

Abstract
Protein Kinase C delta (PKCδ) regulates apoptosis in the mammary gland, however the functional contribution of PKCδ to the development or progression of breast cancer has yet to be determined. Meta-analysis of ErbB2-positive breast cancers shows increased PKCδ expression, and a negative correlation between PKCδ expression and prognosis. Here we present in vivo evidence that PKCδ is essential for the development of mammary gland tumors in a ErbB2-overexpression transgenic mouse model, and in vitro evidence that PKCδ is required for proliferative signaling downstream of the ErbB2 receptor. MMTV-ErbB2 mice lacking PKCδ (δKO) have increased tumor latency compared to MMTV-ErbB2 wild type (δWT) mice, and tumors show a dramatic decrease in Ki-67 staining. To explore the relationship between PKCδ and ErbB2-driven proliferation more directly, we used MCF-10A cells engineered to express a synthetic ligand-inducible form of the ErbB2 receptor. Depletion of PKCδ with shRNA inhibited ligand-induced growth in both 2D (plastic) and 3D (Matrigel) culture, and correlated with decreased phosphorylation of the ErbB2 receptor, reduced activation of Src, and reduced activation of the MAPK/ERK pathway. Similarly, in human breast cancer cell lines in which ErbB2 is overexpressed, depletion of PKCδ suppresses proliferation, Src, and ERK activation. PKCδ appears to drive proliferation through formation of an active ErbB2/PKCδ/Src signaling complex, as depletion of PKCδ disrupts association of Src with the ErbB2 receptor. Taken together, our studies present the first evidence that PKCδ is a critical regulator of ErbB2-mediated tumorigenesis, and suggest further investigation of PKCδ as a target in ErbB2-positive breast cancer.
Keywords: PKCδ, breast cancer, ErbB2, Src
Introduction
The ErbB2 oncogene is amplified or overexpressed in 20–30% of all breast cancers and is associated with decreased survival, faster relapse, and increased lymph node metastasis (1, 2). Overexpression of ErbB2 leads to receptor dimerization and activation of pathways involved in tumor cell proliferation and survival, such as Mitogen Activated Protein Kinase (MAPK), Src, and Phosphatidylinositol 3-kinase (PI3K). Additionally, ErbB2 is the preferred binding partner of other epidermal growth factor receptor (EGFR) family members, thus overexpression of this protein increases receptor interactions, providing an efficient link to mitogenic pathways (3, 4).
The PKC family of serine/threonine kinases consists of 10 isoforms that are structurally related and that regulate many biological functions (5–7). PKCδ is an important regulator of epithelial cell apoptosis, which has led to the speculation that it may play a role in tumorigenesis (8–13). Increased expression of PKCδ has been shown in colon, pancreatic, and high-grade breast tumors (14–16). Conversely, PKCδ expression is decreased in some cancers including bladder and endometrial carcinoma (17, 18). We have recently shown that PKCδ functions as a tumor promoter in Ras-mediated lung tumors in vivo and in K-ras addicted human Non-Small Cell Lung Cancer (NSCLC) cells in vitro through regulation of the Ras/MAPK pathway (19). Likewise, studies from Keshamouni et. al. have implicated PKCδ in regulating Ras signaling downstream of ErbB2 in breast cancer cells in vitro (20). PKCδ has also been shown to positively regulate cell migration in several cell types, including EGFR overexpressing breast cancer cells (21–24).
Src is a major mediator of ErbB2 signaling, and a potential mechanism through which cancer cells can become resistant to ErbB2 therapies (25). PKCδ expression is increased in breast cancer cells resistant to tamoxifen and lapatinib, suggesting that both PKCδ and Src may be necessary for ErbB2 mediated signal transduction (26, 27). Our current studies identify PKCδ as a critical regulator of ErbB2-mediated proliferation, and as a tumor promoter in a MMTV-ErbB2 transgenic mouse model of mammary gland cancer. Meta-analysis of ErbB2-positive human breast cancers reveals a negative correlation between PKCδ expression and prognosis, supporting further investigation of PKCδ as a potential therapeutic target.
Results
Increased expression of PKCδ negatively correlates with prognosis in ErbB2 positive human breast cancer
To explore the contribution of PKCδ to human breast cancer, we used the Oncomine database (28), to interrogate 21 ErbB2 positive human breast cancer data sets (n=> 2,000 patients) for PKCδ mRNA expression. Our analysis shows that PKCδ is significantly overexpressed in ErbB2 positive human breast cancers (Figure 1A, red; P=0.01). Since ErbB2 is associated with a poor prognosis (1, 2), we determined whether increased PKCδ expression effects clinical outcome specifically in patients with ErbB2 positive human breast tumors. Using the GOBO tool (29), we performed a meta-analysis of relapse-free survival in four breast cancer data sets, where 122 patients were identified as ErbB2 positive using the PAM50 array (30). Strikingly, high expression of PKCδ (red line) significantly correlated with decreased relapse-free survival compared to low PKCδ expression (gray line) (P=0.017) (Figure 1B). Similar results were seen when we examined the correlation between PKCδ mRNA expression and relapse-free survival over all molecular subtypes (P=0.0002) (Figure 1C). In contrast, there was no correlation between PKCδ mRNA expression and relapse-free survival in patients with the triple-negative tumor subtype (negative for ER, PR, and ErbB2 (P=0.488) (Figure 1D)), moreover, PKCδ mRNA expression was significantly decreased in 13 triple-negative tumor data sets (Oncomine, data not shown). These results suggest that PKCδ may be a significant factor in the progression of ErbB2 driven breast tumors.
Figure 1. Increased expression of PKCδ negatively correlates with prognosis in ErbB2 positive human breast cancer.
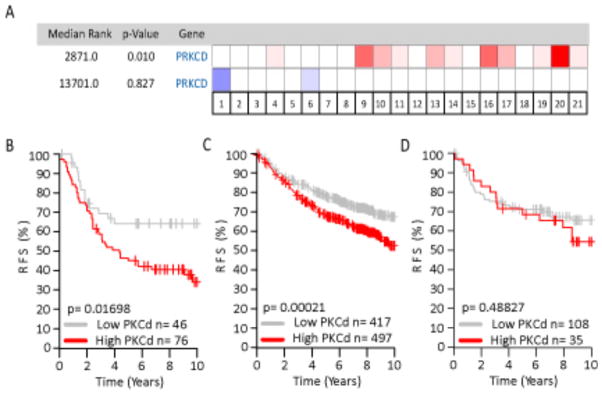
A. Overexpression (red) and underexpression (blue) of PKCδ in 21 human breast cancer data sets. Intensity of the cell color corresponds to the top percentage of ranked genes, with 50% (white) representing no change between tumor sets. The median rank for PKCδ overexpression in ErbB2 positive tumors across each data set is 2,871 (P=0.01), while the underexpression median rank is 13,701 (P=0.827). B, C, and D. Kaplan-Meier analysis, using relapse-free survival as endpoint, for (B) ErbB2 positive tumors (four data sets, n= 122, P=0.017), (C) All molecular subtypes (four data sets, n= 914, P=0.0002), and (D) Triple-negative (ER-/PR-/ErbB2-)(four data sets, n=143, P=0.488), stratified into two quantiles base on high (red line) or low (gray line) PKCδ expression.
Loss of PKCδ increases tumor latency in MMTV-ErbB2 transgenic mice
To directly address the functional role of PKCδ in ErbB2-driven tumors we utilized a transgenic mouse model in which mammary gland tumorigenesis is driven by overexpression of the wild type rat ERBB2 gene under control of the Mouse Mammary Tumor Virus (MMTV) promoter (31, 32). MMTV-ErbB2 mice were crossed with δKO mice to generate MMTV-ErbB2;δWT and MMTV-ErbB2;δKO mice. Both MMTV-ErbB2;δWT and MMTV-ErbB2;δKO mice develop focal mammary tumors consistent with the MMTV-ErbB2 phenotype (31, 32); however, MMTV-ErbB2;δKO mice had a significant delay in tumor onset, with a mean latency of 293 days compared to 243 days in MMTV-ErbB2;δWT mice (P=0.03) (Figure 2A). Mammary gland tumors were never detected in aged δKO mice (>1 year of age; data not shown). Histological analysis showed that both MMTV-ErbB2;δWT and MMTV-ErbB2;δKO tumors were highly vascularized and consisted of uniform solid sheets of cells (Figure 2B, panels a, b).
Figure 2. Loss of PKCδ increases tumor latency in MMTV-ErbB2 transgenic mice.

A. MMTV-ErbB2;δWT (n=10) and δKO (n=7) mice were palpated weekly to detect mammary tumor growth. Graph shows the number of days to tumor detection verses the percentage of tumor-free mice for each genotype; P=0.03. B. MMTV-ErbB2;δWT and MMTV-ErbB2;δKO tumor sections were stained with hematoxylin and eosin (a, b). Histology- 20X, insets depict 60X enlargement of boxed region. C and D. Cleaved caspase-3 (C) and Ki-67 (D) immunohistochemistry on MMTV-ErbB2;δWT and MMTV-ErbB2;δKO tumor sections. Histology- 20X, insets depict 60X enlargement of boxed region. Graphs depict quantification of the percent of cleaved caspase-3 or Ki-67 positive cells over 6–8 fields per tumor, per genotype; n=5–8 MMTV-ErbB2;δWT, 4–7 MMTV-ErbB2;δKO; *P=0.01. E. Tumor lysates from MMTV-ErbB2;δWT (7) and δKO (7) mice were immunoblotted with the following antibodies to determine the levels of ErbB2 receptor phosphorylation: pErbB2 (Y877, Y1221/1221, Y1248); ErbB2; and actin.
To explore the mechanism underlying increased tumor latency in MMTV-ErbB2;δKO mice, we assayed apoptosis and proliferation by immunohistochemistry for cleaved caspase-3 and Ki-67, respectively. Very few apoptotic cells were observed in tumors from either genotype (Figure 2C). In contrast, Ki-67 positive cells were reduced by >50% in MMTV-ErbB2;δKO tumors compared to MMTV-ErbB2;δWT tumors (Figure 2D), suggesting that increased tumor latency is due at least in part to reduced ErbB2-driven proliferation. Consistent with this finding, tumor lysates from MMTV-ErbB2;δKO mice show decreased levels of ErbB2 receptor phosphorylation compared to WT, suggesting that PKCδ regulates ErbB2 signal transduction (Figure 2E).
PKCδ is required for ErbB2-driven proliferation
To explore more directly the contribution of PKCδ to ErbB2-driven proliferation and tumorigenesis, we utilized a derivative of the non-transformed human breast epithelial line, MCF-10A, engineered to express a synthetic ligand-inducible form of the ErbB2 receptor (10A.ErbB2) (33, 34). In this model, the addition of an ErbB2 dimerization ligand induces ErbB2 signaling, resulting in abnormal acinar growth that resembles human ductal carcinoma in situ (35). To ask if PKCδ contributes to this ErbB2-induced morphogenesis, 10A.ErbB2 cells were depleted of PKCδ using lentiviral delivered shRNA targeted to PKCδ (shδ193 and shδ203), or a scrambled control (shSCR), and grown on Matrigel for 6 days (Figure 3A, panels a, b, c). In the absence of the ligand, all cells formed small, round, organized acini typical of normal MCF-10A growth (Figure 3A, panels a, b, c) (36). Acini were then treated with ligand for 3–8 days. Dimerization of ErbB2 resulted in misshapen acini in shSCR, shδ193, and shδ203 cells (Figure 3A, panels g, h, I, m, n, o, insets), however no consistent changes were seen in acini derived from shδ193 and shδ203 cells compared to shSCR cells. In contrast, acinar size appeared to be reduced in shδ193 and shδ203 cells treated with ligand compared to shSCR cells (Figure 3A, panels g, h, i, m, n, o, insets). Indeed, quantification of structure area showed a significant decrease in acinar size in cells depleted of PKCδ as early as 3 days, which persisted through at least 8 days of growth (Figure 3A, panels g, h, i, m, n, o and 2B). In the absence of ligand, there were no significant differences in acinar size between shδ193, shδ203 and shSCR cells, suggesting that PKCδ is required specifically for ErbB2 driven proliferation (Figure 3B).
Figure 3. PKCδ is required for ErbB2-driven proliferation.

For all panels: PKCδ was depleted using lentiviral shRNA constructs (shδ193 and shδ203) and compared to control shRNA (shSCR) as described in Materials and Methods. A. 10A.ErbB2 cells depleted of PKCδ using shRNA (shδ193 and shδ203) were grown on Matrigel for 6 days (a, b, c). Cells were then left untreated (d, e, f; j, k, l) or treated with 1μM ligand for 3–8 days (g, h, i; m, n, o). Representative images of three separate experiments taken at 5X magnification are shown. Inset shows digital enlargement to show structure morphology. B. Quantification of structure area from Figure 2A, using Metamorph software. Representative of three experiments shown; *P=<0.02. D. and E. 10A.ErbB2 cells depleted of PKCδ were plated at equal densities in 3D (D) or 2D (E) growth assays. For 3D growth, cells were plated on Matrigel and grown for 6 days. Ligand was added for 3 days after which cells were tryspinized and counted; *P=<0.0001. For 2D growth, cells were treated with or without ligand for 4 days, trypsinized, and counted; *P=<0.04. B, C, and D. Insets show depletion of PKCδ by immunoblot with actin as a loading control. Boxed region below blots shows relative PKCδ knockdown compared to shSCR controls. Representative graphs of three separate experiments are shown.
To ask if the decrease in acinar area correlated with reduced proliferation, control 10A.ErbB2 cells (shSCR), or 10A.ErbB2 cells depleted of PKCδ (shδ193 or shδ203) were plated on Matrigel (3D culture), or as monolayers on plastic (2D culture) and cell number was quantified. The addition of ligand significantly increased the total cell number of shSCR cells grown as 3D acini (Figure 3C) and as monolayers (Figure 3D) compared to untreated cells. For shSCR cells, the ligand-induced increase in cell number (Figure 3C and D) was considerably more robust than the increase in acinar area (Figure 3B), suggesting that additional mechanisms may contribute to acinar size. In contrast, in ligand-treated cells depleted of PKCδ (shδ193 and shδ203), no increase in cell number was detected under either growth condition (Figure 3C and D), suggesting an absolute requirement for PKCδ in ErbB2-induced proliferation.
Loss of PKCδ attenuates ErbB2 signal transduction
Our studies suggest that PKCδ is necessary to activate ErbB2 proliferative pathways; to determine how PKCδ regulates ErbB2 signaling, we treated 10A.ErbB2 cells in 3D culture (Figure 4A) and 2D culture (Figure 4B) with ligand and analyzed the activation of key signaling molecules. Addition of ligand to 10A.ErbB2 shSCR cells in 3D or 2D culture resulted in robust phosphorylation at two major ErbB2 autophosphorylation sites, Y1221/1222 and Y1248, which couple ErbB2 to the Ras/MAPK pathway through the binding of Shc and companion adaptor proteins, and at Y877, which has been shown to regulate the intrinsic kinase activity of ErbB2 (37–40). In cells depleted of PKCδ (shδ193 and shδ203), ligand-induced autophosphorylation of ErbB2 Y1221/1222 and Y1248 was slightly reduced, while phosphorylation of ErbB2 at Y877 was more dramatically reduced (Figure 4A and 4B, graphs) (40). Of note, total ErbB2 receptor protein was decreased in the absence of ErbB2 dimerization in shSCR cells, consistent with the findings that ErbB2 activation can decrease receptor turnover and stability (41–43).
Figure 4. Loss of PKCδ attenuates ErbB2 signal transduction.

For all panels: PKCδ was depleted using lentiviral shRNA constructs (shδ193 and shδ203) and compared to control shRNA (shSCR) as described in Materials and Methods. A. and B. 10A.ErbB2 cells depleted of PKCδ were plated in 3D culture (A) or 2D culture (B) and then treated for 3 days or 24 hours with ligand respectively. Protein expression and phosphorylation in whole cell lysates was evaluated by immunobloting. The following antibodies were used: pErbB2 (Y877, Y1221/1221, Y1248); ErbB2; pSrc (Y416); Src; pShc (Y239/240); Shc, pErk1/2 (T202/Y204); Erk1/2; PKCδ; and actin. Immunoblots probed for phosphorylated proteins were stripped and probed with antibodies to total protein, as indicated. Blots were probed for actin as a loading control. Relative PKCδ knockdown compared to shSCR is shown in boxed region below blots. Immunoblots were quantified by densitometry; graphs depict the levels of phosphorylated pErbB2 Y877 and pSrc Y416 over total protein levels upon ligand treatment. Representative experiments shown; experiments were repeated a minimum of three times. C. 10A.ErbB2 cells depleted of PKCδ were treated for 24 hours with ligand. Cells were lysed and ErbB2 was immunoprecipitated with anti-HA primary antibody. Immunoprecipitated protein was resolved on an SDS-page gel and probed for HA, PKCδ and Src. Lysates were incubated with a non-specific mouse IgG as a negative control (NEG). Whole cell lysates (WCL) were probed for HA, PKCδ, and Src. Actin was used as a loading control. Representative experiments shown; experiments were repeated a minimum of three times.
Since depletion of PKCδ reduced phosphorylation of ErbB2 Y1221/1222, Y1248, and Y877, phosphorylation of downstream proteins was analyzed to determine signaling pathways regulated by PKCδ. Phosphorylation of ErbB2 has been shown to recruit the adaptor protein Shc, which can also signal through the Ras/MAPK pathway (39, 44, 45). However, only a small decrease in the level of pShc Y239/40 was observed in shδ193 and shδ203 cells compared to shSCR cells in 3D culture (Figure 4A), while no consistent decrease was observed under 2D culture conditions (Figure 4B). Further studies have not detected a difference in the phosphorylation of Shc Y317 or GAB2, both of which have been implicated in downstream signaling through ErbB2 (data not shown). These results suggest that decreased signaling through pShc is unlikely to contribute to the decrease in pERK and proliferation seen with the depletion of PKCδ.
Src is recruited to ErbB2 through sequences surrounding the ErbB2 Y877 residue and is thought to be responsible for phosphorylation at this site (40, 46). (47, 48). Concomitant with reduced phosphorylation of ErbB2 at Y877, activation of Src, as indicated by phosphorylation at Y416, was dramatically suppressed in ligand treated shδ193 and shδ203 cells compared to shSCR cells (Figure 4A and 4B). Quantitation by densitometry showed that in 3D and 2D culture, phosphorylation of Src Y416 was reduced 50–80% in shδ193 and shδ203 cells compared to shSCR cells, which correlated with a 30–60% decrease in ErbB2 phosphorylation at Y877 (Figure 4A and 4B, graphs). Activation of Src regulates proliferative signaling through the Ras/MAPK pathway (49–51). As seen in Figure 4A and B, phosphorylation of ERK is dramatically reduced in cells depleted of PKCδ, suggesting a mechanism by which PKCδ suppresses ErbB2 driven proliferation.
Decreased activation of Src, combined with the reduced phosphorylation of ErbB2 Y877, suggests that PKCδ may be required for interaction of Src with the ErbB2 receptor. To address this possibility, 10A.ErbB2 cells depleted of PKCδ were treated with ligand, immunoprecipitated with antibodies to the HA-tagged ErbB2 receptor, and probed for HA, PKCδ, and Src. In the presence of ligand, Src was found to associate with the ErbB2 receptor in shSCR cells, supporting previous findings that activation of ErbB2 requires Src (Figure 4C). PKCδ was also found to immunoprecipitate with the ErbB2 receptor, and the binding of Src to the ErbB2 receptor was disrupted in cells depleted of PKCδ, demonstrating that PKCδ is required for formation of an active ErbB2 signaling complex.
PKCδ regulates proliferation in ErbB2 overexpressing human breast cancer cells
We next examined the contribution of PKCδ to ErbB2 regulated proliferation in human breast cancer cells that overexpress ErbB2. Depletion of PKCδ with shδ193 or shδ203 in MDA-MB-361, BT-474, and ZR-75-30 cells significantly reduced proliferation compared to shSCR control cells (Figure 5A). The largest decrease in proliferation was observed in BT-474 cells, where proliferation was reduced 59% and 38% in shδ193 or shδ203 cells, respectively. These results correspond to the relative depletion of PKCδ (Figure 5A, PKCδ insets). Analysis of ErbB2 meditated signaling showed that both Src and ERK phosphorylation were decreased in shδ193 and shδ203 cells compared to shSCR in all three cell lines, similar to that seen in 10A.ErbB2 cells (Figure 5B). Interestingly, in all cell lines the total level of Src appeared to be slightly reduced, a result that was not seen in 10A.ErbB2 cells, suggesting that PKCδ may regulate Src stability or turnover in breast cancer cells.
Figure 5. PKCδ regulates proliferation in ErbB2 overexpressing human breast cancer cells.

A. MDA-MB-361, BT-474, and ZR-75-30 cells were depleted of PKCδ as described in Materials and Methods. Cells were plated at equal densities and counted at four time points. Representative experiments shown; experiments were repeated a minimum of three times, p=<0.05. Insets show depletion of PKCδ by immunoblot with actin as a loading control. B. Whole cell lysates from MDA-MB-361, BT-474, and ZR-75-30 cells were resolved on an SDS-page gel and probed with the following antibodies: pSrc (Y416); Src; pERK1/2 (T202/Y204); ERK1/2; PKCδ. Immunoblots probed for phosphorylated proteins were stripped and probed with antibodies to total protein, as indicated. Blots were probed for actin as a loading control. Representative experiments are shown; experiments were repeated a minimum of three times. C., panel a, MDA-MB-361 and BT474 cells with PKCδ knockdown (shδ193 or shδ800) were transduced with an adenovirus encoding either LacZ or a constitutively activated MEK (CA-MEK) MOI=100). Cells were counted on day 5 (BT-474) or day 6 (MDA-MB-361). Inset shows pERK activation upon 48 hours of CA-MEK expression in BT-474 cells. Panel b, MDA-MB-361 cells (SCR and shδ800) were transduced with adenovirus encoding either LacZ or GFP-PKCδWT (MOI=100). On the subsequent day media was replaced and 10uM PD98059 was added to the indicated wells; cells were counted on day 6. Lysates were probed for PKCδ and actin. D. Model: Loss of PKCδ decreases the phosphorylation and binding of Src to the ErbB2 receptor, resulting in decreased ERK signaling and abrogated ErbB2 driven proliferation. See text for detailed discussion.
To determine if PKCδ regulates ErbB2 driven proliferation through the MAPK pathway, MDA-MB-361 and BT-474 cells with PKCδ knockdown were transduced with an adenovirus that expresses constitutively activated MEK (CA-MEK). Expression of CA-MEK activated ERK, and significantly rescued the reduction in proliferation seen with the loss of PKCδ in shδ193 and shδ800 cells (Figure 5C, panel a). Similarly, when PKCδ was re-expressed in MDA-MB-361 cells depleted of PKCδ (shδ800), proliferation was rescued (Figure 5C, panel b), and this rescue was significantly reduced by pre-treatment with the ERK inhibitor, PD98059. Together these results indicate that PKCδ specifically regulates ErbB2 signaling through activation of the MAPK pathway.
Discussion
Our studies reveal PKCδ as a major regulator of ErbB2 mediated proliferation in breast cancer. Analysis of human ErbB2 positive breast cancer tumor samples shows a negative correlation between PKCδ expression and relapse free survival. Using a mouse model of ErbB2-dependent mammary gland tumorigenesis, we show that loss of PKCδ significantly increases tumor latency and suppresses tumor cell proliferation. Further, studies in human breast cancer cell lines support these findings and suggest that PKCδ is essential for activation of Src and ERK downstream of the ErbB2 receptor. Together these findings implicate PKCδ as a tumor promoter and a potential therapeutic target in ErbB2 overexpressing breast cancer.
We show that tumors from MMTV-ErbB2;δKO mice have reduced proliferation and altered ErbB2 activation compared to MMTV-ErbB2;δWT controls (Figure 2), and that PKCδ is necessary for induction of ErbB2 driven proliferation in 2D and 3D culture (Figure 3). An interesting observation is that depletion of PKCδ has a much more dramatic effect on 3D growth of MCF10A.ErbB2 cells, compared to 2D growth. Presumably, 3D culture provides a more complex signaling system by allowing not only cell-cell interactions, but also cell-matrix interactions, that are lacking in cells grown on plastic. We observed a similar phenomenon in lung cancer, where PKCδ was required for lung tumorigenesis in vivo, and for growth of NSCLC lines in soft agar, but not for growth on plastic (19).
Characterization of the proteins involved in ErbB2 signaling in the context of PKCδ depletion revealed a decrease in phosphorylation of the ErbB2 receptor at Y1221/1222, Y1248, and Y877 (Figure 4). We were unable to detect a change in activation of the adaptor protein, Shc; however, loss of PKCδ significantly suppressed Src activation, which likely contributes to the suppression of ERK and downstream proliferation. Phosphorylation at ErbB2 Y877 has been linked to the regulation of ErbB2’s intrinsic kinase activity and loss of this phosphorylation results in a corresponding decrease in ErbB2 Y1221/1222 and Y1248 phosphorylation (40, 46, 52). These results suggests that the ErbB2 receptor may have decreased activity in cells lacking PKCδ, and that decreased auto-phosphorylation of ErbB2 at Y1221/1222 and Y1248 may be secondary to the decrease at ErbB2 Y877. Additionally, total ErbB2 levels decreased in response to EGF starvation (Figure 4A, -Ligand), and addition of the ErbB2 dimerization ligand restored ErbB2 expression. Cells with PKCδ knockdown have decreased expression of the ErbB2 receptor in response to dimerization compared to scrambled controls, indicating that either PKCδ regulates the stability of the receptor or that PKCδ effects ErbB2 translation. Further studies will need to be performed to directly explore this regulation.
Several reports have intimately linked Src with the phosphorylation of ErbB2 Y877 (40, 46). Marcotte et. al. demonstrated that Src associates with the ErbB2 receptor through sequences surrounding ErbB2 Y877 and is required for ErbB2 Y877 phosphorylation (46). Correspondingly, in our studies depletion of PKCδ results in a loss of Src activation (phosphorylation at Y416) and binding to the ErbB2 receptor, suggesting that PKCδ is required for formation of an active ErbB2 signaling complex with Src (Figure 4C). Reduced signaling through ErbB2/Src is likely the mechanism by which depletion of PKCδ suppresses ERK activation and proliferation. It is not clear from our current studies whether ErbB2 or Src are PKCδ kinase substrates; alternatively PKCδ may function as a scaffold in this complex. In NSCLC cell lines ERK activation can be inhibited by expression of a kinase-inactive form of PKCδ, suggesting that the kinase activity of PKCδ is important for activation of this proliferative pathway (19).
Limited studies in human breast cancer tissue, and in human breast cancer cell lines, have suggested that elevated PKCδ may correlate with more aggressive breast cancer (14, 22). Our analysis shows that PKCδ expression is significantly increased in ErbB2 positive verses negative human breast tumors. High expression of PKCδ also significantly correlates with decreased relapse-free survival in ErbB2 positive human breast cancer patients (Figure 1), supporting a role for PKCδ in the regulation of ErbB2 driven tumor formation. Further, a broad analysis of relapse-free survival over all molecular subtypes indicates that high PKCδ expression persistently correlates with a significant negative clinical outcome. Notably, this correlation does not appear to hold true across all subtypes, as our meta-analysis indicates that PKCδ expression is decreased in triple-negative breast cancers with no significant change in relapse-free survival (Figure 1). Interestingly, it has been proposed that ErbB2 overexpressing breast cancer cells can confer therapeutic resistance through an upregulation of Src activation (25). Moreover, Rexer et. al. show that lapatinib resistant cells contain increased levels of PKCδ phosphorylation at Y313, a site known to be phosphorylated by Src, compared to sensitive cells (26). Similarly, our current data indicate that PKCδ increases the occurrence of tumor relapse in ErbB2 positive tumors.
In contrast to numerous studies from our lab and others that demonstrate a pro-apoptotic function for PKCδ (12, 53–55), our current studies indicate a pro-proliferative role for PKCδ in breast cancer. Likewise PKCδ has also been shown to regulate proliferation in murine mammary cells, in mouse models of pancreatic and lung cancer, and in human NSCLC cells (19, 56, 57). Our studies specifically implicate PKCδ in regulating the MAPK pathway downstream of ErbB2 (Figure 5C). We propose that in some oncogenic contexts, the function of PKCδ may switch from pro-apoptotic to pro-survival. In this regard, NSCLC cells addicted to oncogenic K-ras appear to utilize PKCδ as a proliferative signal, while non-K-ras addicted NSCLC cell lines may preferentially utilize PKCδ for apoptosis ((19) and unpublished data).
Taken together, our studies suggest a unique regulatory mechanism by which PKCδ regulates ErbB2 signal transduction (Figure 5D). We show that in cells in which ErbB2 is activated, Src and ErbB2 are found in a complex. This association correlates with increased phosphorylation of ErbB2 at Y877, Y1221/1222, and Y1248 and increased proliferation. Loss of PKCδ disrupts this complex, resulting in decreased association of Src with the ErbB2 receptor, as well as a decrease in ErbB2 and Src activation. Since Src is responsible for the phosphorylation of ErbB2 Y877, a site that has been associated with maintaining ErbB2 activity, loss of Src binding to the ErbB2 receptor ultimately leads to an attenuation of ErbB2 driven proliferation. Together these studies support a critical role for PKCδ in the regulation of ErbB2 signaling and proliferation both in vivo and in vitro and support future investigation of PKCδ as a therapeutic target in human breast cancer.
Materials and Methods
Animal Model
FVB PKCδ KO mice (δKO) have been previously described (19). MMTV-ErbB2 (line 202) transgenic mice (32) were obtained from the Jackson Laboratory, Bar Harbor, ME, USA and crossed to FVB δKO mice. Genotypes were determined by PCR using primers to the ErbB2 transgene (forward primer (Neu-3) 5′-CGGAACCCACATCAGGCC-3′; reverse primer (Neu-4) 5′-TTTCCTGCAGCAGCCTACGC-3′). Animals were maintained at the University of Colorado Denver Anschutz Medical Campus in accordance with Laboratory Animal Care guidelines and protocols and approved by the Institutional Animal Use and Care Committee. Virgin MMTV-ErbB2;δWT and MMTV-ErbB2;δKO mice were palpated weekly for mammary tumors, starting at 120 days of age. MMTV-ErbB2;δWT littermates were used for all studies. Tumors were harvested at 1 cm in diameter, fixed in 10% neutral buffered formalin, processed and paraffin embedded for sectioning.
Cell lines and reagents
MCF10A.ErbB2 (10A.ErbB2) cells were a generous gift from Senthil Muthuswamy and previously described (33, 34). 10A.ErbB2 cells were kept in growth media (Dulbecco’s modified Eagle medium/F12 (DMEM/F12) (HyClone, Thermo Fisher Scientific, Logan, UT, USA) supplemented with 5% donor horse serum (Gibco, Grand Island, NY, USA), 20 ng/mL epidermal growth factor (EGF) (BD Transduction Laboratories, Bedford, MA, USA), 10 mg/mL insulin (Gibco), 1 ng/mL choleratoxin (Thermo Fisher Scientific), 100 mg/mL hydrocortisone, and 200 mg/mL active-G418 (Gibco)). The ErbB2 dimerizers (ligand), AP1510 (Ariad, Cambridge, MA, USA) and B/B homodimer (Clontech, Mountain View, CA, USA) were used at 250–500 nM for 2D cell culture and 1 μM for 3D cell culture. Growth factor reduced Matrigel was purchased from BD Transduction Laboratories. Human breast cancer lines were obtained from the University of Colorado Denver Cancer Center Tissue Culture Core.
Microarray data analysis
Oncomine™ (Compendia Bioscience, Ann Arbor, MI) was used for analysis of mRNA expression data from 21 human breast cancer studies (28). Methods of normalization and statistical calculations are provided on the Oncomine website (www.oncomine.org). Gene expression was compared within ErbB2 positive verses negative tumors. Differential expression of the individual genes analyzed was then ranked by p-value across each data set, with a rank of one being the most significantly changed gene in ErbB2 positive tumors. The median rank of PKCδ was calculated across all 21 data sets and analyzed for significance of overexpression or underexpression. Similar analysis was used to determine PKCδ expression in triple-negative tumors verses all other molecular subtypes. GEO accession numbers or paper references for the 21 datasets are as follows: 1. GSE3143 2. GSE2109 3. GSE5847 4. GSE6861 5. GSE22358 6. GSE25066 7. GSE8465 8. GSE20685 9. GSE18728 10. GSE5460 11. Ma Breast (58) 12. GSE1379 13. GSE14548 14. GSE2603 15. Perou Breast (59) 16. GSE3744 17. GSE5325 18. GSE3893 19. The Cancer Genome Atlas Project (TCGA) Breast http://tcga-data.nci.nih.gov/tcga/ 20. GSE21921 21. GSE3971.
Meta-analysis of PKCδ gene expression and correlation with clinical relapse-free survival was analyzed over four data sets (GSE7390, GSE3494, GSE1456, and GSE6432) encompassing 914 individual tumors using the GOBO tool (http://co.bmc.lu.se/gobo) as previously described (29). ErbB2 enriched tumors were identified from the four data sets (listed above) through the PAM50 gene set signature and represent 122 individual tumors, while basal tumors represent 143 tumors (30).
PKCδ depletion and reconstitution
Stable depletion of PKCδ was achieved using lentiviral constructs containing short hairpin RNA (shRNA) to human PKCδ (pLKO-TRC00010193 (shδ193), pLKO-TRC00010203 (shδ203), or pLKO-TRCN0000284800 (shδ800)) or a shRNA control (pLKO-scrambled (shSCR)) (Open Biosystems, Huntsville, AL, USA or Sigma-Aldrich, St. Louis, MO) as previously described (19). Adenovirus expression of LacZ, PKCδ or CA-MEK was done as previously described (60). Adenovirus transduction efficiency was >95% as verified by visualization of GFP-tagged protein by fluorescent microscopy.
Immunohistochemistry
For immunohistochemistry, sections were exposed to citrate buffer and heat antigen retrieval and then blocked with avidin and biotin. Endogenous peroxidase was quenched with 3% H2O2. Sections were incubated with primary antibody for 1 hour at room temperature (Anti-cleaved caspase-3 and KI-67). Primary antibodies include anti-cleaved caspase-3 (1:100; Cell Signaling Technology, Beverly, MA, USA) and anti-Ki-67 (1:50; DakoCytomation, Carpinteria, CA, USA). Sections were incubated in appropriate secondary for 30 min at room temperature. Sections were incubated with Vectastain ABC Reagent (Vector Laboratories) for an additional 30 minutes at room temperature. The signal was detected with diaminobenzadine substrate (Sigma, St. Louis, MO, USA) and tissues were counterstained with Gill’s hemotoxylin. For quantification of Ki-67 and cleaved caspase-3 staining, tumor sections were scanned using Aperio hardware and software (Vista, CA, USA). Six-eight fields were outlined (per tumor, per genotype), which encompassed the majority of non-border and non-necrotic regions of each tumor section. Staining was quantified and expressed as the percent of positive cells utilizing the Aperio color deconvolution algorithm. n=5–8 MMTV-ErbB2;δWT, 4–7 MMTV-ErbB2;δKO.
3D Morphogenesis Assay
Growth factor reduced Matrigel (40 μl) was plated on eight well chamber slides (#354230, #354118; BD Transduction Laboratories) and allowed to solidify for >15 minutes at 37°C. 10A.ErbB2 cells (5,000 cells/well) were trypsinized and resuspended in plating media (DMEM/F12 supplemented with 2% donor horse serum, 10 mg/ml insulin, 1 ng/ml cholera toxin, 100 mg/ml hydrocortisone, 5 ng/ml EGF, 200 mg/mL active-G418) with 2% Matrigel. On day 5 cells were switched to assay media without EGF and 24 hours later cells were left untreated or treated with ligand (1 μM). Ligand was replenished every two days. Cells were imaged live on day 6, 9, and 14 using the Zeiss Observer.Z1 microscope (Thornwood, NY, USA) and captured using Volocity 6.0 software (Perkin Elmer, Waltham, MA, USA). 5X binary pictures of each condition were then used to quantify structure area using MetaMorph software (Molecular Devices, Sunnyvale, CA, USA); n=>200 structures, representative of 3 experiments shown.
2D and 3D growth curves
For 2D culture growth curves, BT-474, MDA-MB-361, ZR-75-30 and 10A.ErbB2 cells expressing either shRNA to PKCδ (shδ193 and shδ203) or a scrambled control (shSCR) and were plated at equal densities. 10A.ErbB2 cells were plated in growth media. Twenty-four hours after plating, media was changed to assay media and 48 hours after plating cells were treated with ligand in assay media. For 3D growth curves, 10A.ErbB2 cells were grown as described above (3D morphogenesis assay). Three days after the addition of ligand, acini were treated with 0.25% trypsin for 1 hour at 37°C and cells were counted by hemocytometer.
Immunoblot analysis
For 3D cultures, acini were washed in ice cold PBS then treated with lysis buffer for 1 hour at 4°C with continuous shaking. Lysates were spun at 12,500 rpm for 20 minutes at 4°C, and supernatants from 2D and 3D culture was then separated by SDS/PAGE and immunoblotting as previously described (61). Anti-PKCδ (C-20) was purchased from Santa Cruz Biotechnology (sc-937 Santa Cruz, CA, USA). The following antibodies were purchased from Cell Signaling Technologies: Phospho-ErbB2 (Tyr877) (#2241; pErbB2 Y877); phospho-ErbB2 (Tyr1221/1222) (#2249; pErbB2 Y1221/1222); ErbB2 (#2165); phospho-Src Family (Tyr416) (#2101; pSrc); Src (#2108); phospho-Shc (Tyr239/240) (#2434; pShc); Shc (#2432); phospho-ERK1/2 (#9101; pERK1/2); and ERK1/2 (#4695). Phospho-ErbB2 (Tyr1248) (ab47755) and actin-HRP (ab49900) were purchased from Abcam (Cambridge, MA, USA). Densitometry was performed using UVP VisionWorks LS software.
Immunoprecipitation
10A.ErbB2 cells were placed in media without EGF for 24 hours, and then treated with ErbB2 ligand (B/B homodimer, 250 nM) for 2 hours. immunoprecipitation was performed as previously described (61). Five-hundred μg of protein was incubated with primary antibody (2 μg; anti-HA; #MMS-101, Covance) or control (2 μg; anti mouse IgG; #2025; Santa Cruz Biotechnology) for three hours at 4oC. Immunoblots were probed with the following antibodies: anti-HA (#MMS-101, Covance), Src (#8056, Santa Cruz) and PKCδ (#sc-937 Santa Cruz Biotechnology).
Acknowledgments
Grants: Susan G. Komen # BCTR0600812 and NIH # PO1-HD38129 and #R01DE015648
We thank Dr. Senthil Muthuswamy for generously providing the 10A.ErbB2 cells, Drs. Steve Anderson, Heide Ford and Brian Laffin for advice, Dr. Natalie Ahn for the CA-MEK adenovirus and Ariad Inc. for providing the AP1510 compound.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 3.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–55. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones JT, Akita RW, Sliwkowski MX. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett. 1999;447:227–31. doi: 10.1016/s0014-5793(99)00283-5. [DOI] [PubMed] [Google Scholar]
- 5.Reyland ME. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–99. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 7.Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–64. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 8.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. Embo J. 2002;21:6050–60. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeVries-Seimon TA, Ohm AM, Humphries MJ, Reyland ME. Induction of apoptosis is driven by nuclear retention of protein kinase C delta. J Biol Chem. 2007;282:22307–14. doi: 10.1074/jbc.M703661200. [DOI] [PubMed] [Google Scholar]
- 10.Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J Biol Chem. 2001;276:29719–28. doi: 10.1074/jbc.M100273200. [DOI] [PubMed] [Google Scholar]
- 11.Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO. Protein kinase C delta is essential for etoposide-induced apoptosis in salivary gland acinar cells. J Biol Chem. 1999;274:19115–23. doi: 10.1074/jbc.274.27.19115. [DOI] [PubMed] [Google Scholar]
- 12.Humphries MJ, Limesand KH, Schneider JC, Nakayama KI, Anderson SM, Reyland ME. Suppression of apoptosis in the protein kinase Cdelta null mouse in vivo. J Biol Chem. 2006;281:9728–37. doi: 10.1074/jbc.M507851200. [DOI] [PubMed] [Google Scholar]
- 13.Allen-Petersen BL, Miller MR, Neville MC, Anderson SM, Nakayama KI, Reyland ME. Loss of protein kinase C delta alters mammary gland development and apoptosis. Cell Death Dis. 2010;1:e17. doi: 10.1038/cddis.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKiernan E, O’Brien K, Grebenchtchikov N, Geurts-Moespot A, Sieuwerts AM, Martens JW, et al. Protein kinase Cdelta expression in breast cancer as measured by real-time PCR, western blotting and ELISA. Br J Cancer. 2008;99:1644–50. doi: 10.1038/sj.bjc.6604728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans JD, Cornford PA, Dodson A, Neoptolemos JP, Foster CS. Expression patterns of protein kinase C isoenzymes are characteristically modulated in chronic pancreatitis and pancreatic cancer. Am J Clin Pathol. 2003;119:392–402. doi: 10.1309/bkpc9dx98r781b87. [DOI] [PubMed] [Google Scholar]
- 16.Pongracz J, Clark P, Neoptolemos JP, Lord JM. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer. 1995;61:35–9. doi: 10.1002/ijc.2910610107. [DOI] [PubMed] [Google Scholar]
- 17.D’Costa AM, Robinson JK, Maududi T, Chaturvedi V, Nickoloff BJ, Denning MF. The proapoptotic tumor suppressor protein kinase C-delta is lost in human squamous cell carcinomas. Oncogene. 2006;25:378–86. doi: 10.1038/sj.onc.1209065. [DOI] [PubMed] [Google Scholar]
- 18.Varga A, Czifra G, Tallai B, Nemeth T, Kovacs I, Kovacs L, et al. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur Urol. 2004;46:462–5. doi: 10.1016/j.eururo.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Symonds JM, Ohm AM, Carter CJ, Heasley LE, Boyle TA, Franklin WA, et al. Protein kinase C delta is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res. 2011;71:2087–97. doi: 10.1158/0008-5472.CAN-10-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keshamouni VG, Mattingly RR, Reddy KB. Mechanism of 17-beta-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER2 AND PKC-delta. J Biol Chem. 2002;277:22558–65. doi: 10.1074/jbc.M202351200. [DOI] [PubMed] [Google Scholar]
- 21.Iwabu A, Smith K, Allen FD, Lauffenburger DA, Wells A. Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C delta-dependent pathway. J Biol Chem. 2004;279:14551–60. doi: 10.1074/jbc.M311981200. [DOI] [PubMed] [Google Scholar]
- 22.Kiley SC, Clark KJ, Duddy SK, Welch DR, Jaken S. Increased protein kinase C delta in mammary tumor cells: relationship to transformtion and metastatic progression. Oncogene. 1999;18:6748–57. doi: 10.1038/sj.onc.1203101. [DOI] [PubMed] [Google Scholar]
- 23.Kiley SC, Clark KJ, Goodnough M, Welch DR, Jaken S. Protein kinase C delta involvement in mammary tumor cell metastasis. Cancer Res. 1999;59:3230–8. [PubMed] [Google Scholar]
- 24.Kruger JS, Reddy KB. Distinct mechanisms mediate the initial and sustained phases of cell migration in epidermal growth factor receptor-overexpressing cells. Mol Cancer Res. 2003;1:801–9. [PubMed] [Google Scholar]
- 25.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–9. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rexer BN, Ham AJ, Rinehart C, Hill S, de Granja-Ingram NM, Gonzalez-Angulo AM, et al. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011;30:4163–74. doi: 10.1038/onc.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song JS, Swann PG, Szallasi Z, Blank U, Blumberg PM, Rivera J. Tyrosine phosphorylation-dependent and -independent associations of protein kinase C-delta with Src family kinases in the RBL-2H3 mast cell line: regulation of Src family kinase activity by protein kinase C-delta. Oncogene. 1998;16:3357–68. doi: 10.1038/sj.onc.1201886. [DOI] [PubMed] [Google Scholar]
- 28.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 2011;6:e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bouchard L, Lamarre L, Tremblay PJ, Jolicoeur P. Stochastic appearance of mammary tumors in transgenic mice carrying the MMTV/c-neu oncogene. Cell. 1989;57:931–6. doi: 10.1016/0092-8674(89)90331-0. [DOI] [PubMed] [Google Scholar]
- 32.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muthuswamy SK, Gilman M, Brugge JS. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol. 1999;19:6845–57. doi: 10.1128/mcb.19.10.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–88. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 36.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 37.Kwon YK, Bhattacharyya A, Alberta JA, Giannobile WV, Cheon K, Stiles CD, et al. Activation of ErbB2 during wallerian degeneration of sciatic nerve. J Neurosci. 1997;17:8293–9. doi: 10.1523/JNEUROSCI.17-21-08293.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dankort DL, Wang Z, Blackmore V, Moran MF, Muller WJ. Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol Cell Biol. 1997;17:5410–25. doi: 10.1128/mcb.17.9.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dankort D, Jeyabalan N, Jones N, Dumont DJ, Muller WJ. Multiple ErbB-2/Neu Phosphorylation Sites Mediate Transformation through Distinct Effector Proteins. J Biol Chem. 2001;276:38921–8. doi: 10.1074/jbc.M106239200. [DOI] [PubMed] [Google Scholar]
- 40.Xu W, Yuan X, Beebe K, Xiang Z, Neckers L. Loss of Hsp90 association up-regulates Src-dependent ErbB2 activity. Mol Cell Biol. 2007;27:220–8. doi: 10.1128/MCB.00899-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menard S, Tagliabue E, Campiglio M, Pupa SM. Role of HER2 gene overexpression in breast carcinoma. J Cell Physiol. 2000;182:150–62. doi: 10.1002/(SICI)1097-4652(200002)182:2<150::AID-JCP3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 42.Marmor MD, Skaria KB, Yarden Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int J Radiat Oncol Biol Phys. 2004;58:903–13. doi: 10.1016/j.ijrobp.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Zhou X, Agazie YM. Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J Biol Chem. 2009;284:12226–34. doi: 10.1074/jbc.M900020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dankort D, Maslikowski B, Warner N, Kanno N, Kim H, Wang Z, et al. Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol Cell Biol. 2001;21:1540–51. doi: 10.1128/MCB.21.5.1540-1551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schade B, Lam SH, Cernea D, Sanguin-Gendreau V, Cardiff RD, Jung BL, et al. Distinct ErbB-2 coupled signaling pathways promote mammary tumors with unique pathologic and transcriptional profiles. Cancer Res. 2007;67:7579–88. doi: 10.1158/0008-5472.CAN-06-4724. [DOI] [PubMed] [Google Scholar]
- 46.Marcotte R, Zhou L, Kim H, Roskelly CD, Muller WJ. c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol Cell Biol. 2009;29:5858–71. doi: 10.1128/MCB.01731-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muthuswamy SK, Siegel PM, Dankort DL, Webster MA, Muller WJ. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol Cell Biol. 1994;14:735–43. doi: 10.1128/mcb.14.1.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan M, Li P, Klos KS, Lu J, Lan KH, Nagata Y, et al. ErbB2 promotes Src synthesis and stability: novel mechanisms of Src activation that confer breast cancer metastasis. Cancer Res. 2005;65:1858–67. doi: 10.1158/0008-5472.CAN-04-2353. [DOI] [PubMed] [Google Scholar]
- 49.Cheng J, Watkins SC, Walker WH. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in sertoli cells. Endocrinology. 2007;148:2066–74. doi: 10.1210/en.2006-1465. [DOI] [PubMed] [Google Scholar]
- 50.Kraus S, Benard O, Naor Z, Seger R. c-Src is activated by the epidermal growth factor receptor in a pathway that mediates JNK and ERK activation by gonadotropin-releasing hormone in COS7 cells. J Biol Chem. 2003;278:32618–30. doi: 10.1074/jbc.M303886200. [DOI] [PubMed] [Google Scholar]
- 51.Liu D, Lu JS, Yin XL. Role of pp60c-src in mitogen-activated protein kinase activation of vascular smooth muscle cells. Sheng Li Xue Bao. 2000;52:483–6. [PubMed] [Google Scholar]
- 52.Zhang HT, O’Rourke DM, Zhao H, Murali R, Mikami Y, Davis JG, et al. Absence of autophosphorylation site Y882 in the p185neu oncogene product correlates with a reduction of transforming potential. Oncogene. 1998;16:2835–42. doi: 10.1038/sj.onc.1201820. [DOI] [PubMed] [Google Scholar]
- 53.Jackson D, Zheng Y, Lyo D, Shen Y, Nakayama K, Nakayama KI, et al. Suppression of cell migration by protein kinase Cdelta. Oncogene. 2005;24:3067–72. doi: 10.1038/sj.onc.1208465. [DOI] [PubMed] [Google Scholar]
- 54.Lu Z, Hornia A, Jiang YW, Zang Q, Ohno S, Foster DA. Tumor promotion by depleting cells of protein kinase C delta. Mol Cell Biol. 1997;17:3418–28. doi: 10.1128/mcb.17.6.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–9. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 56.Grossoni VC, Falbo KB, Kazanietz MG, de Kier Joffe ED, Urtreger AJ. Protein kinase C delta enhances proliferation and survival of murine mammary cells. Mol Carcinog. 2007;46:381–90. doi: 10.1002/mc.20287. [DOI] [PubMed] [Google Scholar]
- 57.Mauro LV, Grossoni VC, Urtreger AJ, Yang C, Colombo LL, Morandi A, et al. PKC Delta (PKCdelta) promotes tumoral progression of human ductal pancreatic cancer. Pancreas. 2010;39:e31–41. doi: 10.1097/MPA.0b013e3181bce796. [DOI] [PubMed] [Google Scholar]
- 58.Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:5974–9. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 60.Tolwinski NS, Shapiro PS, Goueli S, Ahn NG. Nuclear localization of mitogen-activated protein kinase kinase 1 (MKK1) is promoted by serum stimulation and G2-M progression. Requirement for phosphorylation at the activation lip and signaling downstream of MKK. J Biol Chem. 1999;274:6168–74. doi: 10.1074/jbc.274.10.6168. [DOI] [PubMed] [Google Scholar]
- 61.Adwan TS, Ohm AM, Jones DN, Humphries MJ, Reyland ME. Regulated binding of importin-alpha to protein kinase Cdelta in response to apoptotic signals facilitates nuclear import. J Biol Chem. 2011;286:35716–24. doi: 10.1074/jbc.M111.255950. [DOI] [PMC free article] [PubMed] [Google Scholar]