

Abstract
There is significant variability in individual responses to opioid drugs, which is likely to have a significant genetic component. A number of non-synonymous single-nucleotide polymorphisms (SNPs) in the coding regions of the μ-opioid receptor gene (OPRM1) have been postulated to contribute to this variability. Although many studies have investigated the clinical influences of these μ-opioid receptor variants, the outcomes are reported in the context of thousands of other genes and environmental factors, and we are no closer to being able to predict individual response to opioids based on genotype. Investigation of how μ-opioid receptor SNPs affect their expression, coupling to second messengers, desensitization and regulation is necessary to understand how subtle changes in receptor structure can impact individual responses to opioids. To date, the few functional studies that have investigated the consequences of SNPs on the signalling profile of the μ-opioid receptor in vitro have shown that the common N40D variant has altered functional responses to some opioids, while other, rarer, variants display altered signalling or agonist-dependent regulation. Here, we review the data available on the effects of μ-opioid receptor polymorphisms on receptor function, expression and regulation in vitro, and discuss the limitations of the studies to date. Whether or not μ-opioid receptor SNPs contribute to individual variability in opioid responses remains an open question, in large part because we have relatively little good data about how the amino acid changes affect μ-opioid receptor function.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Keywords: A118G, pharmacogenomics, analgesia, addiction, tolerance, dependence
Introduction
Opioid analgesics are the most important classes of drug used for the treatment of moderate to severe pain. Opioids elicit powerful analgesic effects, yet they are also associated with a number of adverse effects such as respiratory depression, constipation, nausea and sedation (Moore and McQuay, 2005; Dahan et al., 2010; Noble et al., 2010). The development of tolerance to opioid analgesia, coupled with the associated adverse effects, limits the usefulness of opioid therapy in the treatment of long-term and chronic pain. Opioid misuse is also a major social problem in many countries (Dhalla et al., 2011).
There is significant variation between individuals in both the analgesic effect of opioid drugs and the degree of adverse effects experienced. The risk of serious adverse events such as respiratory depression can limit dosing with the result that many individuals receive inadequate pain relief (Skorpen and Laugsand, 2008). Furthermore, as tolerance develops over time, the escalating opioid doses that are required to maintain adequate analgesia can cause intolerable side effects (Corbett et al., 2006). There is also an apparently heritable predisposition towards opioid abuse and addiction (Merikangas et al., 1998). A number of elements may affect final individual response to opioids including drug absorption, distribution and metabolism, as well as the intrinsic efficacy of the drug at the receptor and variation in receptor signalling function, agonist regulation and downstream effector pathways. Genetic factors such as differences in protein sequence, regulatory element function and potentially complex epigenetic regulation of protein expression contribute to variability in all these parameters (Lotsch and Geisslinger, 2005; Skorpen and Laugsand, 2008). Understanding these components could result in the ability to better predict clinical outcomes when prescribing opioid analgesics, reducing the number of patients receiving an inappropriate dose of opioid by potentially limiting the development of tolerance and dependence. Rational dosing would also likely increase the number of patients who benefit from opioid therapy.
Clinically important opioid analgesics act by binding to the μ-opioid receptor (Matthes et al., 1996; Alexander et al., 2013a), making this receptor a prime candidate for contributing to the genetic component of inter-individual differences in opioid response. The μ-opioid receptor is a typical class A GPCR (Alexander et al., 2013a). Many single-nucleotide polymorphisms (SNPs) within the OPRM1 gene have been identified in humans, and a number of these are non-synonymous changes in the coding regions, meaning that there is an amino acid (aa) substitution resulting in an alternative receptor isoform (LaForge et al., 2000; Ikeda et al., 2005; Lotsch and Geisslinger, 2005; Ravindranathan et al., 2009; Fortin et al., 2010). There are good reasons to consider the potential of μ-opioid receptor SNPs to contribute to the clinical variability of opioid responses. GPCR signalling is complex, with the notion of simple, linear and robust rearrangements of protein structure being required for signal transduction no longer accepted. Thus, the possibility that single aa substitutions can lead to subtle or profound changes in the way receptors signal is very real, and has been demonstrated for several GPCRs (Thompson et al., 2008; Zhang and Steinberg, 2013). Furthermore, commonly prescribed opioids such as morphine and buprenorphine have relatively low efficacy, and even modest differences in receptor expression or efficiency of signal transduction could have a significant impact on individual response to these drugs. Finally, clinically used opioids are chemically diverse, and are likely to have subtly different structural features of the μ-opioid receptor determining their signalling – potentially leading to distinct effects of non-synonymous SNPs on different drugs.
An additional level of complexity when considering the functional consequences of SNPs arises from the large number of putative splice variants of the μ-opioid receptor that have been described (Mizoguchi et al., 2012; Pasternak and Pan, 2013). Although the functional role of alternatively spliced OPRM1 transcripts is not yet well established, a single non-synonymous aa change could conceivably have distinct effects on different splice variants of the receptor. For the most part, this remains unexplored.
Many studies have examined potential associations between μ-opioid receptor SNPs and various clinical outcomes, such as the degree of pain relief in response provided by opioids, or the prevalence of substance abuse. These clinical reports are often contradictory and there is no clear consensus as to the effect of any polymorphism on disease susceptibility or the outcomes of drug administration. This is presumably in part due to relatively small sample sizes in most studies, as well as a range of confounding influences such as overall genotype and environment (reviewed in Lotsch and Geisslinger, 2005). Far fewer studies have investigated the molecular consequences of OPRM1 SNPs on receptor function and signalling in vitro, and results from these studies are also conflicting. Nevertheless, in vitro experiments have led to intriguing insights into μ-opioid receptor function, and in this review, we focus on the effects of naturally occurring, non-synonymous SNPs in the coding region of OPRM1 on μ-opioid receptor function. The SNPs considered here, the corresponding aa exchanges and their position on the μ-opioid receptor are summarized in Table 1 and Figure 1.
Table 1.
Summary of non-synonymous μ-opioid receptor variants in the protein coding region, their corresponding OPRM1 SNP, exon and μ-opioid receptor protein domain
| AA exchange | μ-Opioid receptor domain | SNP | Exon |
|---|---|---|---|
| N40D | N-terminus | 118 A > G | 1 |
| A6V | N-terminus | 17 C > T | 1 |
| S42C | N-terminus | 124 T > A | 1 |
| D51N | N-terminus | 151 G > A | 1 |
| G63V | N-terminus | 188 G > T | 1 |
| S66F | N-terminus | 197 C > T | 1 |
| L85I | TM1 | 253 C > A | 1 |
| S147C | TM3 | 440 C > G | 2 |
| N152D | TM3 | 454 C > G | 2 |
| R181C | ICL2 | 541 C > T | 2 |
| N190K | ICL2 | 570 A > T | 2 |
| C192F | TM4 | 575 G > T | 2 |
| R260H | ICL3 | 779 G > A | 3 |
| R265H | ICL3 | 794 G > A | 3 |
| S268P | ICL3 | 802 T > C | 3 |
| D274N | ICL3 | 820 G > A | 3 |
| V293A | TM6 | 877 G > A | 3 |
See references in the text for original reports.
Figure 1.
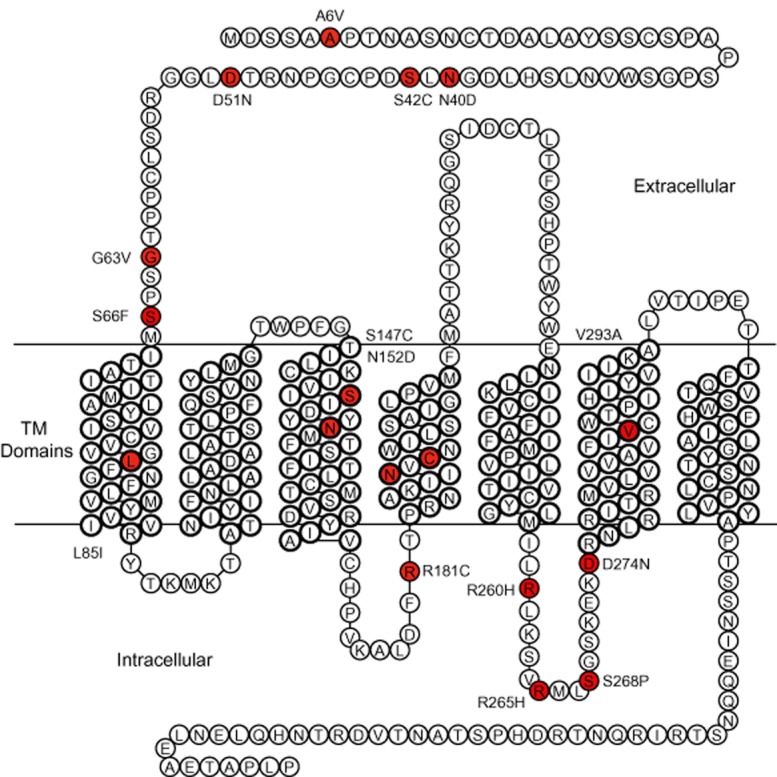
Naturally occurring, non-synonymous OPRM1 variants reported, and their position on the μ-opioid receptor protein. Residues where an aa exchange occurs are indicated in red.
The μ-opioid receptor
The μ-opioid receptor is a class A rhodopsin-like GPCR, with a relatively short extracellular N-terminal domain (66 aa), 7-membrane spanning domains and an intracellular carboxy-terminal ‘tail’ (70 aa) that includes a putative ‘helix 8’ domain tethered to the plasma membrane by a palmitoyl residue (Manglik et al., 2012). Opioid ligands are thought to approach the receptor from the extracellular space, engaging with the receptor by interacting with a binding pocket formed by elements of transmembrane (TM) domains TM3, TM5, TM6 and TM7, and possibly residues in extracellular loop 2 (Serohijos et al., 2011; Manglik et al., 2012). G protein interactions are mediated through intracellular domains, including intracellular loops (ICLs) 2 and 3, and the C-terminal region. The intracellular regions of the μ-opioid receptor, particularly the C-terminal domain, also contain important phosphorylation sites regulating receptor desensitization, internalization and resensitization (Williams et al., 2013).
The μ-opioid receptor modulates a diverse range of physiological systems, including nociception and analgesia, reward and euphoria, immune function, stress responsivity, respiration and gut motility (Jordan and Devi, 1998; Kreek et al., 2005). The most well-characterized signalling pathways of the μ-opioid receptor proceed via activation of heterotrimeric G proteins or β-arrestin (Law et al., 2000). The μ-opioid receptor can couple to a number of different G proteins, including Pertussis toxin-sensitive Gαi/o subunits, the closely related Gαz, and Gα16 (Connor and Christie, 1999). Canonical coupling of the μ-opioid receptor includes Gαi/o inhibition of AC, Gβγ subunit activation of G protein-coupled, inwardly rectifying potassium channels (GIRKs; Alexander et al., 2013b) and inhibition of voltage gated Ca2+ channels (CaV), as well as activation of MAPK. Examples of G protein-independent signalling of μ-opioid receptors include β-arrestin-mediated ERK1/2 activation (Zheng et al., 2010), signal transducer and activator of transcription 5 phosphorylation (Mazarakou and Georgoussi, 2005) and Src-mediated Ras/Raf-1 recruitment (Zhang et al., 2013).
The μ-opioid receptor, like all GPCRs, has many active conformations (Pineyro and Archer-Lahlou, 2007; Kenakin and Miller, 2010; Manglik et al., 2012). In their unbound state, GPCRs constantly oscillate through a range of possible conformational states. Ligands bind to GPCRs and stabilize subsets of conformational states, some of which couple to and activate downstream effectors (agonists), while other are not coupled to effectors, and when ligands bind they prevent downstream signalling (antagonists). The stabilization of subsets of conformations by a ligand may lead to preferential activation of a restricted set of signalling pathways, leading to ligand-specific patterns of signalling and receptor regulation – also known as ligand-biased signalling or functional selectivity. The μ-opioid receptor binds an array of structurally diverse ligands and interacts with many effector and regulatory proteins providing a fertile system for ligand-biased signalling. (Kenakin, 2002; Massotte et al., 2002; Saidak et al., 2006).
The corollary of structurally distinct agonists and effector molecules preferentially coupling via subsets of receptor conformations is that changes in the molecular structure of the receptor itself are likely to affect receptor conformation (Abrol et al., 2013; Cox, 2013). Thus, aa changes resulting from SNPs have the potential to affect μ-opioid receptor signalling globally or in a ligand-dependent manner by affecting the ability of a ligand to bind to the receptor, altering the conformation of the ligand-receptor complex and/or affecting the ability of this complex to couple to G proteins and associated signalling or regulatory pathways.
Functional studies of μ-opioid receptor SNPs
Most functional studies of human (h) μ-opioid receptors use heterologously expressed receptors in an immortalized cell line such as CHO-K1, HEK-293 or AtT-20. The physiological relevance of subtle differences in signalling exhibited by μ-opioid receptor variants in these highly engineered expression systems is difficult to predict, and making direct comparisons between receptor signalling profiles in different expression systems may be problematic as different cell lines vary in the available pool of G proteins, effector molecules and regulatory proteins (e.g. Atwood et al., 2012). Nevertheless, μ-opioid receptors are naturally expressed in a wide variety of cell types, and there is unlikely to be ‘one true path’ for receptor activation and regulation. Thus, studies in diverse systems are probably necessary to capture the possible consequences of variations in receptor structure. However, in order to make comparisons between polymorphic variants meaningful, careful attention needs to be paid to receptor expression levels and the nature of the signalling assays (Connor et al., 2004). While there seems to be functional differences between μ-opioid receptor SNPs and the most common form of the receptor, many variants have been superficially described, and making firm conclusions about the consequences of variations in μ-opioid receptor sequence is limited by the experimental conditions used to study them.
N-terminal domain SNPs
N40D
The N40D variant is the most commonly occurring OPRM1 SNP, with an allelic frequency ranging from 10 to 50% within various populations (Mura et al., 2013). The N40D SNP is in the N-terminal extracellular domain of the μ-opioid receptor, and removes one of five putative asparagine-linked glycosylation sites in this region (Table 1, Singh et al., 1997). First reported by Bergen et al. (1997), many studies have examined associations between the D40 allele and physiological and clinical parameters including nociception, altered response to opioid analgesics, opioid and alcohol dependence and hypothalamic–pituitary–adrenal axis responses (Kreek et al., 2005; Walter and Lotsch, 2009). Most of the association studies report that carriers of the D40 allele have a reduced response to opioids, although some studies have reported the opposite, and others no effect at all (reviewed in Diatchenko et al., 2011). A recent meta-analysis of the clinical effects of the N40D variant in pain management concluded that knowing a patient's allele(s) at position 118 in OPRM1 would have little impact on the treatment (Walter and Lotsch, 2009), although the number of studies available for review was small. The D40 allele has also been associated with an increased, decreased or unchanged susceptibility to drug use and dependence (reviewed in Mague and Blendy, 2010).
Regulation of N40D expression
Regardless of any impact on the function of the μ-opioid receptor, the possibility that the nucleotide or aa substitutions may affect μ-opioid receptor expression levels needs to be considered. There is some evidence for reduced μ-opioid receptor expression associated with the G118 allele (or its murine orthologue). It was reported that in the cortex and the pons from the brains of A118G heterozygotes, there was significantly less G118 mRNA (1.5–2.5-fold) than A118 mRNA (Zhang et al., 2005). A similar reduction in mRNA was found in a knock-in mouse model with an orthologous A112 to G112 mutation (Mague et al., 2009). A potential explanation for the reduced levels of G118 mRNA was provided by Oertel et al. (2012), who deduced that the G118 allele has an extra methylation site introduced by the guanine nucleotide, which was suggested to inhibit compensatory up-regulation of μ-opioid receptor mRNA in chronic opioid users. It is possible that this epigenetic regulation also results in lower levels of G118 mRNA in basal conditions.
The loss of the potential glycosylation site, N40, may also contribute to lower cell surface receptor levels for the D40 allele, although this has not been consistently reported (Zhang et al., 2005; Oertel et al., 2009). In mice, the molecular mass of μ-opioid receptors in 112G/G animals (55 kDa) is lower than 112A/A mice (62 kDa), whereas the molecular mass of deglycosylated μ-opioid receptors was identical (42 kDa) for both variants, indicating less glycosylation in 112G/G mice (Huang et al., 2012). The G/G mice also have lower μ-opioid receptor expression compared with A/A mice (Mague et al., 2009; Wang et al., 2012). Findings of lower expression extend to cells lines (Zhang et al., 2005; Huang et al., 2012), with a shorter half-life of D40 (12 h) compared with N40 (28 h) in CHO cells. Enzymatic deglycosylation of the μ-opioid receptor also decreased receptor expression in HEK-293 cells by 90% (Kroslak et al., 2007). Decreased mRNA stability, potential epigenetic repression and incomplete glycosylation could all contribute to reduced D40 receptor expression, potentially providing a mechanism for greater opioid requirement in D40 carriers (Mura et al., 2013).
Second messenger coupling
The consequences of the N40D substitution on the signalling profile of the μ-opioid receptor are not well understood, and despite the intense research into the clinical effects of the D40 variant, only a handful of functional studies in cells have been performed on this variant (Table 2). The first reported functional consequences of a μ-opioid receptor SNP was a threefold increase in the affinity of β-endorphin for the D40 variant and a threefold increase in the potency of β-endorphin to activate GIRK channels co-expressed with D40 in Xenopus laevis oocytes (Bond et al., 1998). No differences in binding or signalling were reported for other opioids including ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), endomorphins 1 and 2, and enkephalins. Unfortunately, these provocative results were based on very limited quantification of the cellular responses to activation of the N40 and D40 alleles, and no statistical analysis was included. Subsequent studies looking at different signalling pathways in other expression systems have failed to find differences in β-endorphin potency at N40 and D40 (Befort et al., 2001; Beyer et al., 2004; Kroslak et al., 2007).
Table 2.
Summary of the key findings about μ-opioid receptor SNP signalling
| MOPr variant | Key observations | pEC50 WT | pEC50 SNP | Bmax WT | Bmax Var | Reference |
|---|---|---|---|---|---|---|
| N40D | Unchanged agonist affinity. Similar DAMGO stimulated GTPγS activation. | 7.0 – DAM | 6.7 – DAM | 5.5 pmol·mg−1 | 6.1 pmol·mg−1 | Befort et al., 2001 |
| Similar cAMP inhibition. Reduced D40 expression. | 9.1 – Mor 8.8 – M-6-G 7.9 – β-end | 9.0 – Mor 8.8 – M-6-G 7.8 – β-end | 4.8 pmol·mg−1 | 0.63 pmol mg−1 | Beyer et al., 2004 | |
| Three times increased β-endorphin affinity for D40 than WT, and three times increased potency for GIRK activation in D40 expressing Xenopus oocytes. | Not provided | Not provided | Not provided | Not provided | Bond et al., 1998 | |
| Different N/D40 stimulated PKA activity and ERK1/2 phosphylation after chronic morphine treatment. | N/A (1 μM morphine only) | N/A (1 μM morphine only) | 835 fmol·mg–1 | 830 fmol mg−1 | Deb et al. 2010 | |
| Similar inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.8 – DAM 8.9 – End-1 8.5 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 | |
| Decreased agonist potency to inhibit AC in D40-HEK293 and D40-AV-12 cells | 8.6 – DAM 8.4 – Mor 8.3 – Meth 8.4 – β-End | 8.1 – DAM* 7.8 – Mor* 7.8 – Meth* 8.1 – β-End | Not provided | 66% of WT | Kroslak et al., 2007 | |
| Decreased morphine potency for CaV inhibition in mouse trigeminal ganglion cells expressing ‘humanized’ D40. | 7.3 – Mor 7.2 – Fent | 6.6 – Mor* 7.0 – Fent* | Not provided | Similar to WT | Mahmoud et al., 2011 | |
| Increased DAMGO and morphine potency for CaV inhibition at D40 expressing rat SCG cells. | 7.5 – DAM 7.1 – Mor 7.1 – M-6-G 7.1 – End-1 | 7.8 – DAM* 7.4 – Mor* 7.1 – M-6-G 7.1 – End-1 | Not provided | Not provided | Margas et al., 2007 | |
| Decreased D40 expression in SII region of cortex in post-mortem brain. SII region-specific decrease in DAMGO efficacy in D40 carriers. | 5.9 – DAM | 6.0 – DAM | 97 fmol·mg–1 | 114 fmol mg−1 | Oertel et al., 2009 | |
| No difference in DAMGO potency at D40 for CaV inhibition in ‘humanized’ mouse trigeminal ganglion cells. | 7.2 – DAM 6.3 – β-End | 7.1 – DAM 6.2 – β-End | Not provided | Similar to WT | Ramchandani et al., 2011 | |
| Increased DAMGO potency for Cav2.2 inhibition in D40-HEK-293 cells. | 8.6 – DAM | 9.5 – DAM* | Not provided | Not provided | Lopez Soto & Raingo, 2012 | |
| Lower mRNA levels of G118 allele for heterozygous A118G carriers in post-mortem brain. Decreased G118 mRNA and 10-fold decreased D40 expression in CHO-K1 cells. | N/A | N/A | Not provided | Not provided | Zhang et al., 2005 | |
| A6V | Similar inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.6 – DAM 8.7 – End-1 8.2 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 |
| Unchanged agonist efficacy and potency for intracellular Ca release at A/V6 on MOR1A backbone. | 7.5 – DAM 7.4 – Mor | 7.9 – DAM 7.3 – Mor | 5.6 pmol·mg−1 | 5.8 pmol·mg−1 | Ravindranathan et al., 2009 | |
| S42C | Decreased agonist potency for intracellular Ca release at C42 on MOR1A backbone. | 7.5 – DAM 7.4 – Mor | >6.8 – DAM* >6.8 – Mor* | 2.7 pmol·mg−1 | 5.8 pmol·mg−1 | Ravindranathan et al., 2009 |
| D51N | Similar inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.6 – DAM 8.8 – End-1 8.4 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 |
| G63V | Similar inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 9.0 – DAM 8.9 – End-1 8.5 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 |
| S66F | Decreased potency of DAMGO and endormorphin 1 at F66 for inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.2 – DAM* 8.3 – End-1* 7.7 – L-Enk* | Not provided | Similar to WT | Fortin et al., 2010 |
| L85I (L83I) | Increased morphine stimulated endocytosis in I83-HEK293 cells. Decreased agonist efficacy in inhibition of AC and ERK phosphorylation. | 6.7 – DAM 6.7 – Mor | 6.5 – DAM 6.9 – Mor | 1.8 pmol mg−1 | 2.7 pmol mg−1 | Cooke et al., 2014 |
| L85I | Increased morphine stimulated endocytosis in I85-HEK293 cells. Increased AC super activation in I85 HEK-293 cells. No change in agonist potency. | 7.5 – DAM 7.4 – Mor | 7.9 – DAM 7.7 – Mor | 5.6 pmol mg−1 | 5.2 pmol mg−1 | Ravindranathan et al., 2009 |
| S147C | Decreased agonist potency for inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.3 – DAM* 8.4 – End-1* 7.9 – L-Enk* | Not provided | Similar to WT | Fortin et al., 2010 |
| Increased agonist potency for intracellular Ca release at C147 on MOR1A backbone. | 7.5 – DAM 7.4 – Mor | 7.9 – DAM* 8.3 – Mor* | 5.6 pmol·mg−1 | 5.0 pmol·mg−1 | Ravindranathan et al., 2009 | |
| N152D | Decrease in morphine affinity for D152 in COS cells. | N/A | N/A | 5.5 pmol·mg−1 | 1.9 pmol·mg−1 | Befort et al., 2001 |
| R181C | HEK-293 cells expressing C181 failed to signal via DAMGO or morphine. | 7.5 – DAM 7.4 – Mor | N/A | 5.6 pmol·mg−1 | 3.5 pmol·mg−1 | Ravindranathan et al., 2009 |
| N190K | Decreased K190 expression in HEK-293 cells. Treatment with naloxone and naltrexone both increased K190 expression and inhibition of cAMP-stimulated CRE transcription. | Not provided | Not provided | Not provided | N/A | Fortin et al., 2010 |
| N192F | Decreased agonist potency at F192 for intracellular calcium release in HEK-293 cells expressing F192 on MOR1A backbone. | 7.5 – DAM 7.4 – Mor | >6.8 – DAM* >6.8 – Mor* | 5.6 pmol·mg−1 | 4.4 pmol·mg−1 | Ravindranathan et al., 2009 |
| R260H | Decreased basal GTPγS activity at H260 in HEK293 cells. Slight decrease in morphine stimulated GTPγS at H260, and slight decrease in affinity of H260 for CaM. | 8.4 – Mor | 8.6 – Mor | 3.5 pmol·mg−1 | 3.9 pmol·mg−1 | Wang et al., 2001 |
| R265H | Decreased basal GTPγS activity at H260 in COS cells. Slight decrease in maximal DAMGO stimulated GTPγS at H260. | 7.0 – DAM | 6.9 – DAM | 5.5 pmol·mg−1 | 4.6 pmol·mg−1 | Befort et al., 2001 |
| Decreased agonist potency for inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.0 – DAM* 8.1 – End-1* 7.6 – L-Enk* | Not provided | Similar to WT | Fortin et al., 2010 | |
| Decreased basal GTPγS activity at H265 in HEK293 cells. Slight decrease in maximal morphine stimulated GTPγS at H265. Decreased affinity of H265 for CaM binding, and decreased desensitization following morphine pretreatment. | 8.4 – Mor | 8.5 – Mor | 3.5 pmol·mg−1 | 4.2 pmol·mg−1 | Wang et al., 2001 | |
| S268P | No of agonist-stimulated GTPγS binding in COS cells. Decreased agonist potency and efficacy at P268 for inhibition of cAMP accumulation. | 7.2 – DAM 6.5 – β-End 6.2 – Mor | 6.4 – DAM* 5.9 – β-End* 5.8 – Mor* | 5.5 pmol·mg−1 | 3.6 pmol·mg−1 | Befort et al., 2001 |
| Decreased potency of DAMGO and endomorphin-1 for inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.2 – DAM* 8.4 – End-1* 7.9 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 | |
| Decreased GTPγS binding, slower desensitization and decreased AC inhibition in response to DAMGO. | N/A | N/A | 643 fmol·mg–1 | 340 fmol·mg–1 | Koch et al., 2000 | |
| Decreased morphine potency at P268 for inhibition of cAMP accumulation. | 7.0 – Mor | 6.3 – Mor* | 3.5 pmol·mg−1 | 4.5 pmol·mg−1 | Wang et al., 2001 | |
| D274N | Increased agonist potency for inhibition of cAMP-stimulated CRE transcription. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 9.1 – DAM* 8.3 – End-1* 8.6 – L-Enk* | Not provided | Similar to WT | Fortin et al., 2010 |
| V293I | Unchanged in agonist potency for inhibition of cAMP accumulation. | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | 8.8 – DAM 8.8 – End-1 8.4 – L-Enk | Not provided | Similar to WT | Fortin et al., 2010 |
P < 0.05, from original publications. Abbreviations: β-End, β-endorphin; DAM, DAMGO; End-1, endomorphin 1; Fent, fentanyl; L-ENK, [Leu]5enkephalin; Meth, methadone; Mor, morphine; MOR1A, μ-opioid receptor 1A splice variant; SCG, superior cervical ganglion.
N40 inhibition of AC has been examined in several studies in HEK 293 cells (Beyer et al., 2004; Kroslak et al., 2007; Fortin et al., 2010). Unfortunately, these studies did not use N40 and D40 cell lines with equivalent receptor expression, and receptor reserve for AC inhibition was not assessed. Beyer et al. (2004) found no differences in the effects of morphine, morphine-6-glucuronide (M-6-G) or β-endorphin to inhibit acute cyclic adenosine monophosphate (cAMP) accumulation despite sevenfold lower expression of D40 than N40 in their cells. Fortin et al. (2010) also found no difference in how DAMGO, endomorphin 1 or leu-enkephalin modified cAMP-dependent gene transcription in cells acutely transfected with D40 and N40 constructs. This strategy produced apparently equivalent levels of receptor expression (measured by elisa), but the absolute levels were not reported. By contrast, Kroslak et al. (2007) reported a decreased potency of morphine, methadone and DAMGO, but not β-endorphin to inhibit cAMP accumulation in cells expressing D40, however, this was associated with a 66% lower expression of D40 compared with N40. It is difficult to explain the differences between these studies, particularly in the absence of information about relative efficacy. Studies using cAMP-dependent gene expression assays measure μ-opioid receptor activity after prolonged incubation with agonist, and the response measured reflects the integrated outcome of acute inhibition of AC as well as agonist-dependent uncoupling, internalization and possible recycling or degradation of the receptor, any of which could be altered by the D40 polymorphism (Connor et al., 2004; Dang and Christie, 2012). Likewise, possible differences in the acute regulation of D40 and N40 variants of the μ-opioid receptor over the time course of acute cAMP accumulation assays could also confound their outcomes (Connor et al., 2004).
The activity of N40 and D40 have also been compared by measuring inhibition of Ca2+ channels in acutely transfected sympathetic neurons (Margas et al., 2007), HEK293 cells (Lopez Soto and Raingo, 2012) as well as mice ‘humanized’ with A118 and G118 knock-in (Mahmoud et al., 2011; Ramchandani et al., 2011). Opioid receptor inhibition of CaV is via direct Gβγ-subunit inhibition of channel gating. In both HEK293 cells and sympathetic neurons, DAMGO inhibited CaV more potently in cells expressing the D40 variant, with morphine also being more potent at D40 in sympathetic neurons (Margas et al., 2007; Lopez Soto and Raingo, 2012; Table 2). Interestingly, the potency of endomorphin 1 and M-6-G was not different between N40 and D40 in sympathetic neurons. Although the relative expression levels of each receptor were not determined in these studies, the selective enhancement of DAMGO and morphine coupling to CaV in sympathetic neurons suggest that there may be genuine differences in N40 and D40 signalling via this pathway. By contrast, trigeminal neuron CaV from ‘humanized’ N40 and D40 mice was inhibited in an essentially identical manner by DAMGO (Ramchandani et al., 2011) and fentanyl (Mahmoud et al., 2011), but morphine was less potent and had a lower efficacy in the neurons from the D40 mice (Mahmoud et al., 2011). These results are essentially opposite to those found in the acutely transfected cell lines. There is no ready explanation for these differences, although differences in receptor expression cannot be ruled out. It is likely that HEK293 cells, rat sympathetic neurons and mouse trigeminal neurons express different complements of Gα and βγ subunits, which may also contribute to the observed differences. Finally, it should be noted that the humanized N40/D40 μ-opioid receptors are hybrids, with only the first exon of the human receptor inserted into mouse, meaning that the receptors are human/mouse chimeras. The receptors had a similar affinity for DAMGO (Ramchandani et al., 2011), but their signalling properties have not been well characterized.
Deb et al. (2010) expressed N40 and D40 variants of the μ-opioid receptor in the mouse neuroblastoma cell line Neuro2A, with radioligand binding experiments indicating similar levels of receptor expression. Using measurements of PKA activity and phosphorylated ERK1/2 (pERK) levels in response to a single concentration of morphine (1 μM) applied for 5 min or 6 days, the investigators found differences in PKA and pERK levels between N40 and D40 expressing cells after 6 days only. Unfortunately, the basal PKA activity and acute agonist-stimulated ERK phosphorylation differed significantly between the cell lines, making sensible interpretations difficult. The differences in the signalling responses of the cells could be due to the expression of the different opioid receptor variants, or could have arisen due to variations in the phenotype of different Neuro2A cells at the time of clonal selection.
A6V
The A6V variant is located at the N-terminus of μ-opioid receptor (Table 1). A6V is quite common in some populations, but not others, having been reported at allelic frequencies ranging from less than 1% in Caucasian and east Asian populations (Rommelspacher et al., 2001; Tan et al., 2003) to upwards of 20% in African-American and northern Indian populations (Crowley et al., 2003; Kapur et al., 2007). Few studies have investigated the clinical effects of this polymorphism. Crystal et al. (2012) reported an association between the T/T genotype in African-American women and the risk of alcohol, cocaine, tobacco but not opioid use. Other studies have demonstrated a trend towards a higher frequency of V6 in individuals with substance abuse; however, these studies have lacked sufficient statistical power due to small sample sizes, and the confounding factor of overall genotype (Berrettini et al., 1997; Rommelspacher et al., 2001; Comptom et al., 2003; Crowley et al., 2003).
There are no studies comparing acute A6 and V6 signalling on the predominant isoform of the μ-opioid receptor. In an assay of cAMP-dependent gene transcription, no difference in potency was found for DAMGO, endomorphin 1 or leu-enkephalin in HEK293 cells expressing A6 and V6 (Table 2, Fortin et al., 2010). The A6V variant was studied on the MOR1A splice variant sequence expressed in HEK293 cells, where DAMGO but not morphine showed a higher maximum effect at V6- than A6-MOR1A in an assay of intracellular Ca release catalysed by a transiently transfected chimeric G protein. No differences in internalization of the V6-MOR1A receptor in response to DAMGO and morphine were observed (Ravindranathan et al., 2009). The significance of these findings for more naturalistic coupling of the μ-opioid receptor are unclear, but suggest that further work is warranted.
S42C, D51N, G63V and S66F
Other rare polymorphisms within the N-terminal domain of the μ-opioid receptor have been identified within the population, but no clinical or phenotypic information is available (Table 1). The S42C variant resulted in reduced receptor expression and coupling to intracellular calcium release when assayed on the MOR1A splice variant background (Ravindranathan et al., 2009, Table 2).
Several extracellular domain polymorphisms for which there is no phenotypic data were identified on the GPCR Natural Variant database (Kazius et al. 2008) and examined in a cAMP-dependent gene transcription assay (Fortin et al., 2010). Neither D51N nor G63V showed any differences to wild-type (WT) μ-opioid receptors in this assay. However, the S66F variant showed a reduction in the potency of DAMGO and endomorphin 1, but not leu-enkephalin to inhibit AC (Table 2; Fortin et al., 2010).
TM domain SNPs
L85I (TM1)
The TM helices of μ-opioid receptors are important elements of the ligand-binding pocket, and they are essential for transmitting information from the extracellular surface to the intracellular signalling domains and also participate in the formation of oligomers (Serohijos et al., 2011; Manglik et al., 2012). The L85I variant, in TM1 (Table 1), was first reported by Ravindranathan et al. (2009). Although there is no information about the phenotype of people carrying the I85 allele, it has an interesting functional profile in vitro. Both DAMGO and morphine have a moderately lower efficacy in signalling assays measuring I85 (or I83 – the rat orthologue) activity; however, morphine displays an enhanced capacity to promote internalization of the I85/I83 variant (Ravindranathan et al., 2009; Cooke et al., 2014). Co-expression of the I85 and L85 receptors results in morphine promoting internalization of both variants, suggesting that they may form functional dimers (Ravindranathan et al., 2009).
Previous studies have shown that the WT-μ-opioid receptor internalizes relatively poorly in response to morphine, and there is also evidence that high-efficacy agonists such as DAMGO and etorphine appear to induce receptor desensitization by different mechanisms to partial agonists such as morphine (Ueda et al., 2001; Borgland et al., 2003; Johnson et al., 2006; Kelly et al., 2008; Bailey et al., 2009). Intriguingly, while morphine activated-μ-opioid receptor has been shown to be a poor substrate for GPCR kinase (GRK) subtypes 2/3 phosphorylation, which is required for endocytosis (Doll et al., 2012), internalization of the I83 μ-opioid receptor was significantly attenuated with overexpression of a GRK2 dominant negative mutant, suggesting this variant is better able to recruit GRK2 (Cooke et al., 2014). Hierarchical, multi-site phosphorylation is required for efficient μ-opioid receptor endocytosis (El Kouhen et al., 2001), and while morphine induces phosphorylation of the S375 residue on the C-terminus of the μ-opioid receptor, DAMGO also efficiently stimulates phosphorylation of T370 after S375 phosphorylation, resulting in receptor internalization (Doll et al., 2011; Grecksch et al., 2011). Morphine-stimulated internalization of I83 was not due to enhanced phosphorylation of S375 compared with the WT-μ-opioid receptor, but T370 phosphorylation was not investigated (Cooke et al., 2014). The observations that the I83/85 μ-opioid receptor show apparently decreased signalling efficacy compared with enhanced receptor trafficking in response to morphine increase the likelihood that distinct receptor conformations underlie each of these processes. It will be interesting to see whether it is possible to further define the structural elements in the region of L85 that are involved in μ-opioid receptor signalling and phosphorylation, and whether it will be possible to independently manipulate these properties of the agonist/receptor complex.
Compensatory changes in cell signalling processes are associated with chronic μ-opioid receptor activation, one of the most well described of these is the up-regulation of AC activity that results in ‘superactivation’ of AC upon opioid withdrawal (Sharma et al., 1975; Avidor-Reiss et al., 1996; Whistler et al., 1999). It has also been suggested that these compensatory changes are limited by agonist-induced receptor internalization (Wang et al., 2003). Ravindranathan et al. tested the I85 variant for changes in AC superactivation. Cells expressing the L/I85 μ-opioid receptor variant were chronically treated with morphine for 14 h. Upon morphine withdrawal, cells expressing the I85 μ-opioid receptor showed a significantly lower level of cAMP compared with WT cells (2.5- and 1.5-fold cAMP levels of morphine naive cells respectively). Upon a 4 h ‘acute’ rechallenge with 10 nM morphine, cAMP levels were again significantly lower in the I85 expressing cells, indicating a reduction in AC superactivation and morphine tolerance.
S147C and N152D (TM3)
Computational modelling and X-ray crystallography studies have shown TM domains 3, 5 and 6 to be of particular importance in the formation of the ligand-binding pocket of the μ-opioid receptor (Serohijos et al., 2011; Manglik et al., 2012). Two polymorphisms in TM3 have been detected within the population, S147C and N152D (Table 1), both occurring at frequencies of <1% (Bergen et al., 1997; Uhl et al., 1999; Befort et al., 2001; Ravindranathan et al., 2009). No information on the clinical phenotype of C147 or D152 carriers is available, and limited functional studies have been published. When expressed on the MOR1A splice variant backbone C147 appeared to support an increased efficacy and potency for DAMGO and morphine to stimulate intracellular calcium release when compared with S147 (Ravindranathan et al., 2009); however, when expressed on the WT-μ-opioid receptor backbone, C147 was modestly less effective at supporting agonist-mediated inhibition of cAMP-dependent gene transcription (Fortin et al., 2010). Whether this discrepancy is because of the different receptor backgrounds or whether it hints at a reciprocal change in the capacity of μ-opioid receptors to activate different signalling pathways remains unknown. The N152D SNP appears to have reduced affinity for morphine but not opioid peptides. Unfortunately, it was not possible to measure receptor activity, apparently due to low overall expression (Befort et al., 2001).
C192F (TM4)
One SNP in TM4 of OPRM1 has been identified, C192F (Ravindranathan et al., 2009). When expressed on the MOR1A splice variant backbone, C192F showed significant decreases in DAMGO and morphine potency to mobilize calcium in HEK293 cells transfected with an engineered G protein. No phenotypic information is available.
V293I (TM6)
Shi et al. (2002) detected a V293I aa exchange in μ-opioid receptors. I293 was reported to signal in an identical manner to V293 (Fortin et al., 2010) and there is no clinical information about this phenotype.
ICL SNPs
The ICL domains of the μ-opioid receptor form major elements of the cytoplasmic interface between the receptor and intracellular effector proteins. ICL2 and 3 have been shown to be of key importance in G protein coupling of GPCRs, as well as being involved in regulatory processes such as receptor phosphorylation, uncoupling and internalization (Lefkowitz, 1998). GPCRs with the ICL2 and ICL3 domains deleted cannot couple to G proteins but can retain their ligand-binding properties, and there are a number of examples of ICL SNPs affecting selectivity of G protein coupling (Capeyrou et al., 1997; Visiers et al., 2001; Goldfeld et al., 2011; Zheng et al., 2013).
ICL2 contains the highly conserved E/DRY motif, mutations in which have been shown to reduce μ-opioid receptor agonist efficacy, and also to increase the constitutive activity of μ-opioid receptors (Li et al., 2001; Clayton et al., 2010). Mutations in ICL2 have also been shown to affect receptor uncoupling and desensitization (Celver et al., 2001; 2004). ICL3 is highly conserved among all opioid receptor types and has been shown to be involved in basal and agonist-stimulated G protein coupling, β-arrestin recruitment and contains multiple phosphorylation consensus sequences (Merkouris et al., 1996; Georgoussi et al., 1997; Wang, 1999). Mutations within the ICL3 of the μ-opioid receptor have been shown to differentially affect agonist potency and efficacy (Chaipatikul et al., 2003). In addition to their role in acute signalling and short-term regulatory processes, the intracellular domains of GPCRs may be of importance in long-term adaptations to chronic opioid exposure, and contribute to the development of opioid tolerance (Chavkin et al., 2001; Koch and Hollt, 2008; Williams et al., 2013).
R181C (ICL2)
The R181C variant was reported by Ravindranathan et al. (2009). Interestingly, C181 appears to have an unchanged affinity for DAMGO, but it fails to promote calcium mobilization or be internalized in response to agonist. Whether the receptor is unable to signal to all effectors remains to be established.
N190K (ICL2)
The rare N190K variant is located at the base of TM4, and was originally reported as an ICL2 SNP (Table 1; Fortin et al., 2010). Total K190 expression in HEK293 cells is lower than N190, but cell surface expression is almost absent. DAMGO fails to signal through K190, although it is not clear if this is because of the inaccessibility of the intracellular receptor or a change in the transduction of peptide agonist signals. Interestingly, treatment of the K190 variant with the non-peptide, cell permeable opioid receptor ligands naltrexone, naloxone, buprenorphine or β-chlornaltrexamine (β-CNA; 10 μM, 18 h) increased cell surface receptor expression, with naltrexone treatment producing levels similar to WT-μ-opioid receptor. Small, membrane-permeable ligands can ‘rescue’ misfolded or immature GPCR, including opioid receptors, by stabilizing a more native-type conformation in the endoplasmic reticulum and allowing the protein to enter the appropriate secretory pathway (Petäjä-Repo et al., 2002; Ulloa-Aguirre et al., 2004; Chen et al., 2006). Fascinatingly, naloxone and naltrexone were also apparently agonists at K190, producing significant inhibition of cAMP-stimulated reporter gene transcription after prolonged treatment (Fortin et al., 2010). This suggests that K190 is not misfolded/misconfigured to such a degree that it cannot recognize G proteins, but that it nonetheless has an aberrant native conformation.
Four rare, naturally occurring SNPs present on ICL3 have been described, R260H (Bond et al., 1998), R265H (Hoehe et al., 2000; Befort et al., 2001; Wang et al., 2001), S268P (Hoehe et al., 2000) and D274N (Wang et al., 2001; Table 1). The importance of ICL3 in the regulation and signalling of μ-opioid receptors has prompted investigation of the functional consequences of ICL3 polymorphisms, despite their rarity within the population.
R260H, R265H, S268P
The R260H and R265H variant receptors exhibited very similar ability to bind opioids and be activated by morphine or DAMGO, with minor differences in agonist-stimulated GTPγS binding potentially accounted for by small differences in receptor expression or the proportion of receptors on the cell surface. An intriguing finding was that basal GTPγS activity was significantly lower in cells expressing H260 or H265, suggesting a lower constitutive activity (Befort et al., 2001; Wang et al., 2001).
Assays of cAMP accumulation have provided inconsistent results with respect to H260 or H265 signalling. Wang et al. (2001) found no differences in morphine potency or efficacy for inhibition of forskolin-stimulated radiolabelled cAMP accumulation in cells expressing WT-μ-opioid receptors, H260 or H265 while Befort et al. (2001) also found no differences between H265 and WT in a cAMP response element (CRE) reporter gene assay (see Table 2). By contrast, Fortin et al. (2010) used a different CRE reporter assay and found a decrease in potency of DAMGO, endomorphin-1 and leu-enkephalin signalling through both H260 and H265. It is difficult to directly compare these studies as Fortin et al. (2010) did not quantify receptor expression, but the relatively high levels of receptor expression in the cells used by Wang et al. (2001) and Befort et al. (2001) could conceivably reduce the sensitivity of the assay to detect differences in agonist potency at the variant receptors.
A third ICL3 variant, S268P, results in the loss of a putative Ca2+/calmodulin (CaM)-dependent PK II (CaMK II) phosphorylation site (Koch et al., 2000) and insertion of an aa, proline, that is likely to significantly disrupt the structure of ICL3. Most studies have found that P268 or the rat orthologue S266P (Koch et al., 2000) have a significantly reduced signalling capacity, although the extent of this depends somewhat on the assay conditions used (Koch et al., 2000, Befort et al., 2001; Wang et al., 2001; Fortin et al., 2010; Table 2). The reduction in signalling does not seem to be associated with a change in ligand affinity for the receptor (Koch et al., 2000; Befort et al., 2001), but it is unclear what the relative contributions of the loss of the potential phosphorylation site or the introduction of the proline residue are to the observed in vitro phenotype.
Mutations in ICL3 of the μ-opioid receptor affect the signalling of the receptor, but changes in the signalling profile of the μ-opioid receptor resulting from ICL3 SNPs are likely to be expressed in situations other than acute μ-opioid receptor signalling because ICL domains of GPCRs interact with effectors involved in receptor regulation and adaptive processes such as receptor down-regulation (Lefkowitz, 1998). The ICL3 domain of the μ-opioid receptor has multiple consensus phosphorylation sites, as well as a putative CaM-binding domain (Wang et al., 1999; Koch et al., 2000). Sustained exposure to high concentrations of agonist produces down-regulation of receptor protein in cell lines, and the ICL3 variants R260H, R265H and S268P were down-regulated (∼80%) to a similar degree to WT receptors by 10 μM DAMGO (Befort et al., 2001). Functionally, P268 μ-opioid receptor-mediated inhibition of AC desensitized with a similar time course to P268, while desensitization of P268-mediated activation of GIRK was slower and incomplete when compared with P268 when the proteins were expressed in Xenopus oocytes (Koch et al., 2000).
In addition to phosphorylation sites, μ-opioid receptor ICL3 contains a putative CaM-binding site. It has been suggested that CaM competes with G protein coupling at ICL3, and regulates basal μ-opioid receptor signalling (Wang et al., 1999). Wang et al. (2001) investigated the interaction of CaM with μ-opioid receptor ICL3 domains by incubating CaM with short peptides derived from ICL3 sequences as well as full-length μ-opioid receptors purified from transfected HEK293 cells. The ICL3 H260 peptide showed a marginal reduction of CaM binding, but the ICL3 H265 and P268 peptides bound CaM significantly less well. A similar pattern was observed in Western blots of full-length μ-opioid receptor variants bound to CaM. The broader significance of these findings has not been firmly established.
D274N
The D274N variant has received much less attention than ICL3 variants discussed earlier. It was originally reported by Wang et al. (2001), but not investigated until the study of Fortin et al. (2010). DAMGO and leu-enkephalin showed a slight increase in potency for inhibition of cAMP accumulation at N274, while endomorphin 1 potency was significantly increased when compared with WT-μ-opioid receptors in HEK293 cells (see Table 2). No change in DAMGO efficacy was observed. These results are in direct contrast to other ICL3 variants, where receptor signalling tended to be reduced.
Limitations of extant functional SNP studies
The interpretation of studies of opioid receptor function in vitro, and the extent to which fruitful comparisons can be made between studies are subject to several important caveats. These extend beyond the everyday differences in the way that laboratories perform studies, and can limit the confidence we have in our understanding of the impact non-synonymous SNPs have on OPRM1 function. Firstly, many studies do not quantify receptor expression, either in the whole cell or on the cell surface. While it is unrealisitic to expect ‘physiological’ expression levels (whatever they may be) in all expression systems, high levels of receptor can lead to significant receptor reserve or exaggerated coupling to effectors not normally accessed by the receptor. Receptor reserve is an important issue that has apparently rarely been considered, and even modest differences in receptor expression could significantly affect the signalling profile of important partial agonists such as morphine, and spare receptors may mask subtle differences between variant signalling.
Secondly, the techniques used to measure μ-opioid receptor activation in many studies do not reflect acute, real-time, naturalistic signalling of the μ-opioid receptor. The μ-opioid receptor undergoes rapid desensitization and internalization following agonist exposures of 5–10 min (Connor et al., 2004). Thus, the reporter gene assays used for facile quantification of μ-opioid receptor function measure the summed effects of μ-opioid receptor activation, desensitization, internalization and resensitization, and this may obscure differences between variants at any of these points. Clonal selection of transformed cells during establishment of cell lines expressing variants may contribute to signalling differences observed between variants, and this is rarely controlled for with experiments on endogenous GPCR in each cell line used. These shortcomings are common to many studies of cell signalling in heterologous systems, and to an extent come with the territory, but they are especially important to consider and try and minimize given the potentially subtle nature of changes produced by SNPs.
The μ-opioid receptor is expressed in a wide variety of human cell types, and subtle changes in μ-opioid receptor signalling arising from SNPs are likely to differ between tissue and cell type. As such, it is difficult to lay out an ‘ideal’ strategy for investigating functional consequences of μ-opioid receptor SNPs. In reality, studies undertaken in a variety of heterologous expression systems are probably useful for capturing the range of signalling and regulatory differences that may be produced by μ-opioid receptor variants (e.g. Charfi et al., 2013). However, simple measures that might enable more confident assertions that differences seen might represent more than just an experimental quirk would include using similar expression systems when attempting to make direct comparisons between the effects of changes in μ-opioid receptor sequence and/or the effects of multiple ligands, controlling for receptor expression and reserve, and examining as many effectors as possible in similar conditions. Practical steps towards this include the use of cell lines with defined recombination sites to allow the construction of multiple clones on an isogenic background (e.g. FlpIn cells, Knapman et al., 2014) and the use of inducible expression systems or transient transfections to minimize the effects of prolonged expression of μ-opioid receptors on cell phenotype and perhaps gain some ability to titrate the amount of cell surface receptor (e.g. Fortin et al., 2010; Knapman et al., 2014). It is always useful to use assays that capture the kinetics of drug/receptor/second messenger activity, rather than simply endpoint assays (e.g. Johnson et al., 2006; Cawston et al., 2013; Knapman et al., 2013, 2014; Tudashki et al., 2014), and it is also important to have a system where changes in efficacy can be readily determined, whether by use of pharmacological tools or by choosing cell lines where there are a minimum of spare receptors. Defining receptor reserve using irreversible antagonists such as β-funaltrexamine or β-CAN, and then fitting data to operational models (e.g. Borgland et al., 2003; Rivero et al., 2012; Kelly, 2013) can allow for precise determination of rank orders of agonist efficacy and uncover differences in signalling across different effectors in the same cell, enabling a more complete characterization of the consequences of changes in receptor sequence. All these ideas have been extensively reviewed in the context of defining ligand bias and allostery at GPCRs, and there is no reason they should not be applied when it is the receptor that changes rather than the ligand (Kenakin and Christopoulos, 2013).
Future studies
Areas of great importance for opioid receptor function remain largely unexplored for most SNPs. In particular, the efficiency of coupling of SNPs to the range of possible μ-opioid receptor signalling pathways has barely been touched on, as have possible ligand-specific changes in this coupling. Several studies have examined the trafficking of μ-opioid receptor variants in response to a limited range of agonists (Ravindranathan et al., 2009; Cooke et al., 2014), but the effect of μ-opioid receptor SNPs on the rapid desensitization of signalling that precedes receptor internalization remains unknown. The way in which μ-opioid receptor SNPs may affect the occurrence or function of putative μ-opioid receptor dimers has received limited attention (Ravindranathan et al., 2009), even though most carriers of variant μ-opioid receptor alleles will be heterozygous for the WT receptor.
Understanding how μ-opioid receptor SNPs affect cellular signalling is important for predicting the potential clinical or phenotypic consequences of these variants in humans. However, understanding other aspects of μ-opioid receptor function such as the regulation of gene expression in response to environmental or epigenetic factors, and the function of μ-opioid receptors in the wide range of human cells that normally express it, are equally important and more difficult to achieve. Nevertheless, understanding the consequences of expressing a particular μ-opioid receptor variant should one day contribute to a more personalized approach to opioid prescription. The ability to predict the effects of specific opioid drugs in individuals, including side effects and the development of tolerance, would minimize the risk of serious adverse events associated with opioid overdose, while maximizing therapeutic benefits and ensuring individuals receive adequate pain relief. Such prediction would necessarily involve determining the genotype of multiple proteins involved in opioid ligand distribution and metabolism, as well as effectors downstream of the μ-opioid receptor, but a key element would be knowing what version of the μ-opioid receptor a patient had, and knowing which of the many opioid analgesics available had the best pharmacodynamic profile at that variant.
Acknowledgments
A. K. supported by a MQRES Scholarship. M. C. supported by a NH & MRC Project Grant No. 1011979.
Glossary
Abbreviations
- aa
amino acid
- β-CNA
β-chlornaltrexamine
- CaM
calmodulin
- CaMK II
Ca2+/calmodulin-dependent PK II
- CaV
voltage gated Ca channels
- CRE
cAMP response element
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin;
- GIRK
G protein gated, inwardly rectifying potassium channel
- GRK
GPCR kinase;
- ICL
intracellular loop
- M-6-G
morphine-6-glucuronide
- OPRM1
μ-opioid receptor 1 gene
- pERK
phosphorylated ERK1/2
- SNP
single-nucleotide polymorphism
- TM
transmembrane
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- Abrol R, Kim SK, Bray JK, Trzaskowski B, Goddard WA., 3rd Conformational ensemble view of G protein-coupled receptors and the effect of mutations and ligand binding. Methods Enzymol. 2013;520:31–48. doi: 10.1016/B978-0-12-391861-1.00002-2. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G-protein couple receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2012;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Nevo I, Levy R, Pfeuffer T, Vogel Z. Chronic opioid treatment induces adenylyl cyclase V superactivation. Involvment of Gβγ. J Biol Chem. 1996;271:21309–21315. doi: 10.1074/jbc.271.35.21309. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, et al. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befort K, Filliol D, Decaillot FM, Gaveriaux-Ruff C, Hoehe MR, Kieffer BL. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J Biol Chem. 2001;276:3130–3137. doi: 10.1074/jbc.M006352200. [DOI] [PubMed] [Google Scholar]
- Bergen AW, Kokoszka J, Peterson R, Long JC, Virkkunen M, Goldman D. μ-opioid receptor gene variants: lack of association with alcohol dependence. Mol Psychiatry. 1997;2:490–494. doi: 10.1038/sj.mp.4000331. [DOI] [PubMed] [Google Scholar]
- Berrettini WH, Hoehe MR, Ferraro TN, DeMaria PA, Gottheil E. Human mu-opioid receptor gene polymorphisms and vulnerability to substance abuse. Addict Biol. 1997;2:303–308. doi: 10.1080/13556219772598. [DOI] [PubMed] [Google Scholar]
- Beyer A, Koch T, Schroder H, Schulz S, Hollt V. Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization and resensitization of the human mu-opioid receptor. J Neurochem. 2004;89:553–560. doi: 10.1111/j.1471-4159.2004.02340.x. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters β-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- Capeyrou R, Riond J, Corbani M, Lepage JF, Bertin B, Emorine LJ. Agonist-induced signaling and trafficking of the μ-opioid receptor: role of serine and threonine residues in the third cytoplasmic loop and C-terminal domain. FEBS Lett. 1997;415:200–205. doi: 10.1016/s0014-5793(97)01124-1. [DOI] [PubMed] [Google Scholar]
- Cawston EE, Redmond WJ, Breen C, Grimsey N, Connor M, Glass M. Real-time characterisation of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonin 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the mu-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Chaipatikul V, Loh HH, Law PY. Ligand-selective activation of μ-opioid receptor: demonstrated with deletion and single amino acid mutations of third intracellular loop domain. J Pharmacol Exp Ther. 2003;305:909–918. doi: 10.1124/jpet.102.046219. [DOI] [PubMed] [Google Scholar]
- Charfi I, Nagi K, Mnie-Filali O, Thibault D, Balboni G, Schiller PW, et al. Ligand- and cell-dependent determinants of internalization and cAMP modulation by delta opioid receptor (DOR) agonists. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1461-7. doi: 10.1007/s00018-013-1461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, McLaughlin JP, Celver JP. Regulation of opioid receptor function by chronic agonist exposure: constitutive activity and desensitization. Mol Pharmacol. 2001;60:20–25. doi: 10.1124/mol.60.1.20. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chen C, Wang Y, Liu-Chen LY. Ligands regulate cell surface level of the human kappa opioid receptor by activation-induced down-regulation and pharmacological chaperone-mediated enhancement: differential effects of nonpeptide and peptide agonists. J Pharmacol Exp Ther. 2006;319:765–775. doi: 10.1124/jpet.106.107987. [DOI] [PubMed] [Google Scholar]
- Clayton CC, Bruchas MR, Lee ML, Chavkin C. Phosphorylation of the μ-opioid receptor at tyrosine 166 (Tyr3.51) in the DRY motif reduces agonist efficacy. Mol Pharmacol. 2010;77:339–347. doi: 10.1124/mol.109.060558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comptom P, Geschwind DH, Alarcon M. Association between human mu-opioid receptor gene polymorphism, pain tolerance, and opioid addiction. Am J Med Genet B Neuropsychiatr Genet. 2003;121B:76–82. doi: 10.1002/ajmg.b.20057. [DOI] [PubMed] [Google Scholar]
- Connor M, Christie MD. Opioid receptor signaling mechanisms. Clin Exp Pharmacol Physiol. 1999;26:493–499. doi: 10.1046/j.1440-1681.1999.03049.x. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. Mu-opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke AE, Oldfield S, Krasel C, Mundell SJ, Henderson G, Kelly E. Morphine-induced internalization of the L83I mutant of the rat μ-opioid receptor. Brit J Pharmacol. 2014 doi: 10.1111/bph.12709. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AD, Henderson G, McKnight AT, Paterson SJ. 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol. 2006;147(Suppl. 1):S153–S162. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B. Recent developments in the study of opioid receptors. Mol Pharmacol. 2013;83:723–728. doi: 10.1124/mol.112.083279. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Oslin DW, Patkar AA, Gottheil E, DeMaria PA, Jr, O'Brien CP, et al. A genetic association study of the mu opioid receptor and severe opioid dependence. Psychiatr Genet. 2003;13:169–173. doi: 10.1097/00041444-200309000-00006. [DOI] [PubMed] [Google Scholar]
- Crystal HA, Hamon S, Randesi M, Cook J, Anastos K, Lazar J. A C17T polymorphism in the mu opiate receptor is associated with quantitative measures of drug use in African American women. Addict Biol. 2012;17:181–191. doi: 10.1111/j.1369-1600.2010.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Aarts L, Smith TW. Incidence, reversal and prevention of opioid-induced respiratory depression. Anesthesiology. 2010;112:226–238. doi: 10.1097/ALN.0b013e3181c38c25. [DOI] [PubMed] [Google Scholar]
- Dang VC, Christie MJ. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br J Pharmacol. 2012;165:1704–1716. doi: 10.1111/j.1476-5381.2011.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb I, Chakraborty J, Gangopadhyay PK, Choudhury SR, Das S. Single-nucleotide polymorphism (A118G) in exon 1 of OPRM1 gene causes alteration in downstream signaling by mu-opioid receptor and may contribute to the genetic risk for addiction. J Neurochem. 2010;112:486–496. doi: 10.1111/j.1471-4159.2009.06472.x. [DOI] [PubMed] [Google Scholar]
- Dhalla IA, Persaud N, Juurlink DN. Facing up to the prescription opioid crisis. BMJ. 2011;343:d5142. doi: 10.1136/bmj.d5142. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Robinson JE, Maixner W. Elucidation of mu-opioid gene structure: how genetics can help predict therapeutic response to opioids. Eur J Pain Suppl. 2011;5:433–448. doi: 10.1016/j.eujps.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Konietzko J, Poll F, Koch T, Hollt V, Schulz S. Agonist-selective patterns of mu-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. 2011;164:298–307. doi: 10.1111/j.1476-5381.2011.01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Poll F, Peuker K, Loktev A, Gluck L, Schulz S. Deciphering μ-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol. 2012;167:1259–1270. doi: 10.1111/j.1476-5381.2012.02080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370 and Ser375 residues within the carboxyl tail differentially regulates μ-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- Fortin JP, Ci L, Schroeder J, Goldstein C, Montefusco MC, Peter I, et al. The μ-opioid receptor variant N190K is unresponsive to peptide agonists yet can be rescued by small-molecule drugs. Mol Pharmacol. 2010;78:837–845. doi: 10.1124/mol.110.064188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgoussi Z, Merkouris M, Mullaney I, Megaritis G, Carr C, Zioudrou C, et al. Selective interactions of mu-opioid receptors with Pertussis toxin-sensitive G proteins: involvement of the third intracellular loop and the c-terminal tail in coupling. Biochim Biophys Acta. 1997;1359:263–274. doi: 10.1016/s0167-4889(97)00097-9. [DOI] [PubMed] [Google Scholar]
- Goldfeld DA, Zhu K, Beuming T, Friesner RA. Successful prediction of the intra- and extracellular loops of four G-protein coupled receptors. Proc Natl Acad Sci USA. 2011;109:9665–9671. doi: 10.1073/pnas.1016951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecksch G, Just S, Pierstorff C, Imhof AK, Gluck L, Doll C, et al. Analgesic tolerance to high-efficacy agonists but not to morphine is diminished in phosphorylation-deficient S375A mu-opioid receptor knock-in mice. J Neurosci. 2011;31:13890–13896. doi: 10.1523/JNEUROSCI.2304-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehe MR, Kopke K, Wendel B, Rohde K, Flachmeier C, Kidd KK, et al. Sequence variability and candidate gene analysis in complex disease: association of mu opioid receptor gene variation with substance dependence. Hum Mol Genet. 2000;9:2895–2908. doi: 10.1093/hmg/9.19.2895. [DOI] [PubMed] [Google Scholar]
- Huang P, Chen C, Mague SD, Blendy JA, Liu-Chen LY. A common single nucleotide polymorphism A118G of the μ opioid receptor alters its N-glycosylation and protein stability. Biochem J. 2012;441:379–386. doi: 10.1042/BJ20111050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Ide S, Han W, Hayashida M, Uhl GR, Sora I. How individual sensitivity to opiates can be predicted by gene analyses. Trend Pharmacol Sci. 2005;26:311–317. doi: 10.1016/j.tips.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Jordan B, Devi LA. Molecular mechanisms of opioid receptor signal transduction. Br J Anaesth. 1998;81:12–19. doi: 10.1093/bja/81.1.12. [DOI] [PubMed] [Google Scholar]
- Kapur S, Sharad S, Singh RA, Gupta AK. A118G polymorphism in mu opioid receptor gene (oprm1): association with opiate addiction in subjects of Indian origin. J Integr Neurosci. 2007;6:511–522. doi: 10.1142/s0219635207001635. [DOI] [PubMed] [Google Scholar]
- Kazius J, Wurdinger K, van Iterson M, Kok J, Back T, Ijzerman AP. GPCR NaVa database: natural variants in human G protein-coupled receptors. Hum Mut. 2008;29:39–44. doi: 10.1002/humu.20638. [DOI] [PubMed] [Google Scholar]
- Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153(Suppl. 1):S379–S388. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E. Efficacy and ligand bias at the μ-opioid receptor. Br J Pharmacol. 2013;169:1430–1446. doi: 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapman A, Santiago M, Du Y-P, Bennallack P, Christie MJ, Connor M. A continuous, fluorescence based assay of μ-opioid receptor activation in AtT-20 cells. J Biomol Screen. 2013;18:269–276. doi: 10.1177/1087057112461376. [DOI] [PubMed] [Google Scholar]
- Knapman A, Abogadie F, McIntrye P, Connor M. A real time, fluorescence-based assay for measuring μ-opioid receptor modulation of adenylyl cyclase activity in Chinese hamster ovary cells. J Biomol Screen. 2014;19:223–231. doi: 10.1177/1087057113501391. [DOI] [PubMed] [Google Scholar]
- Koch T, Hollt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol Ther. 2008;117:199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Koch T, Kroslak T, Averbeck M, Mayer P, Schroder H, Raulf E, et al. Allelic variation S268P of the human mu-opioid receptor affects both desensitization and G protein coupling. Mol Pharmacol. 2000;58:328–334. doi: 10.1124/mol.58.2.328. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Bart G, Lilly C, LaForge KS, Nielsen DA. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol Rev. 2005;57:1–26. doi: 10.1124/pr.57.1.1. [DOI] [PubMed] [Google Scholar]
- Kroslak T, LaForge KS, Gianotti RJ, Ho A, Nielsen DA, Kreek MJ. The single nucleotide polymorphism A118G alters functional properties of the human mu opioid receptor. J Neurochem. 2007;103:77–87. doi: 10.1111/j.1471-4159.2007.04738.x. [DOI] [PubMed] [Google Scholar]
- LaForge KS, Yuferov V, Kreek MJ. Opioid receptor and peptide gene polymorphisms: potential implications for addictions. Eur J Pharmacol. 2000;410:249–268. doi: 10.1016/s0014-2999(00)00819-0. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein couple receptors III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- Li J, Huang P, Chen C, de Riel JK, Weinstein H, Liu-Chen LY. Constitutive activation of the mu-opioid receptor by mutation of D3.49(164), but not D3.32(147): D3.49(164) is critical for stabilization of the inactive form of the receptor and for its expression. Biochemistry. 2001;40:12039–12050. doi: 10.1021/bi0100945. [DOI] [PubMed] [Google Scholar]
- Lopez Soto EJ, Raingo J. A118G mu opioid receptor polymorphism increases inhibitory effects on CaV2.2 channels. Neurosci Lett. 2012;523:190–194. doi: 10.1016/j.neulet.2012.06.074. [DOI] [PubMed] [Google Scholar]
- Lotsch J, Geisslinger G. Are μ-opioid receptor polymorphisms important for clinical opioid therapy? Trends Mol Med. 2005;11:82–89. doi: 10.1016/j.molmed.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Mague SD, Blendy JA. OPRM1 SNP (A118G): involvement in disease development, treatment response, and animal models. Drug Alcohol Depend. 2010;108:172–182. doi: 10.1016/j.drugalcdep.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Isiegas C, Huang P, Liu-Chen LY, Lerman C, Blendy JA. Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proc Natl Acad Sci USA. 2009;106:10847–10852. doi: 10.1073/pnas.0901800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud S, Thorsell A, Sommer WH, Heilig M, Holgate JK, Bartlett SE, et al. Pharmacological consequence of the A118G μ opioid receptor polymorphism on morphine- and fentanyl-mediated modulation of Ca2+ channels in humanized mouse sensory neurons. Anesthesiology. 2011;115:1054–1062. doi: 10.1097/ALN.0b013e318231fc11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margas W, Zubkoff I, Schuler HG, Janicki PK, Ruiz-Velasco V. Modulation of Ca2+ channels by heterologously expressed wild-type and mutant human μ opioid receptors (hMORs) containing the the A118G single-nucleotide polymorphism. J Neurophysiol. 2007;97:1058–1067. doi: 10.1152/jn.01007.2006. [DOI] [PubMed] [Google Scholar]
- Massotte D, Brillet K, Kieffer BL, Milligan G. Agonists activate Gi1α or Gi2α fused to the human mu opioid receptor differently. J Neurochem. 2002;81:1372–1382. doi: 10.1046/j.1471-4159.2002.00946.x. [DOI] [PubMed] [Google Scholar]
- Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Mazarakou G, Georgoussi Z. STAT5A interacts with and is phosphorylated upon activation of the mu-opioid receptor. J Neurochem. 2005;93:918–931. doi: 10.1111/j.1471-4159.2005.03069.x. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Stolar M, Stevens DE, Goulet J, Preisig MA, Fenton B, et al. Familial transmission of substance use disorders. Arch Gen Psychiatry. 1998;55:973–979. doi: 10.1001/archpsyc.55.11.973. [DOI] [PubMed] [Google Scholar]
- Merkouris M, Dragatsis I, Megaritis G, Konidakis G, Zioudrou C, Milligan G, et al. Identification of the critical domains of the delta-opioid receptor involved in G protein coupling using site-specific synthetic peptides. Mol Pharmacol. 1996;50:985–993. [PubMed] [Google Scholar]
- Mizoguchi H, Watanabe C, Sakurada T, Sakurada S. New vistas in opioid control of pain. Curr Opin Pharmacol. 2012;12:87–91. doi: 10.1016/j.coph.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomized trials of oral opioids. Arthritis Res Ther. 2005;7:R1046–R1051. doi: 10.1186/ar1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mura E, Govoni S, Racchi M, Carossa V, Ranzani GN, Allegri M, et al. Consequences of the 118A > G polymorphism in the OPRM1 gene: translation from bench to bedside? J Pain Res. 2013;6:331–353. doi: 10.2147/JPR.S42040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M, Treadwell JR, Tregear SJ, Coates VH, Wiffen PJ, Akafomo C, et al. Long-term opioid management for chronic noncancer pain. Cochrane Database Syst Rev. 2010;(1) doi: 10.1002/14651858.CD006605.pub2. CD006605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel BG, Kettner M, Scholich K, Renne C, Roskam B, Geisslinger G, et al. A common human μ-opioid receptor genetic variant diminishes the receptor signaling efficacy in brain regions processing the sensory information of pain. J Biol Chem. 2009;284:6530–6535. doi: 10.1074/jbc.M807030200. [DOI] [PubMed] [Google Scholar]
- Oertel BG, Doehring A, Roskam B, Kettner M, Hackmann N, Ferreiros N, et al. Genetic-epigenetic interaction modulates μ-opioid receptor regulation. Hum Mol Genet. 2012;21:4751–4760. doi: 10.1093/hmg/dds314. [DOI] [PubMed] [Google Scholar]
- Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petäjä-Repo UE, Hogue M, Bhalla S, Laperrière A, Morello JP, Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J. 2002;21:1628–1637. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineyro G, Archer-Lahlou E. Ligand-specific receptor states: implications for opiate receptor signaling and regulation. Cell Signal. 2007;19:8–19. doi: 10.1016/j.cellsig.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry. 2011;16:809–817. doi: 10.1038/mp.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranathan A, Joslyn G, Robertson M, Schuckit MA, Whistler JL, White RL. Functional characterization of human variants of the mu-opioid receptor gene. Proc Natl Acad Sci USA. 2009;106:10811–10816. doi: 10.1073/pnas.0904509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell S, McArdle CA, et al. Endomorphin 2: a biased agonist at the μ-opioid receptor. Mol Pharmacol. 2012;82:178–188. doi: 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelspacher H, Smolka M, Schmidt LG, Samochowiec J, Hoehe MR. Genetic analysis of the mu-opioid receptor in alcohol-dependent individuals. Alcohol. 2001;24:129–135. doi: 10.1016/s0741-8329(01)00139-2. [DOI] [PubMed] [Google Scholar]
- Saidak Z, Blake-Palmer K, Hay DL, Northup JK, Glass M. Differential activation of G-proteins by μ-opioid receptor agonists. Br J Pharmacol. 2006;147:671–680. doi: 10.1038/sj.bjp.0706661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serohijos AWR, Yin S, Ding F, Gauthier J, Gibson DG, Maixner W, et al. Structural bias for mu-opioid receptor binding and activation. Structure. 2011;19:1683–1690. doi: 10.1016/j.str.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Nirenberg M, Klee WA. Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci USA. 1975;72:590–594. doi: 10.1073/pnas.72.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Hui L, Xu Y, Wang F, Huang W, Hu G. Sequence variations in the mu-opioid receptor gene (OPRM1) associated with human addiction to heroin. Hum Mutat. 2002;19:459–460. doi: 10.1002/humu.9026. [DOI] [PubMed] [Google Scholar]
- Singh VK, Bajpai K, Biswas S, Haq W, Khan MY, Mathur KB. Molecular biology of opioid receptors. Neuroimmunomodulation. 1997;4:285–297. doi: 10.1159/000097349. [DOI] [PubMed] [Google Scholar]
- Skorpen F, Laugsand EA. Variable response to opioid treatment: any genetic predictors in sight? Palliat Med. 2008;22:310–327. doi: 10.1177/0269216308089302. [DOI] [PubMed] [Google Scholar]
- Tan EC, Tan CH, Karupathivan U, Yap EP. Mu opioid receptor gene polymorphisms and heroin dependence in Asian populations. Neuroreport. 2003;14:569–572. doi: 10.1097/00001756-200303240-00008. [DOI] [PubMed] [Google Scholar]
- Thompson MD, Percy ME, McIntyre WB, Cole DE. G protein-coupled receptors disrupted in human genetic disease. Methods Mol Biol. 2008;448:109–137. doi: 10.1007/978-1-59745-205-2_7. [DOI] [PubMed] [Google Scholar]
- Tudashki HB, Robertson DN, Schiller PW, Pineyro G. Endocytic profiles of δ-opioid receptor ligands determine the duration of rapid but not sustained cAMP responses. Mol Pharmacol. 2014;85:148–161. doi: 10.1124/mol.113.089003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Inoue M, Matsumoto T. Protein kinase C-mediated inhibition of mu-opioid receptor internalization and its involvement in the development of acute tolerance to peripheral mu-agonist analgesia. J Neurosci. 2001;21:2967–2973. doi: 10.1523/JNEUROSCI.21-09-02967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl GR, Sora I, Wang Z. The mu opiate receptor as a candidate gene for pain: polymorphisms, variations in expression, nociception, and opiate responses. Proc Natl Acad Sci USA. 1999;96:7752–7755. doi: 10.1073/pnas.96.14.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- Visiers I, Hassan SA, Weinstein H. Differences in conformational properties of the second intracellular loop (IL2) in 5HT(2C) receptors modified by RNA editing can account for G protein coupling efficiency. Protein Eng. 2001;14:409–414. doi: 10.1093/protein/14.6.409. [DOI] [PubMed] [Google Scholar]
- Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain. 2009;146:270–275. doi: 10.1016/j.pain.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Wang D, Sadee W, Quillan JM. Calmodulin binding to G protein coupling domain of opioid receptors. J Biol Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- Wang D, Quillan JM, Winans K, Lucas JL, Sadee W. Single nucleotide polymorphisms in the human μ opioid receptor gene alter basal G protein coupling and calmodulin binding. J Biol Chem. 2001;276:34624–34630. doi: 10.1074/jbc.M104083200. [DOI] [PubMed] [Google Scholar]
- Wang HL. A conserved arginine in the distal third intracellular loop of the mu-opioid receptor is required for G protein activation. J Neurochem. 1999;72:1307–1314. doi: 10.1046/j.1471-4159.1999.0721307.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li JG, Huang P, Xu W, Liu-Chen LY. Differential effects of agonists on adenylyl cyclase superactivation mediated by the kappa opioid receptors: adenylyl cylcase superactivation is independent of agonist-induced phosphorylation, desensitization, internalization, and down-regulation. J Pharmacol Exp Ther. 2003;307:1127–1134. doi: 10.1124/jpet.103.055814. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Huang P, Ung A, Blendy JA, Liu-Chen LY. Reduced expression of the μ-opioid receptor in some, but not all, brain regions in mice with OPRM1 A112G. Neuroscience. 2012;205:178–184. doi: 10.1016/j.neuroscience.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, et al. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Steinberg SF. S49G and R389G polymorphisms of the β-adrenergic receptor influence signaling via the cAMP-PKA and ERK pathways. Physiol Genomics. 2013;45:1186–1192. doi: 10.1152/physiolgenomics.00087.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Loh HH, Law PY. A novel noncanonical signaling pathway for the μ-opioid receptor. Mol Pharmacol. 2013;84:844–853. doi: 10.1124/mol.113.088278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–32624. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- Zheng C, Chen L, Chen X, He X, Yang J, Shi Y, et al. The second intracellular loop of the human cannabinoid CB2 receptor governs G protein coupling in coordination with the carboxyl terminal domain. PLoS ONE. 2013;8:e63262. doi: 10.1371/journal.pone.0063262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Loh HH, Law PY. Agonist-selective signaling of G protein-coupled receptor: mechanisms and implications. IUBMB Life. 2010;62:112–119. doi: 10.1002/iub.293. [DOI] [PMC free article] [PubMed] [Google Scholar]