

Abstract
Opioids activate GPCRs to produce powerful analgesic actions but at the same time induce side effects and generate tolerance, which restrict their clinical use. Reducing this undesired response profile has remained a major goal of opioid research and the notion of ‘biased agonism’ is raising increasing interest as a means of separating therapeutic responses from unwanted side effects. However, to fully exploit this opportunity, it is necessary to confidently identify biased signals and evaluate which type of bias may support analgesia and which may lead to undesired effects. The development of new computational tools has made it possible to quantify ligand-dependent signalling and discriminate this component from confounders that may also yield biased responses. Here, we analyse different approaches to identify and quantify ligand-dependent bias and review different types of confounders. Focus is on δ opioid receptor ligands, which are currently viewed as promising agents for chronic pain management.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Keywords: δ opioid receptor, bias, regulation, operational model, functional selectivity
Introduction
Opioids are the most effective analgesics known but their clinical use is limited by a compromise between maintaining analgesic efficacy on the one hand and controlling side effects and development of tolerance on the other (Dworkin, 2009). Not surprisingly then, improving the side effects profile and reducing the potential for analgesic tolerance have remained major goals in opioid receptor research. Within this context the notion of ‘biased agonism’ has raised considerable interest as a means of separating desired actions from undesired effects of opioid analgesics. This new pharmacological concept refers to the ability of certain receptor ligands to stabilize the receptor into conformations that distinctively engage specific signalling partners, thus directing pharmacological stimuli towards desired responses. As such, biased ligands may constitute a powerful means of separating analgesic actions from undesired effects of opioid analgesics. However, to fully exploit this opportunity, we must be able to confidently identify ligands of interest and evaluate which of their responses contribute to analgesic efficacy and which support undesired actions. The development of new quantification tools (Rajagopal et al., 2011; Kenakin et al., 2012; Rajagopal, 2013) has not only made this identification possible, but should allow us to verify novel hypotheses with respect to the type of signals responsible for desired and undesired effects of opioids. Here, we review how to identify and quantify ligand-dependent bias and also examine the extent to which an imbalance in signalling versus internalization responses may be a desirable property as a predictor of ligand potential for generating tolerance. Focus will be on δ opioid receptor ligands (for nomenclature see Alexander et al., 2013), which are currently viewed as promising agents for chronic pain management (Gaveriaux-Ruff and Kieffer, 2011a; Gaveriaux-Ruff et al., 2011b)
Biased responses versus biased agonism
‘Biased agonism’ is a term that describes a specific type of signalling event in which different ligands acting at the same receptor may distinctively engage responses by different signalling partners (Urban et al., 2007). Initial reports of this type of signalling behaviour characteristically revealed a reversal in the rank order of ligand potency and/or maximal responses across different functional assays (Meller et al., 1992; Spengler et al., 1993; Berg et al., 1998). An example of this type of behaviour for δ opioid receptor agonists is given in Figure 1, where maximal responses by morphine and Tyr-Ticpsi [CH2-NH]Cha-Phe-OH (TICP) do not maintain the same rank order in cAMP (Figure 1A) and ERK (Figure 1B) cascades (Audet et al., 2005). These observations are in sharp contrast with what is observed when comparing δ opioid receptor ligands with respect to their relative intrinsic activities for inhibiting cAMP production and inducing conformational changes that lead to Gαi1β1γ2 activation (Figure 2) (Audet et al., 2008). Indeed, maximal cAMP inhibition and changes in BRET at the interface of Gαi1 and Gβ1γ2 subunits (Figure 2B) not only maintained the same rank order but were also correlated (Figure 2C).
Figure 1.
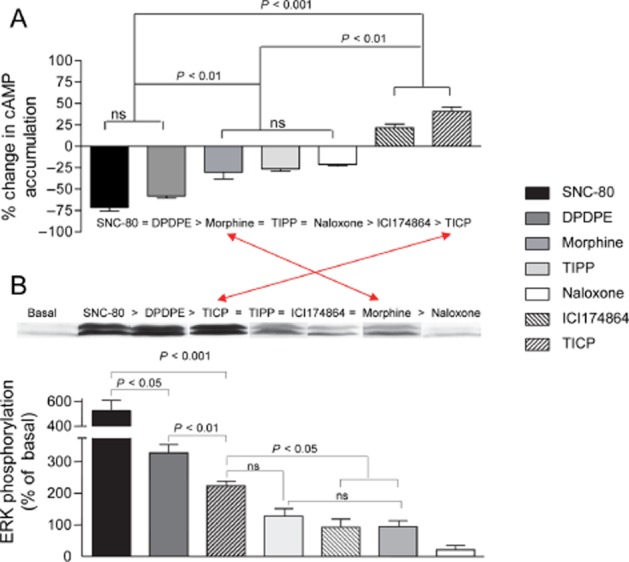
Maximal responses elicited by δ opioid receptor ligands do not maintain the same rank order in cyclase and ERK cascades. HEK293 cells expressing δ opioid receptor-Flag were treated with maximal effective concentrations (10 μM) of indicated ligands and (A) cAMP accumulation or (B) ERK activation were assessed. Drug effects are expressed as % change with respect to signals obtained in the absence of ligand and correspond to mean ± SEM of at least seven experiments carried out in triplicates. Rank orders are given for each graph; arrows indicate reversal in rank order in Emax values for morphine and TICP. Statistical differences that appear in the figure were established using one-way anova followed by Tukey's post hoc test. Immunoblots above the histogram bars correspond to representative examples of results obtained for each of the indicated drugs (Modified from Audet et al., 2005).
Figure 2.
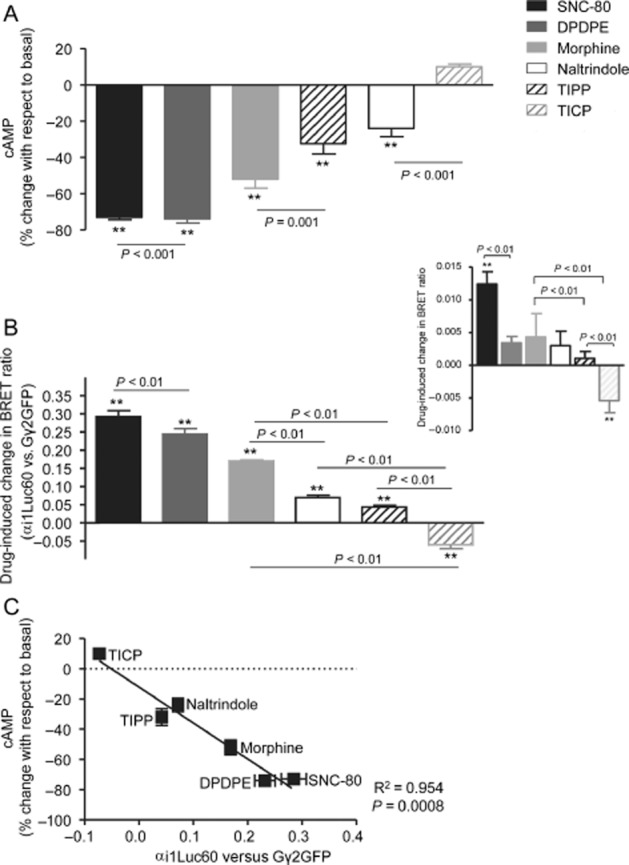
Ligand-induced changes in cAMP production and conformational rearrangements undergone at the Gαi1/Gβ1γ2 interface are proportional. (A) HEK293 cells expressing δ opioid receptor-Flag were treated with maximal effective concentrations (10 μM) of indicated ligands and cAMP accumulation assessed in radiometric assays. Drug effects are expressed as % change with respect to signals obtained in the absence of ligand and correspond to mean ± SEM of seven independent experiments. (B) HEK 293 cells were transfected with recombinant plasmids for δ opioid receptors, a BRET pair constituted by the Gαi1 subunit tagged with the donor at position 60 (Gαi1-Luc60), the Gγ2 subunit tagged with the acceptor at the N-terminus (GFP-Gγ2) together with untagged complementary Gβ1 subunits. Results are expressed as the difference between measures obtained in presence and absence of ligand and correspond to mean ± SEM of six experiments carried out in duplicates. Statistical comparisons were done by one-way anova using Dunnett's correction to compare drug effects with basal conditions. Fisher's ‘least significance difference’ adjustment was used in order to assess differences among drugs. Inset: shows conformational changes induced by different ligands at the interface between δ opioid receptor-GFP and Gαi1-Luc60. (C) Correlational analysis of responses shown in (A) and (B). (Modified from Audet et al., 2008).
According to classical receptor theory, ligands that produce different maximal responses impart pharmacological stimuli of different magnitude upon the receptor (Stephenson, 1993). Stimuli of different magnitude result in the accumulation of different amounts of a single active state of the receptor such that the more efficacious the ligand the greater the accumulation of this single active conformation, which is considered responsible for all responses controlled by the receptor. An essential prediction of this model is that ligands of different efficacy should conserve their rank order of maximal effect (Emax; and potency) at all responses assessed (Kenakin, 2002a). Hence, the direct correlation between the amplitude of conformational rearrangements elicited by Gαi1β1γ2 activation and the magnitude of cAMP inhibition is consistent with this notion, and if we were to evaluate bias from these observations, we would conclude that there is none for the responses in question. By contrast, different amounts of a single active state cannot account for one drug being more efficacious than the other in one type of response but less efficacious in another readout. Consequently, the idea that GPCRs could adopt multiple, agonist-specific receptor conformations was introduced in order to explain changes in rank order of Emax values across different responses (Kenakin, 1994; Leff, 1996; Urban et al., 2007; Onaran and Costa, 2012). In the case of δ opioid receptors, the existence of agonist-specific receptor states has been proposed from functional assays and corroborated by different biophysical methods such as BRET (Audet et al., 2008; 2012,) or plasmon wave resonance (Alves et al., 2003; 2004,).
A common observation attributed to the existence of ligand-specific conformations is the non-linear relationship observed between relative Emax values of opioid receptor ligands in signalling responses and in responses related to receptor regulation (Alvarez et al., 2002; Pradhan et al., 2010; Raehal et al., 2011; Charfi et al., 2013). For example, disproportion between Emax values for maximal G-protein activation and maximal β-arrestin recruitment by δ and μ opioid receptor ligands was shown to conform to a hyperbolic function (Molinari et al., 2010). A similar type of graph describes the relationship between maximal cAMP inhibition and internalization by δ opioid receptor ligands (Figure 3) indicating a clear imbalance between partial agonist ability to induce maximal cAMP inhibition and δ opioid receptor sequestration (Charfi et al., 2013). The question is whether such a deflection from a proportional correlation corresponds to ligand bias. As we saw from previous examples, a change in rank order of Emax values constitutes a fair indication of ligand-specific signalling. However, if we look at maximal effects in Figure 3, there is no significant change in the rank order of maximal cAMP and internalization responses for any of the ligands studied. Instead, we observe a uniform mitigation of internalization as compared with cyclase inhibition by partial agonists. Bias that similarly affects a specific response by different ligands has been called system bias, and should be distinguished from ligand-specific signals (Kenakin et al., 2012; Onaran and Costa, 2012; Kenakin and Christopoulos, 2013b). For example, Molinari et al. (2010) propose that the need for β-arrestin to diffuse from the cytosol to the membrane could be a system factor that reduces coupling efficiency between β-arrestin recruitment and receptor activation. The situation is quite different for G-protein activation where coexistence of heterotrimeric subunits and receptors at the membrane makes them immediately available to translate receptor occupancy into response (Gales et al., 2005; Rebois et al., 2006; Molinari et al., 2010; Onaran and Costa, 2012). Similar considerations apply to observational bias, which refers to how different assay conditions may influence the way ligand actions are perceived. For example, observational bias may explain why in some assays maximal responses by full and partial agonists are similar to one another while they are well discriminated in others. Figure 4 shows an example of this phenomenon when cAMP inhibition by the full agonist [D-Pen(2), D-Pen(5)]-enkephalin (DPDPE) and the partial agonist morphine were monitored in a radiometric (Figure 4A) and a BRET-based assay (Figure 4B). It is clear from the figure that in the radiometric assay DPDPE does not produce a measurable change in cAMP levels until the drug reaches a concentration of 10 nM while cAMP inhibition is already evident at 0.1 nM in the BRET-based assay. Consistent with these observations, responses observed at the 10 μM concentration were similar for both drugs in BRET assays but morphine's response was smaller than that of DPDPE in the radiometric one. In the latter assay, cAMP levels were measured 20 min after agonist exposure (Charfi et al., 2013) while BRET readings were completed only 6 min after drugs were introduced into the medium (Tudashki et al., 2014). This implies that the process of desensitization has more weight in radiometric than BRET assays. Thus, in the more desensitized system, where receptor coupling to downstream effectors has been reduced the most, maximal tested concentrations produced a smaller effect for morphine than for DPDPE. In the BRET assay, where desensitization has less time to develop, the 10 μM response is similar for both drugs. Assay conditions also influence the relative potency of both drugs. In the BRET assay, EC50 for DPDPE (−9.8 ± 0.2 M) was 3.6 orders of magnitude smaller than that of morphine (−6.2 ± 0.2 M), but in the radiometric assay, EC50 values for DPDPE were right-shifted (−8.22 ± 0.11 M) and the difference with morphine (−6.04 ± 0.14 M) was of 2.2 orders of magnitude. These differences may be interpreted as the result of two factors: (i) desensitization reduced DPDPE's potency without modification of its maximal response in the radiometric assay and (ii) even if in the BRET assay DPDPE and morphine produce similar maximal responses, DPDPE's efficacy translated into a much more potent response.
Figure 3.
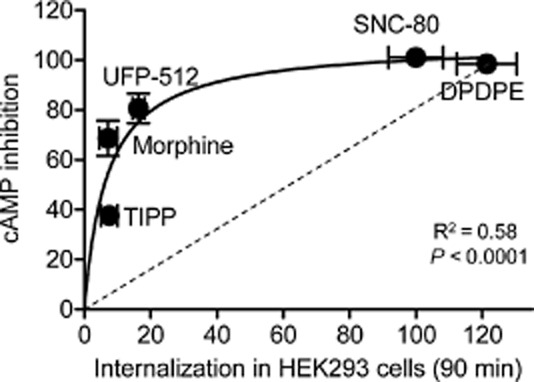
The relationship between maximal cAMP inhibition and internalization elicited by δ opioid receptor ligands is not proportional. HEK293 cells expressing δ opioid receptor-Flag were treated with maximal effective concentrations (10 μM) of the indicated ligands, and cAMP inhibition (n = 6–9) or receptor internalization (n = 9–12) were assessed using a radiometric or an ELISA-based assay. Data were fit to a single site hyperbola and goodness of fit is indicated in the figure. (Modified with permission from Charfi et al., 2013).
Figure 4.
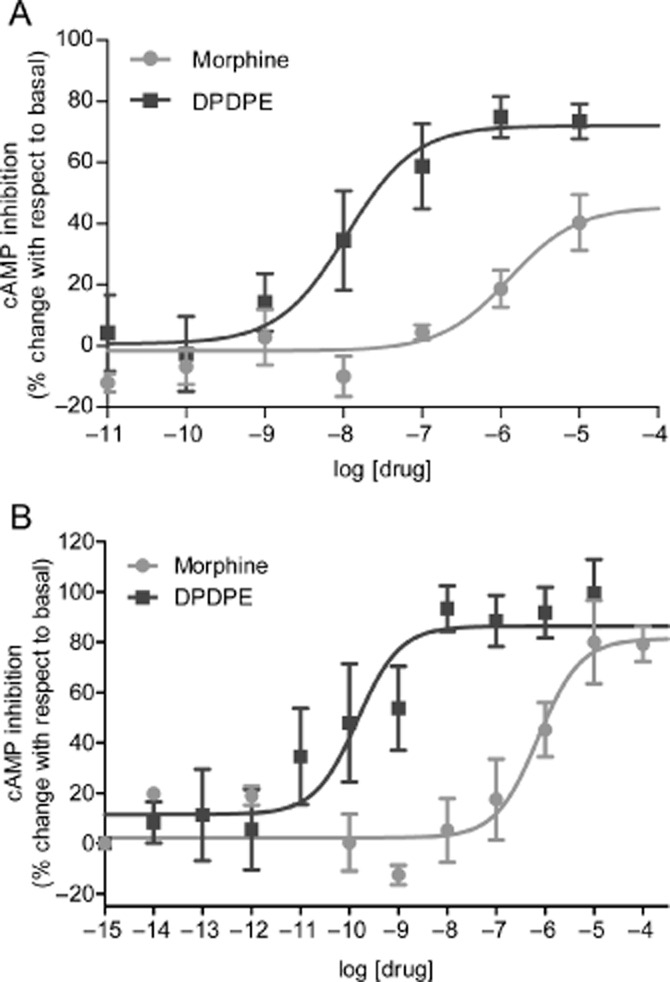
Concentration-response curves showing how observational bias may influence relative Emax for cAMP inhibition by δ opioid receptor agonists. (A) cAMP levels were monitored in a radiometric assays as in the previous figure and inhibition of cAMP production was expressed as % change with respect to cells that were not exposed to ligand. Values correspond to mean ± SEM of six to nine independent experiments carried out in triplicate. Statistical analyses using two-way anova showed an effect of drug (P < 0.0001), and effect of concentration (P < 0.0001) and an interaction (P < 0.0001) (Modified with permission from Charfi et al., 2013). (B) HEK293 cells were transfected with δ opioid receptors and a BRET-based cAMP biosensor that undergoes conformational changes upon cAMP binding. Results are expressed as % of maximal BRET change produced by DPDPE and correspond to mean ± SEM of four to eight independent experiments. (Modified with permission from Bagheri Tudashki et al., 2013). Statistical analyses using two-way anova showed an effect of drug (P < 0.0001), an effect of concentration (P < 0.0001) and an interaction (P < 0.0001).
Taken together, the examples analysed in this section indicate that: (i) response bias and ligand bias are not the same, (ii) although ligand-specific signalling may favour a specific signalling pathway, system and observational bias may also contribute to the overall imbalance in maximal responses, (iii) because of these influences comparison of maximal effects does not allow us to identify ligand bias unless there is a reversal in the rank order of Emax values in the pathways of interest and (iv) isolating the ligand-determined component within biased responses is essential if this pharmacological property is to be exploited in rational design of therapeutic agents (Kenakin, 2007; Pineyro and Archer-Lahlou, 2007; Kenakin and Miller, 2010). As discussed in the following section, shortcomings associated with maximal responses may be controlled by using a whole range of concentrations to assess bias.
Identifying and quantifying biased agonism from experimental data
As a single active state of the receptor cannot account for a reversal in rank order of EC50/Emax values for different responses, whenever this experimental observation is verified it may be considered evidence that the signals in question result from ligand-specific receptor states (Kenakin, 1995; 2002b,; Onaran and Costa, 2012). However, despite being quite specific, this way of identifying biased agonists may not be sufficiently sensitive. For example, if bias is given by a reversal in rank order of potency this could be missed when only Emax changes are monitored. Ideally, a method to identify biased ligands should be one that simultaneously evaluates positional parameters and maximal responses. In addition, the method should allow meaningful quantification of ligand-dependent bias independent of system and assay confounders.
Kenakin and Christopoulos (2013a) have used the operational model by Black and Leff (1983) to develop an analytical tool that fulfils such requirements (Kenakin, 2010; Kenakin et al., 2012). In the operational model, fractional response (E/Emax) at different agonist concentrations ([A]) may be calculated from the equation:
where E is drug effect, Emax is the maximal response allowed by the system and n describes the efficiency of the system to transduce receptor occupation into response. By fitting dose-response data to this equation, Kenakin and Christopoulos define two ligand-related parameters: (i) efficacy (τ) of the agonist to couple receptor occupancy to a specific response and (ii) ‘functional affinity’ (KA) of the ligand (Kenakin et al., 2012; Kenakin and Christopoulos, 2013b) (Figure 5A and B). In this model, KA is derived from functional and not binding data and it is defined as a parameter representing the tendency of the ligand to interact with the receptor state(s) mediating the response of interest (Kenakin et al., 2012; Kenakin and Christopoulos, 2013b). Intuitively, its meaning may be considered in terms of the allosteric properties of GPCRs; that is if biased ligands stabilize the receptor into different conformations that preferentially interact with distinct signalling partners, allosteric properties of the receptor imply that the reciprocal is also true and therefore immediate downstream effectors condition agonist affinity for the receptor (De Lean et al., 1980; Lee et al., 1986; Costa and Herz, 1989; Kenakin and Miller, 2010; Kenakin, 2012). In other words, the affinity for efficacious ligands is determined at least in part by the signalling partners with which the receptor associates. Thus, taking both parameters as indicators of drug response, bias may result from ligands displaying distinct relative efficacies (τ) at different pathways, as well as from their differential affinity for the receptor state(s) mediating the response of interest (KA). Whatever the combination, Kenakin and Christopoulos (2013b) propose that all possibilities are contemplated by calculating a consolidated transduction coefficient (log τ/KA), which describes the efficiency with which a drug evokes a particular effect. This single numerical value can be used to compare ligands across different readouts, to determine whether they display bias and to quantify its magnitude (Kenakin et al., 2012; Kenakin and Christopoulos, 2013b) (Figure 5C). However, it is important to note that together with an estimation of ligand efficacy, τ values also incorporate system-dependent variables (Black and Leff, 1983). As the latter may vary across different responses, these variations need to be taken into consideration when estimating the magnitude of bias using transduction coefficients. To do so, log(τ/KA) ratios are normalized to a common standard. This can be done by expressing test drug efficiency ratios in relation to the efficiency of the chosen standard, yielding normalized transduction coefficients or Δlog(τ/KA) values (Figure 5D). Δlog(τ/KA) is calculated by subtracting the transduction coefficient of the standard (log(τ/KA)STAN) from that of the test drug (log(τ/KA)TEST). Normalized transduction coefficients can be used to statistically compare ligand signalling efficiencies in different pathways, and to determine how much more (or less) efficient any ligand may be at inducing response in one readout with respect to another. This comparison is established by subtracting normalized transduction coefficients for each pathway (e.g. Δlog(τ/KA)INTERN–Δlog(τ/KA)CYCLASE) to establish a bias factor (ΔΔlog(τ/KA)) (Figure 5E). (Gregory et al., 2012; Charfi et al., 2013; Deng et al., 2013). Because bias factors result from the comparison of transduction coefficients that were normalized to a common standard, bias is always expressed with respect to this standard, and varies accordingly. This difference is evident in Figure 6 where internalization versus cyclase signalling bias of δ opioid receptor agonists was obtained using different standards. This difference is evident in Figure 6 where internalization versus cyclase signalling bias of δ opioid receptor agonists was obtained using different standards. Given that opioid analgesics compete with endogenous opioids to produce their effects, the use of enkephalins as standards would allow us to directly compare drugs of interest to the neuromodulator(s) they replace.
Figure 5.
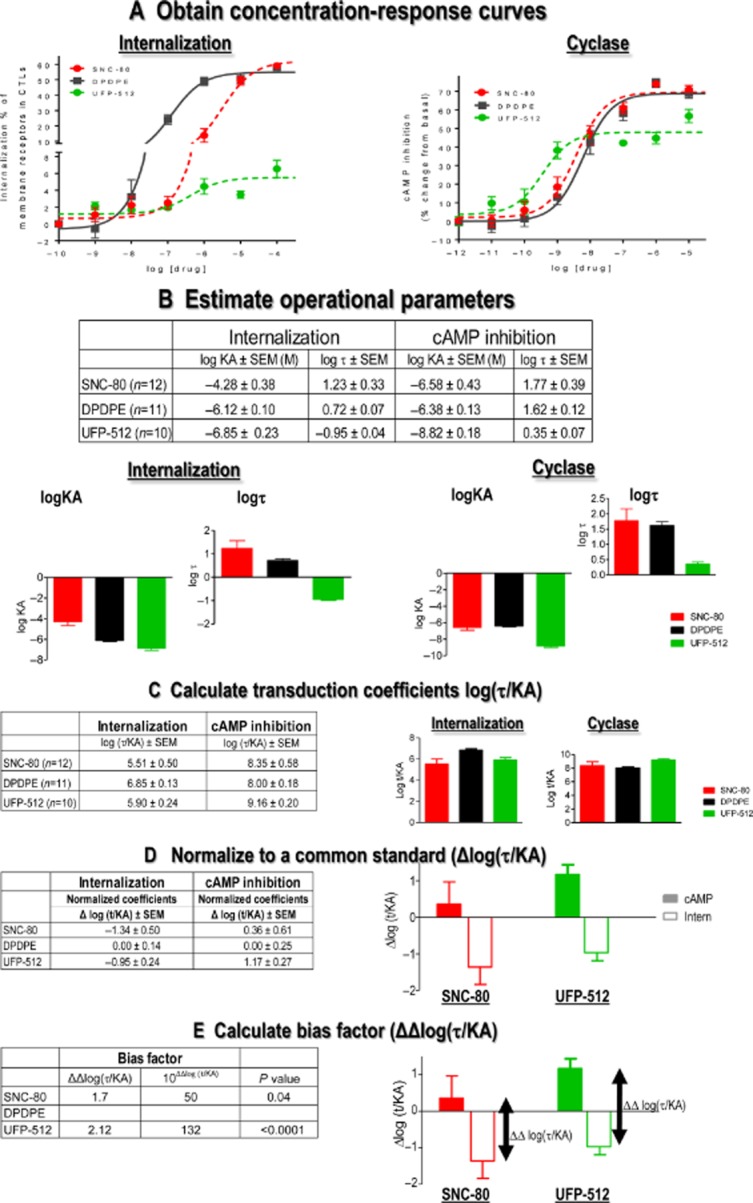
Quantification of signalling versus internalization bias for δ opioid receptor agonists. Concentration-response curves describing modulation of cAMP production or δ opioid receptor internalization by the indicated ligands were obtained in HEK293 cells (A). Data were analysed with the operational model to obtain efficacy (τ) and conditional affinity (KA) values (B) that were then used in the calculation of transduction coefficients log(τ/KA) (C). Note that τ values for SNC-80 are higher than those for those than UFP-512 both in cyclase and internalization responses. However, because UFP-512's KA values are smaller than those obtained for SNC-80, log(τ/KA) ratios of both ligands are not different in either response. Internalization and cyclase transduction coefficients for all ligands were then normalized to DPDPE both in cyclase and internalization readouts to yield corresponding Δlog(τ/KA)CYCLASE and Δlog(τ/KA)INTERN (D). The difference between these coefficients ΔΔlog(τ/KA) corresponds to the bias factor. In this case, these factors indicate that compared with DPDPE, SNC-80 is 80-fold and UFP-512 132-fold more efficient in engaging cyclase inhibition over internalization (E). Statistical significance of these differences was established using two-tailed Student's t-test to compare normalized transduction coefficients (Δlog(τ/KA)) obtained in the two assays (Modified with permission from Charfi et al., 2013).
Figure 6.
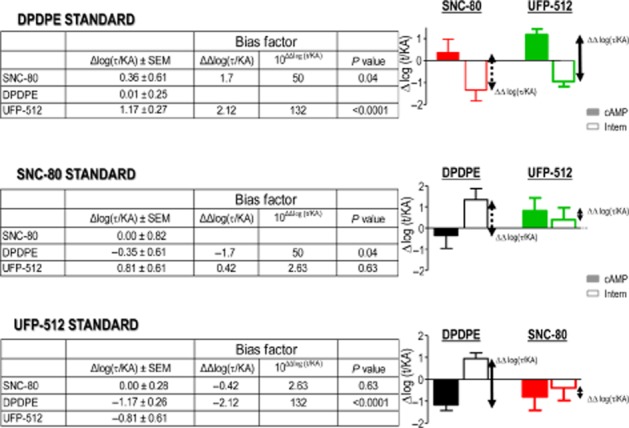
Bias factors heavily depend on the standard chosen for normalization. Transduction coefficients generated in Figure 5C were normalized to DPDPE (as before), SNC-80 or UFP-512 as indicated. Note the difference in the bias profile of SNC-80 when either DPDPE or UFP-512 was used as the standard. Also note that internalization versus cyclase bias displayed by SNC-80 when DPDPE was the standard is of same magnitude but opposite as the bias displayed by DPDPE when SNC-80 was the standard.
The operational model has also been used with alternative assumptions to quantify bias (Rajagopal et al., 2011; Rivero et al., 2012; Rajagopal, 2013). This alternative approach admits that ligand-specific conformations distinctively engage different effectors but assumes independence between ligand affinity for the receptor and its association with the signalling partners that support different responses of interest (Rajagopal et al., 2011). Thus, the main difference between the two quantification approaches is that one of the methods does not admit allosteric interactions between ligand-occupied receptors and immediate signalling partners. In keeping with this assumption, the method proposed by Rajagopal et al. (2011) provides the operational model with a fixed affinity value obtained in binding assays and which is assumed to be the same for all responses. As a consequence, the method solely relies on operational efficacies (τ) to measure whether a ligand displays bias in producing two or more responses. This approach has been applied to μ opioid receptor agonists, revealing a significant bias for endomorphin 2 in G-protein activation versus β-arrestin 2 recruitment (Rivero et al., 2012; Kelly, 2013). In contrast, a single affinity state did not adequately represent δ opioid receptor ligand responses in cyclase and internalization assays (Charfi et al., 2013). This is illustrated in Figure 7, where cyclase and internalization data for DPDPE, SNC-80 and UFP-512 were fitted to the operational model, but this time fixing KA in both responses to a single affinity value (corresponding to binding affinity). In this case, only cyclase data points could be reasonably fit to the model, confirming that a single affinity state could not account for functional responses in both readouts. Based on these observations, it seems that at least some aspects of ligand diversity are better represented by allowing the receptor to adopt distinct functional affinities for different responses (Kenakin and Christopoulos, 2013a).
Figure 7.
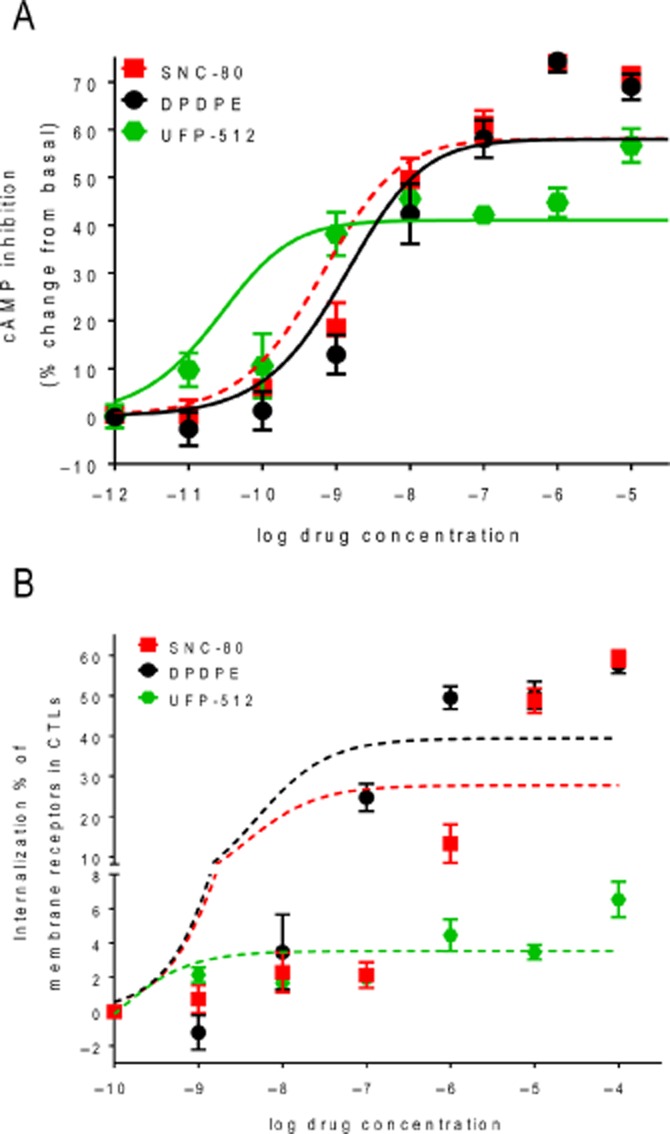
A single affinity state could not account for cyclase and internalization responses of δ opioid receptor agonists. (A) δ opioid receptor internalization and (B) cAMP accumulation data shown in Figure 5 were fit with the operational model, except that KA values for the agonists were fixed from published references as follows: KASNC-80: 5.75 nM; KAUFP-512: 100 pM; and KADPDPE: 12.02 nM. (Modified with permission from Charfi et al., 2013).
The use of functional affinities as a means of describing ligand ability to bias pharmacological stimulus is quite recent (Kenakin and Miller, 2010; Kenakin, 2012; Kenakin and Christopoulos, 2013a,b,). By contrast, the concept that affinity is conditioned by ligand efficacy and the association of the receptor to downstream signalling partners has been accepted for many years as a central notion of the ternary complex model (De Lean et al., 1980; Kent et al., 1980). In this model, the ligand, the receptor and a nucleotide binding protein X (today identified as the G-protein) were proposed to associate into a complex where the ligand's intrinsic activity directly determined the affinity of the agonist-receptor complex for the G-protein. Conversely, the presence of the G-protein warranted higher affinity between the receptor and agonist ligands (De Lean et al., 1980; Kent et al., 1980). In the case of δ opioid receptors, the use of plasmon waveguide resonance spectroscopy (PWRS) has not only validated this prediction (Alves et al., 2003) but has also shown that the association with different Gα subunits distinctively modifies receptor affinity for different agonists (Alves et al., 2004; Hruby et al., 2010). The studies showed that δ opioid receptor affinity for DPDPE was ∼10-fold higher when the receptor was associated to Gαi2 than Gαi1, but the conformation stabilized by morphine displayed 10 times higher affinity for Gαi1 than for Gαi2 (Alves et al., 2004). This change in agonist affinity when the receptor is associated with different Gα subunits confirms that downstream signalling partners condition receptor affinity. Consequently, observations obtained with PWRS provide direct experimental support for the use of the operational model as proposed by Kenakin and Christopoulos (2013b), where KA values are not fixed but conditioned by receptor association with different effectors. Recent crystallization studies carried out on β2-adrenoceptors further underscore the extent to which allosteric interactions with heterotrimeric G-proteins may influence agonist-receptor association (Nygaard et al., 2013). These studies revealed that agonist binding by itself was not enough to fully stabilize an active conformation of the receptor, requiring the presence of the corresponding Gαβγ heterotrimer (Chung et al., 2011; Rasmussen et al., 2011b) or of surrogate antibodies (Rasmussen et al., 2011a) to consolidate an active state and make it amenable for crystallization.
There is now considerable evidence that in addition to G-proteins, GPCRs may signal via non-canonical pathways, which involve β-arrestin interaction with the receptor (Shenoy and Lefkowitz, 2003). As with the canonical complex, β-arrestin interaction with the receptor leads to its stabilization into conformations that display high affinity for the agonist. This has been observed in reconstitution systems where the addition of purified β-arrestin increased agonist affinity for biogenic amine receptors (Gurevich et al., 1997) and in live cells, where fusion of peptide-binding receptors to β-arrestin was shown to produce a similar increase in agonist affinity (Martini et al., 2002; Jorgensen et al., 2005). Recent characterization of the sodium allosteric site in δ opioid receptors indicates that, at least in the case of this receptor, the switch between G-protein and β-arrestin signalling is modulated by sodium-coordinating residues in transmembrane domains II, III and VII (Fenalti et al., 2014).
Biased signalling by δ opioid receptor agonists
Although spectroscopic studies provide clear evidence that association with different agonists stabilized purified δ opioid receptors into conformations that distinctively interact with Gαi/o subunits (Alves et al., 2004; Hruby et al., 2010), the drawback is that PWRS gives no information about the physiological consequences of these distinct interactions. Studies in live cells have provided this type of information, showing that δ opioid receptors behave as pleiotropic receptors capable of activating different Gα subunits (Allouche et al., 1999; Alves et al., 2004; Pineyro and Archer-Lahlou, 2007). In most cases in which ligand ability to activate different G-proteins was assessed, experiments were done at maximal effective concentration and, as we previously saw, for maximal responses to unequivocally indicate the existence of bias, there must be a clear reversal in ligand rank order to activate the different subunits (Kenakin, 2002a,b,). Results fulfilling such conditions have been obtained in SK-N-BE neuroblastoma cells endogenously expressing δ opioid receptors. Herein, the rank order of maximal GTPγS binding by DPDPE was αi2 > αo and was reversed to αo > αi2 for deltorphin I (Allouche et al., 1999). Etorphine, a non-selective μ/δ opioid receptor agonist, did not stimulate αo but activated αi2 much more than αi3. As the SK-N-BE neuroblastoma cell line used in this study only expressed δ opioid receptors (Polastron et al., 1994), the specific profile of Gα activation by these ligands cannot be attributed to concomitant activation of other opioid receptor subtypes therefore indicating that ligand-specific δ opioid receptor responses may be observed at endogenous levels of expression.
The way Gα and Gβγ subunits reorganize with respect to one another upon activation of the G-protein by the receptor may also be ligand-specific. In particular, SNC-80, DPDPE and morphine reduced the distance between position 60 of the Gαi1 subunit and the N-terminus of Gγ while the same regions were drawn apart by TICP (Figure 2B) (Audet et al., 2008). Moreover, ligand differences in conformational rearrangements could be similarly traced upstream in the way the receptor C-terminus interacted with Gαi (Figure 2B; inset), and downstream as functionally selective responses in cAMP and MAPK pathways (Audet et al., 2008). SNC-80, DPDPE and morphine inhibited cAMP production and promoted ERK activation while TICP behaved as an ‘inverse agonist’ that enhanced cAMP production but also stimulated the MAPK cascade (Audet et al., 2005; 2008). Thus, it is possible to conclude that the conformation stabilized by TICP was active for ERK but inactive for cyclase signalling, while the conformations stabilized by the other ligands were active in both pathways.
DPDPE and H-Tyr-Tic-Phe-Phe-OH (TIPP), a tetrapeptide related to TICP (Schiller et al., 1999), were also found to engage ERK activity through different mechanisms suggesting ligand-specific modulation of the MAPK pathway by δ opioid receptor ligands. The mechanism triggered by DPDPE involved promoting a Gβγ-PLCβ3-cSrc complex that led to Raf1-mediated stimulation of the MAPK while its activation by TIPP relied upon β-arrestin 1/2 (Xu et al., 2010). However, despite these striking differences, some caution is warranted before concluding that the two mechanisms were triggered by ligand-specific conformations. Indeed, together with the characterization of the two activation pathways, the authors showed that TIPP was a partial agonist that failed to induce δ opioid receptor phosphorylation at Ser363. Mutating Ser for Ala at this position abolished phosphorylation by DPDPE together with the ligand's ability to engage the PLCβ3/Src/Raf1 pathway, causing it to shift to the β-arrestin 1/2 pathway (Hong et al., 2009; Xu et al., 2010). Thus, as the two different modalities of ERK activation were determined by differential phosphorylation of Ser363 and given that the partial agonist failed to induce the required phosphorylation for one of the pathways to be engaged, we cannot exclude the possibility that the differences between TIPP and DPDPE were not simply related to the full agonist producing δ opioid receptor phosphorylation while the partial agonist did not. Making this distinction is of importance to exploit the observed differences for therapeutic use.
Ligand-specific regulation of δ opioid receptors
Activation by agonists not only promotes signalling, but it also triggers homologous desensitization. The process is initiated by receptor phosphorylation and is followed by β-arrestin recruitment, receptor internalization and its ulterior sorting to degradation or reinsertion at the membrane. As for signalling, numerous reports point to the presence of biased responses along the different steps of the regulatory cascade. Some examples of this type of behaviour by δ opioid receptor agonists are considered in terms of identifying the specific contribution of ligand-specific signalling to observed response bias.
Ligand-specific patterns of δ opioid receptor phosphorylation
Phosphorylation usually involves more than one amino acid and the specific pattern of Ser/Thr residues that undergoes this type of modification has been considered a means of transferring the information codified in ligand-specific conformations to downstream regulatory proteins (Liggett, 2011; Just et al., 2013). For example, μ opioid receptors have been shown to undergo hierarchical, ligand-specific phosphorylation patterns of their C-terminus and the combination of phosphorylated residues was predictive of distinct internalization profiles by different agonists (Just et al., 2013). The δ opioid receptor C-tail also undergoes hierarchical phosphorylation of its Ser/Thr residues, Ser363 being critical for the process (Guo et al., 2000; Kouhen et al., 2000) and Thr358 contributing as an accessory (Guo et al., 2000). Ser363 is phosphorylated by highly efficacious agonists like DPDPE (Guo et al., 2000; Kouhen et al., 2000), SNC-80 (Pradhan et al., 2009), etorphine, deltorphin II (Marie et al., 2008) and (+)BW373U86 (Bradbury et al., 2009). In contrast, maximal effective concentrations of partial agonists like TIPP and morphine fail to produce any phosphorylation at this site (Bradbury et al., 2009; Xu et al., 2010). Given that failure to induce Ser363 phosphorylation corresponds with low intrinsic activity, these differences do not provide sufficient information as to whether post-translational modification at this residue is ligand-specific. Like morphine and TIPP, ARM100390 fails to produce Ser363 phosphorylation (Pradhan et al., 2009), while the effect of TAN-67 is present but minimal (Bradbury et al., 2009). What distinguishes these two ligands from morphine and TIPP is that GTPγS binding by TAN-67 and ARM100390 reached similar maximal responses as full agonists (Bradbury et al., 2009; Pradhan et al., 2009). Thus, the question is whether these four ligands that fail to induce Ser363 phosphorylation but differ in their maximal GTPγS responses display biased signalling in these two possible outcomes. One alternative is that the disproportion between GTPγS binding and phosphorylation elicited by TAN-67 and ARM100390 as compared with morphine and TIPP could be caused by factors such as the non-linear relationship between receptor occupancy and drug response (see quantification section). Using the operational model to control for this type of confounder, we found that ARM100390s transduction coefficient (log(Tau/KA)) for Gαo protein activation was ∼eightfold lower than that of SNC-80 but ∼1000 higher than that of TIPP (unpublished observation), indicating that both, ARM100390 and TIPP behaved as partial agonists as compared with SNC-80. By comparing G-protein activation by TAN-67 and SNC-80, Quock et al. (1997) arrived to a similar conclusion, namely that TAN-67 also behaved as a partial agonist and was almost 10-fold less efficacious than the standard SNC-80. In this case, the relationship between occupancy and response was taken into account by using the equation derived by Ehlert (1985), which allows us to estimate relative efficacies from empirical data such as the drug's half-maximal response (0.5 × Emaxdrug/Emaxsystem), its affinity for the receptor (Kd) and its potency (EC50):
Hence, after controlling for system factors TAN-67 and ARM100390 displayed partial signalling efficacy, but it is not possible to rule out that disproportion in GTPγS versus phosphorylation responses is not simply due to more efficient stimulus-response coupling for the signalling than phosphorylation event. Quantification of bias for G-protein activation versus Ser363 phosphorylation should yield an unequivocal answer as to whether this residue is involved in ligand-specific regulation of δ opioid receptor signalling.
Unlike functional approaches, mutagenesis studies have clearly established that δ opioid receptor phosphorylation can be ligand-specific. These studies show that SNC-80, but not DPDPE, phosphorylates and down-regulates a receptor truncated at Gly338 (Okura et al., 2003), observations that have been taken as an indication that more phosphorylation sites are accessible for protein kinases in δ opioid receptors bound to SNC-80 than DPDPE (Varga et al., 2004). While the idea that DPDPE and SNC-80 stabilize δ opioid receptors into different conformations has been verified (Audet et al., 2012), the region and identity of the residues involved in this difference remains to be determined. The third intracellular loop seems a likely candidate as it contains numerous Ser/Thr residues which contribute to β-arrestin binding (Cen et al., 2001a,b,) and this response is also distinctively engaged by DPDPE and SNC-80 (Audet et al., 2012).
Agonist-specific internalization of δ opioid receptors
Maximal internalization and signalling responses elicited by δ opioid receptor agonists are not proportional, an observation that has been repeatedly obtained in heterologous expression systems (Bradbury et al., 2009; Charfi et al., 2013), cultured neurons (Charfi et al., 2013) and in vivo brain samples (Pradhan et al., 2009). Although this type of observation may be interpreted as the result of ligand-specific responses in the absence of a clear reversal in the rank order, the imbalance in signalling versus internalization Emax values could also be due to differences in stimulus-response coupling or to system factors. Transduction coefficients, on the other hand, allow a controlled quantification of bias. Data in Figure 5 illustrate how Emax values and transduction coefficients distinctively inform us with respect to signalling versus internalization bias by different DOP agonists. In the example, in question Emax values reveal no disproportion between maximal internalization and cAMP inhibition induced by DPDPE or SNC-80, but relative cyclase inhibition by UFP-512 was much higher than its relative internalization response. From these data, no bias would be predicted for DPDPE or SNC-80, but Emax differences for UFP-512 responses would be frequently interpreted as an indication of preferential cyclase versus internalization signalling. In contrast, bias factors calculated from transduction coefficients (Figure 5E) indicate that both UFP-512 and SNC-80 are capable of preferentially engaging cAMP over internalization. Bias for UFP-512 is mainly determined by low internalization efficacy and high ‘conditional affinity’ for cyclase-inhibiting receptor states. Bias for SNC-80 is associated to its low ‘conditional affinity’ for internalizing conformations of the receptor, a property that would have been overlooked by only comparing maximal responses.
The preferential engagement of cyclase over sequestration observed for SNC-80 (Charfi et al., 2013) is in contrast with internalization bias predicted from the in vivo actions of this ligand (Pradhan et al., 2009). Different reasons could explain this divergence. Firstly, the signalling response monitored in one study was G-protein activation (Pradhan et al., 2009) and cyclase modulation in the other (Charfi et al., 2013). Secondly, both reports compared SNC-80 with different standards. Thirdly, quantification of internalization was different, relying on kinetic parameters in the in vivo study (Pradhan et al., 2009) and endpoint measurements in vitro (Charfi et al., 2013). Fourthly, one set of conclusions was based on comparison of maximal responses (Pradhan et al., 2009) and the other relied on transduction coefficients (Charfi et al., 2013). Fifthly, cellular backgrounds used in both studies were different, involving HEK cells in the in vitro study (Charfi et al., 2013) and brain tissue in the other (Pradhan et al., 2009). Importantly, not all regulatory proteins involved in δ opioid receptor internalization are conserved across these cellular phenotypes. For example, while β-arrestins contribute to δ opioid receptor internalization in HEK cells and neurons, PKCs and G protein–coupled receptor kinase 2 play a role in the latter but not the former (Charfi et al., 2013). At the same time, the molecular basis of biased responses is given by the distinct way ligands may interact with the receptor when the latter is associated with different signalling partners (Kenakin and Miller, 2010; Kenakin et al., 2012; Kenakin, 2012). Thus, the very existence of cell-specific internalization partners implies that ligand bias may not be conserved across different cell types or even in different neuronal populations mediating different effects of opioids. If this is the case, cellular background constitutes a source of variation that cannot be simply controlled by the use of the operational model, but should be taken into account if ligand bias is to be used in the rational design of novel therapeutic agents.
Distinct in vivo internalization profiles of δ opioid receptor agonists have been considered of predictive value with respect to ligand potential for generating tolerance (Pradhan et al., 2010). However, tolerance seems to be related not only to internalization but also to the specific type of response assessed. For example, repeated administration of the non-internalizing agonist ARM100390 over a 6-day period developed tolerance for analgesic but not for locomotor or anxiolytic actions (Pradhan et al., 2010). Over a similar period of time, the internalizing agonist SNC-80 produced tolerance to all of these effects (Jutkiewicz et al., 2005; Pradhan et al., 2010) but not to antidepressant-like effects associated with its administration (Jutkiewicz et al., 2005). Numerous possibilities may explain why sequestration profiles fail to predict the occurrence of tolerance for all responses mediated by a given agonist. At the molecular level, one possible reason could be that neurons, which mediate different responses rely upon different proteins for internalization, and consequently the ability of a given ligand to engage δ opioid receptor sequestration may vary in different neuronal populations. Alternatively, internalization may not equally limit all responses mediated by the same ligand. Thus, if δ opioid receptors in different neuronal populations produce their effects through different effectors, internalization may shut down one type of signal but not the other. For instance, by separating the receptor from Ca2+ channels or removing both from the membrane (Altier et al., 2006) internalization may limit depolarization. However, internalization may not necessarily shut down cyclase (Bagheri Tudashki et al., 2013) or MAPK signals (McLennan et al., 2008). Finally, the predictive value of internalization with respect to tolerance may also be influenced by the fact that δ opioid receptors internalized by different ligands do not necessarily follow the same post-endocyctic pathway (Marie et al., 2003; Audet et al., 2012). For example, deltorphin II and DPDPE are more efficient than SNC-80 to induce internalization (Bagheri Tudashki et al., 2013; Charfi et al., 2013), but unlike the latter they do not develop acute analgesic tolerance (Beaudry et al., 2009; Bradbury et al., 2009). A major difference between DPDPE and SNC-80 is that δ opioid receptors activated by DPDPE efficiently recycle back to the membrane, while those activated by SNC-80 are targeted for degradation (Lecoq et al., 2004; Audet et al., 2012). As mentioned in the preceding section, DPDPE stabilizes δ opioid receptors into a conformation that is exclusively phosphorylated at the C-terminus while the conformation stabilized by SNC-80 also induces phosphorylation outside the C-terminal domain (Okura et al., 2003). While receptors activated by SNC-80 remain locked in a stable association with β-arrestin 2, those activated by DPDPE establish a transient interaction with this regulatory protein which allows them to recycle back to the membrane once the complex dissociates (Audet et al., 2012).
In conclusion, here, we have revised evidence indicating that the presence of biased responses may not always correspond to ligand bias. Making the difference between both situations is essential to understand how ligand-specific responses may support desired and undesired effect of opioids in view of exploiting functional selectivity with therapeutic purposes.
Acknowledgments
This work was supported by grants to G. P. from the Natural Sciences and Engineering Research Council of Canada (NSERC) [311997] and from the Canadian Institutes of Health Research (CIHR) [MOP 79432]. I. C. holds a studentship from Ste-Justine Hospital Research Center.
Glossary
- DPDPE
[D-Pen(2), D-Pen(5)]-enkephalin
- Emax
maximal effect
- PWRS
plasmon waveguide resonance spectroscopy
- SNC-80
(1)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3- methoxybenzyl]-N,N-diethylbenzamide
- TICP
Tyr-Ticpsi [CH2-NH]Cha-Phe-OH
- TIPP
H-Tyr-Tic-Phe-Phe-OH
- UFP-512
H-Dmt-Tic-NH-CH(CH2-COOH)-Bid
Conflict of interest
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouche S, Polastron J, Hasbi A, Homburger V, Jauzac P. Differential G-protein activation by alkaloid and peptide opioid agonists in the human neuroblastoma cell line SK-N-BE. Biochem J. 1999;342(Pt 1):71–78. [PMC free article] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, et al. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, et al. mu-Opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves ID, Salamon Z, Varga E, Yamamura HI, Tollin G, Hruby VJ. Direct observation of G-protein binding to the human delta-opioid receptor using plasmon-waveguide resonance spectroscopy. J Biol Chem. 2003;278:48890–48897. doi: 10.1074/jbc.M306866200. [DOI] [PubMed] [Google Scholar]
- Alves ID, Ciano KA, Boguslavski V, Varga E, Salamon Z, Yamamura HI, et al. Selectivity, cooperativity, and reciprocity in the interactions between the delta-opioid receptor, its ligands, and G-proteins. J Biol Chem. 2004;279:44673–44682. doi: 10.1074/jbc.M404713200. [DOI] [PubMed] [Google Scholar]
- Audet N, Paquin-Gobeil M, Landry-Paquet O, Schiller PW, Pineyro G. Internalization and Src activity regulate the time course of ERK activation by delta opioid receptor ligands. J Biol Chem. 2005;280:7808–7816. doi: 10.1074/jbc.M411695200. [DOI] [PubMed] [Google Scholar]
- Audet N, Gales C, Archer-Lahlou E, Vallieres M, Schiller PW, Bouvier M, et al. Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing delta-opioid receptors and heterotrimeric G proteins. J Biol Chem. 2008;283:15078–15088. doi: 10.1074/jbc.M707941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet N, Charfi I, Mnie-Filali O, Amraei M, Chabot-Dore AJ, Millecamps M, et al. Differential association of receptor-Gβγ complexes with β-arrestin2 determines recycling bias and potential for tolerance of δ opioid receptor agonists. J Neurosci. 2012;32:4827–4840. doi: 10.1523/JNEUROSCI.3734-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri Tudashki H, Robertson D, Schiller P, Pineyro G. Endocytic profiles of delta-opioid receptor (DOR) ligands determine the duration of rapid but not sustained cAMP responses. Mol Pharmacol. 2013;85:148–161. doi: 10.1124/mol.113.089003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudry H, Proteau-Gagne A, Li S, Dory Y, Chavkin C, Gendron L. Differential noxious and motor tolerance of chronic delta opioid receptor agonists in rodents. Neuroscience. 2009;161:381–391. doi: 10.1016/j.neuroscience.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bradbury FA, Zelnik JC, Traynor JR. G protein independent phosphorylation and internalization of the delta-opioid receptor. J Neurochem. 2009;109:1526–1535. doi: 10.1111/j.1471-4159.2009.06082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen B, Xiong Y, Ma L, Pei G. Direct and differential interaction of beta-arrestins with the intracellular domains of different opioid receptors. Mol Pharmacol. 2001a;59:758–764. doi: 10.1124/mol.59.4.758. [DOI] [PubMed] [Google Scholar]
- Cen B, Yu Q, Guo J, Wu Y, Ling K, Cheng Z, et al. Direct binding of beta-arrestins to two distinct intracellular domains of the delta opioid receptor. J Neurochem. 2001b;76:1887–1894. doi: 10.1046/j.1471-4159.2001.00204.x. [DOI] [PubMed] [Google Scholar]
- Charfi I, Nagi K, Mnie-Filali O, Thibault D, Balboni G, Schiller PW, et al. Ligand- and cell-dependent determinants of internalization and cAMP modulation by delta opioid receptor (DOR) agonists. Cell Mol Life Sci. 2013;71:1529–1546. doi: 10.1007/s00018-013-1461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, et al. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci U S A. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Deng H, Sun H, Fang Y. Label-free cell phenotypic assessment of the biased agonism and efficacy of agonists at the endogenous muscarinic M receptors. J Pharmacol Toxicol Methods. 2013;68:323–333. doi: 10.1016/j.vascn.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin RH. Introduction: recommendations for the diagnosis, assessment, and treatment of neuropathic pain. Am J Med. 2009;122(10 Suppl):S1–S2. doi: 10.1016/j.amjmed.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol Pharmacol. 1985;28:410–421. [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, et al. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, et al. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Delta opioid receptor analgesia: recent contributions from pharmacology and molecular approaches. Behav Pharmacol. 2011a;22:405–414. doi: 10.1097/FBP.0b013e32834a1f2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Nozaki C, Nadal X, Hever XC, Weibel R, Matifas A, et al. Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain. 2011b;152:1238–1248. doi: 10.1016/j.pain.2010.12.031. [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, et al. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol. 2012;82:860–875. doi: 10.1124/mol.112.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wu Y, Zhang W, Zhao J, Devi LA, Pei G, et al. Identification of G protein-coupled receptor kinase 2 phosphorylation sites responsible for agonist-stimulated delta-opioid receptor phosphorylation. Mol Pharmacol. 2000;58:1050–1056. doi: 10.1124/mol.58.5.1050. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Hong MH, Xu C, Wang YJ, Ji JL, Tao YM, Xu XJ, et al. Role of Src in ligand-specific regulation of delta-opioid receptor desensitization and internalization. J Neurochem. 2009;108:102–114. doi: 10.1111/j.1471-4159.2008.05740.x. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Alves I, Cowell S, Salamon Z, Tollin G. Use of plasmon waveguide resonance (PWR) spectroscopy for examining binding, signaling and lipid domain partitioning of membrane proteins. Life Sci. 2010;86:569–574. doi: 10.1016/j.lfs.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Martini L, Schwartz TW, Elling CE. Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: a high-affinity receptor phenotype. Mol Endocrinol. 2005;19:812–823. doi: 10.1210/me.2004-0312. [DOI] [PubMed] [Google Scholar]
- Just S, Illing S, Trester-Zedlitz M, Lau EK, Kotowski SJ, Miess E, et al. Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol Pharmacol. 2013;83:633–639. doi: 10.1124/mol.112.082875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, Woods JH. Differential behavioral tolerance to the delta-opioid agonist SNC80 ([(+)-4-[(alphaR)-alpha-[(2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl]-(3-me thoxyphenyl)methyl]-N,N-diethylbenzamide) in Sprague-Dawley rats. J Pharmacol Exp Ther. 2005;315:414–422. doi: 10.1124/jpet.105.088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E. Efficacy and ligand bias at the mu-opioid receptor. Br J Pharmacol. 2013;169:1430–1446. doi: 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. On the definition of efficacy. Trends Pharmacol Sci. 1994;15:408–409. doi: 10.1016/0165-6147(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2002a;42:349–379. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002b;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Being mindful of seven-transmembrane receptor ‘guests’ when assessing agonist selectivity. Br J Pharmacol. 2010;160:1045–1047. doi: 10.1111/j.1476-5381.2010.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Measurements of ligand bias and functional affinity. Nat Rev Drug Discov. 2013a;12:483. doi: 10.1038/nrd3954-c2. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013b;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent RS, De Lean A, Lefkowitz RJ. A quantitative analysis of beta-adrenergic receptor interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol Pharmacol. 1980;17:14–23. [PubMed] [Google Scholar]
- Kouhen OM, Wang G, Solberg J, Erickson LJ, Law PY, Loh HH. Hierarchical phosphorylation of delta-opioid receptor regulates agonist-induced receptor desensitization and internalization. J Biol Chem. 2000;275:36659–36664. doi: 10.1074/jbc.M006788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq I, Marie N, Jauzac P, Allouche S. Different regulation of human delta-opioid receptors by SNC-80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] and endogenous enkephalins. J Pharmacol Exp Ther. 2004;310:666–677. doi: 10.1124/jpet.103.063958. [DOI] [PubMed] [Google Scholar]
- Lee TW, Sole MJ, Wells JW. Assessment of a ternary model for the binding of agonists to neurohumoral receptors. Biochemistry. 1986;25:7009–7020. doi: 10.1021/bi00370a038. [DOI] [PubMed] [Google Scholar]
- Leff P. The two-state model of agonist action: challenges to pharmacological receptor theory. Proc West Pharmacol Soc. 1996;39:67–68. [PubMed] [Google Scholar]
- Liggett SB. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci Signal. 2011;4:pe36. doi: 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- Marie N, Lecoq I, Jauzac P, Allouche S. Differential sorting of human delta-opioid receptors after internalization by peptide and alkaloid agonists. J Biol Chem. 2003;278:22795–22804. doi: 10.1074/jbc.M300084200. [DOI] [PubMed] [Google Scholar]
- Marie N, Aguila B, Hasbi A, Davis A, Jauzac P, Allouche S. Different kinases desensitize the human delta-opioid receptor (hDOP-R) in the neuroblastoma cell line SK-N-BE upon peptidic and alkaloid agonists. Cell Signal. 2008;20:1209–1220. doi: 10.1016/j.cellsig.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Martini L, Hastrup H, Holst B, Fraile-Ramos A, Marsh M, Schwartz TW. NK1 receptor fused to beta-arrestin displays a single-component, high-affinity molecular phenotype. Mol Pharmacol. 2002;62:30–37. doi: 10.1124/mol.62.1.30. [DOI] [PubMed] [Google Scholar]
- McLennan GP, Kiss A, Miyatake M, Belcheva MM, Chambers KT, Pozek JJ, et al. Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem. 2008;107:1753–1765. doi: 10.1111/j.1471-4159.2008.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller E, Puza T, Diamond J, Lieu HD, Bohmaker K. Comparative effects of receptor inactivation, 17 beta-estradiol and pertussis toxin on dopaminergic inhibition of prolactin secretion in vitro. J Pharmacol Exp Ther. 1992;263:462–469. [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Gro C, Riitano D, Ambrosio C, et al. Morphine-like opiates selectively antagonize receptor-arrestin interactions. J Biol Chem. 2010;285:12522–12535. doi: 10.1074/jbc.M109.059410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okura T, Varga EV, Hosohata Y, Navratilova E, Cowell SM, Rice K, et al. Agonist-specific down-regulation of the human delta-opioid receptor. Eur J Pharmacol. 2003;459:9–16. doi: 10.1016/s0014-2999(02)02823-6. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Costa T. Where have all the active receptor states gone? Nat Chem Biol. 2012;8:674–677. doi: 10.1038/nchembio.1024. [DOI] [PubMed] [Google Scholar]
- Pineyro G, Archer-Lahlou E. Ligand-specific receptor states: implications for opiate receptor signalling and regulation. Cell Signal. 2007;19:8–19. doi: 10.1016/j.cellsig.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Polastron J, Mur M, Mazarguil H, Puget A, Meunier JC, Jauzac P. SK-N-BE: a human neuroblastoma cell line containing two subtypes of delta-opioid receptors. J Neurochem. 1994;62:898–906. doi: 10.1046/j.1471-4159.1994.62030898.x. [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, et al. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS ONE. 2009;4:e5425. doi: 10.1371/journal.pone.0005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, et al. Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci. 2010;30:16459–16468. doi: 10.1523/JNEUROSCI.3748-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quock RM, Hosohata Y, Knapp RJ, Burkey TH, Hosohata K, Zhang X, et al. Relative efficacies of delta-opioid receptor agonists at the cloned human delta-opioid receptor. Eur J Pharmacol. 1997;326:101–104. doi: 10.1016/s0014-2999(97)83488-7. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S. Quantifying biased agonism: understanding the links between affinity and efficacy. Nat Rev Drug Discov. 2013;12:483. doi: 10.1038/nrd3954-c1. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, et al. Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol. 2011;80:367–377. doi: 10.1124/mol.111.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebois RV, Robitaille M, Gales C, Dupre DJ, Baragli A, Trieu P, et al. Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. J Cell Sci. 2006;119(Pt 13):2807–2818. doi: 10.1242/jcs.03021. [DOI] [PubMed] [Google Scholar]
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, et al. Endomorphin-2: a biased agonist at the mu-opioid receptor. Mol Pharmacol. 2012;82:178–188. doi: 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller PW, Weltrowska G, Berezowska I, Nguyen TM, Wilkes BC, Lemieux C, et al. The TIPP opioid peptide family: development of delta antagonists, delta agonists, and mixed mu agonist/delta antagonists. Biopolymers. 1999;51:411–425. doi: 10.1002/(SICI)1097-0282(1999)51:6<411::AID-BIP4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375(Pt 3):503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, et al. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- Stephenson RP. A modification of receptor theory. Br J Pharmacol Chemother. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudashki HB, Robertson DN, Schiller PW, Pineyro G. Endocytic profiles of delta-opioid receptor ligands determine the duration of rapid but not sustained cAMP responses. Mol Pharmacol. 2014;85:148–161. doi: 10.1124/mol.113.089003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Varga EV, Navratilova E, Stropova D, Jambrosic J, Roeske WR, Yamamura HI. Agonist-specific regulation of the delta-opioid receptor. Life Sci. 2004;76:599–612. doi: 10.1016/j.lfs.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Xu C, Hong MH, Zhang LS, Hou YY, Wang YH, Wang FF, et al. Serine 363 of the {delta}-opioid receptor is crucial for adopting distinct pathways to activate ERK1/2 in response to stimulation with different ligands. J Cell Sci. 2010;123(Pt 24):4259–4270. doi: 10.1242/jcs.073742. [DOI] [PubMed] [Google Scholar]