM, Simon J, Assefa D, Wallis W, et al. 3D MRI provides improved visualization and detection of aortic arch plaques compared to transesophageal echocardiography. J Magn Reson Imaging. 2012;36:604–611. doi: 10.1002/jmri.23679. [DOI] [PubMed] [Google Scholar]
- 53.Yildiz M, Yildiz BS, Soy M, Tutkan H. Impairment of arterial distensibility in premenopausal women with systemic lupus erythematosus. Kardiol Pol. 2008;66:1194–1199. [PubMed] [Google Scholar]
- 54.Bjarnegråd N, Bengtsson C, Brodszki J, Sturfelt G, Nived O, et al. Increased aortic pulse wave velocity in middle aged women with systemic lupus erythematosus. See comment in PubMed Commons below. Lupus. 2006;15:644–650. doi: 10.1177/0961203306071402. [DOI] [PubMed] [Google Scholar]
- 55.Qureshi G, Salciccioli L, Lee S, Qureshi M, Kapoor A, et al. QPV interval as a measure of arterial stiffness in women with systemic lupus erythematosus. Clin Cardiol. 2009;32:154–158. doi: 10.1002/clc.20301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sabio JM, Vargas-Hitos J, Zamora-Pasadas M, Mediavilla JD, Navarrete N, et al. Grupo Lupus Virgen de las Nieves. Metabolic syndrome is associated with increased arterial stiffness and biomarkers of subclinical atherosclerosis in patients with systemic lupus erythematosus. J Rheumatol. 2009;6:2204–11. doi: 10.3899/jrheum.081253. [DOI] [PubMed] [Google Scholar]
- 57.Reference Values for Arterial Stiffness’ Collaboration Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J. 2010;31:2338–2350. doi: 10.1093/eurheartj/ehq165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhew EY, Ramsey-Goldman R. Premature atherosclerotic disease in systemic lupus erythematosus--role of inflammatory mechanisms. Autoimmun Rev. 2006;5:101–105. doi: 10.1016/j.autrev.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 59.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 60.Doria A, Shoenfeld Y, Wu R, Gambari PF, Puato M, et al. Risk factors for subclinical atherosclerosis in a prospective cohort of patients with systemic lupus erythematosus. Ann Rheum Dis. 2003;62:1071–1077. doi: 10.1136/ard.62.11.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang CY, Lu LJ, Li FH, Li HL, Gu YY, et al. Evaluation of risk factors that contribute to high prevalence of premature atherosclerosis in Chinese premenopausal systemic lupus erythematosus patients. J Clin Rheumatol. 2009;15:111–116. doi: 10.1097/RHU.0b013e31819d8489. [DOI] [PubMed] [Google Scholar]
- 62.Petri M, Perez-Gutthann S, Spence D, Hochberg MC. Risk factors for coronary artery disease in patients with systemic lupus erythematosus. Am J Med. 1992;93:513–519. doi: 10.1016/0002-9343(92)90578-y. [DOI] [PubMed] [Google Scholar]
- 63.Petri M. Detection of coronary artery disease and the role of traditional risk factors in the Hopkins Lupus Cohort. Lupus. 2000;9:170–175. doi: 10.1191/096120300678828226. [DOI] [PubMed] [Google Scholar]
- 64.Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–2337. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 65.Bilora F, Boccioletti V, Girolami B, Zanon E, Armani M, et al. Are antiphospholipid antibodies an independent risk factor for atherosclerosis? Clin Appl Thromb Hemost. 2002;8:103–113. doi: 10.1177/107602960200800205. [DOI] [PubMed] [Google Scholar]
- 66.Nuttall SL, Heaton S, Piper MK, Martin U, Gordon C. Cardiovascular risk in systemic lupus erythematosus--evidence of increased oxidative stress and dyslipidaemia. Rheumatology (Oxford) 2003;42:758–762. doi: 10.1093/rheumatology/keg212. [DOI] [PubMed] [Google Scholar]
- 67.Molad Y, Levin-Iaina N, Vaturi M, Sulkes J, Sagie A. Heart valve calcification in young patients with systemic lupus erythematosus: a window to premature atherosclerotic vascular morbidity and a risk factor for all-cause mortality. Atherosclerosis. 2006;185:406–12. doi: 10.1016/j.atherosclerosis.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 68.Palmieri V, Migliaresi P, Orefice M, Lupo T, Di Minno MN, et al. High prevalence of subclinical cardiovascular abnormalities in patients with systemic lupus erythematosus in spite of a very low clinical damage index. Nutr Metab Cardiovasc Dis. 2009;19:234–40. doi: 10.1016/j.numecd.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 69.Gamal YM, Elmasry OA, El Hadidi IS, Soliman OK. Proximal aortic stiffness is increased in systemic lupus erythematosus activity in children and adolescents. ISRN Pediatr. 2013;2013:765253. doi: 10.1155/2013/765253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roldan PC, Schevchuck A, Macias L, Greene ER, Qualls CR, et al. Premature aortic and carotid atherosclerosis in systemic lupus erythematosus: which one is first or worse? J Am Coll Cardiology. 2013;6:973–83. [Google Scholar]
- 71.Tessitore E, Rundek T, Jin Z, Homma S, Sacco RL, et al. Association between carotid intima-media thickness and aortic arch plaques. J Am Soc Echocardiogr. 2010;23:772–777. doi: 10.1016/j.echo.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsuda M, Miyazaki D, Tojo K, Tazawa K, Shimojima Y, et al. Intestinal angina due to atherosclerosis in a 45-year-old systemic lupus erythematosus patient. Intern Med. 2010;49:2175–2178. doi: 10.2169/internalmedicine.49.3769. [DOI] [PubMed] [Google Scholar]
- 73.da Rocha MC, Vilar MJ, Freire EA, Santiago MB. Arterial occlusion in systemic lupus erythematosus: a good prognostic sign? Clin Rheumatol. 2005;24:602–605. doi: 10.1007/s10067-005-1111-z. [DOI] [PubMed] [Google Scholar]
- 74.Wang SH, Chang YS, Liu CJ, Lai CC, Chen TJ, et al. Incidence and risk analysis of aortic aneurysm and aortic dissection among patients with systemic lupus erythematosus: a nationwide population-based study in Taiwan. Lupus. 2014 doi: 10.1177/0961203314523868. [DOI] [PubMed] [Google Scholar]
- 75.Kurata A, Kawakami T, Sato J, Sakamoto A, Muramatsu T, et al. Aortic aneurysms in systemic lupus erythematosus: a meta-analysis of 35 cases in the literature and two different pathogeneses. Cardiovasc Pathol. 2011;20:e1–7. doi: 10.1016/j.carpath.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 76.Soltész P, Kerekes G, Dér H, Szücs G, Szántó S, et al. Comparative assessment of vascular function in autoimmune rheumatic diseases: considerations of prevention and treatment. Autoimmun Rev. 2011;10:416–425. doi: 10.1016/j.autrev.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 77.Mendez-Fernandez Y, Major A. Sizing up stability: combination therapy with Apo-AI peptide mimetics and statins in systemic lupus erythematosus-mediated atherosclerosis. Arthritis Res Ther. 2010;12:139. doi: 10.1186/ar3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woo JM, Lin Z, Navab M, Van Dyck C, Trejo-Lopez Y, et al. Treatment with apolipoprotein A-1 mimetic peptide reduces lupus-like manifestations in a murine lupus model of accelerated atherosclerosis. Arthritis Res Ther. 2010;12:R93. doi: 10.1186/ar3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mok CC, Wong CK, To CH, Lai JP, Lam CS. Effects of rosuvastatin on vascular biomarkers and carotid atherosclerosis in lupus: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken) 2011;63:875–883. doi: 10.1002/acr.20440. [DOI] [PubMed] [Google Scholar]
- 80.Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195–205. doi: 10.1136/ard.2007.070367. [DOI] [PubMed] [Google Scholar]
- 81.Wahl DG, Bounameaux H, de Moerloose P, Sarasin FP. Prophylactic antithrombotic therapy for patients with systemic lupus erythematosus with or without antiphospholipid antibodies: do the benefits outweigh the risks? A decision analysis. Arch Intern Med. 2000;160:2042–2048. doi: 10.1001/archinte.160.13.2042. [DOI] [PubMed] [Google Scholar]
- 82.Richez C, Richards RJ, Duffau P, Weitzner Z, Andry CD, et al. The effect of mycophenolate mofetil on disease development in the gld.apoE (−/−) mouse model of accelerated atherosclerosis and systemic lupus erythematosus. PLoS One. 2013;8:e61042. doi: 10.1371/journal.pone.0061042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Akhavan PS, Su J, Lou W, Gladman DD, Urowitz MB, et al. The early protective effect of hydroxychloroquine on the risk of cumulative damage in patients with systemic lupus erythematosus. J Rheumatol. 2013;40:831–841. doi: 10.3899/jrheum.120572. [DOI] [PubMed] [Google Scholar]
- 84.Wallace DJ, Gudsoorkar VS, Weisman MH, Venuturupalli SR. New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat Rev Rheumatol. 2012;8:522–533. doi: 10.1038/nrrheum.2012.106. [DOI] [PubMed] [Google Scholar]
- 85.Wozniacka A, Lesiak A, Narbutt J, McCauliffe DP, Sysa-Jedrzejowska A. Chloroquine treatment influences proinflammatory cytokine levels in systemic lupus erythematosus patients. Lupus. 2006;15:268–75. doi: 10.1191/0961203306lu2299oa. [DOI] [PubMed] [Google Scholar]
- 86.Borba EF, Bonfá E. Longterm beneficial effect of chloroquine diphosphate on lipoprotein profile in lupus patients with and without steroid therapy. J Rheumatol. 2001;28:780–785. [PubMed] [Google Scholar]
- 87.Jung H, Bobba R, Su J, Shariati-Sarabi Z, Gladman DD, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 2010;62:863–868. doi: 10.1002/art.27289. See comment in PubMed Commons below. [DOI] [PubMed] [Google Scholar]
- 88.Juárez-Rojas JG, Medina-Urrutia AX, Jorge-Galarza E, Caracas-Portilla NA, Posadas-Sánchez R, et al. Pioglitazone improves the cardiovascular profile in patients with uncomplicated systemic lupus erythematosus: a double-blind randomized clinical trial. Lupus. 2012;21:27–35. doi: 10.1177/0961203311422096. [DOI] [PubMed] [Google Scholar]