

Abstract
BACKGROUND AND PURPOSE
Tapentadol is a novel analgesic that combines moderate μ-opioid receptor agonism and noradrenaline reuptake inhibition in a single molecule. Both mechanisms of action are involved in producing analgesia; however, the potency and efficacy of tapentadol in individual neurons has not been characterized.
EXPERIMENTAL APPROACH
Whole-cell patch-clamp recordings of G-protein-coupled inwardly rectifying K+ (KIR3.x) currents were made from rat locus coeruleus neurons in brain slices to investigate the potency and relative efficacy of tapentadol and compare its intrinsic activity with other clinically used opioids.
KEY RESULTS
Tapentadol showed agonist activity at μ receptors and was approximately six times less potent than morphine with respect to KIR3.x current modulation. The intrinsic activity of tapentadol was lower than [Met]enkephalin, morphine and oxycodone, but higher than buprenorphine and pentazocine. Tapentadol inhibited the noradrenaline transporter (NAT) with potency similar to that at μ receptors. The interaction between these two mechanisms of action was additive in individual LC neurons.
CONCLUSIONS AND IMPLICATIONS
Tapentadol displays similar potency for both µ receptor activation and NAT inhibition in functioning neurons. The intrinsic activity of tapentadol at the μ receptor lies between that of buprenorphine and oxycodone, potentially explaining the favourable profile of side effects, related to μ receptors.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Keywords: tapentadol, noradrenalin reuptake inhibitor, opioid receptor agonist, locus coeruleus, GIRK channels, KIR3.x
Introduction
Opioid analgesics are the mainstay of drug therapy for management of acute and chronic severe pain but their utility is limited by on-target side effects, as well as development of tolerance and dependence (Christie, 2008; Williams et al., 2013). One approach to limit side effects, tolerance and dependence clinically is to combine a μ-opioid receptor agonist (nomenclature follows Alexander et al., 2013a) and another analgesic drug with a different mode of action, which can enhance overall analgesic actions and produce opioid-sparing effects (Tzschentke et al., 2007; Schröder et al., 2011).
Tapentadol is a centrally acting analgesic that combines μ receptor agonism with a similar potency for noradrenaline transporter (NAT) inhibition in a single molecule (Tzschentke et al., 2007). The rationale behind this particular combination effect is based on findings that both opioid and noradrenergic pathways contribute to production of analgesia in different ways, and can synergistically interact at spinal and supraspinal levels (Ossipov et al., 1982; Fairbanks and Wilcox, 1999). Although tapentadol exhibits approximately 50-fold lower binding affinity for μ receptors than morphine, its analgesic potency is comparable with morphine for acute and chronic pain in animal models (Tzschentke et al., 2007; Schiene et al., 2011), and it has been widely shown that tapentadol has a favourable tolerability in humans, particularly with respect to gastrointestinal side effects (Hartrick et al., 2009; Tzschentke et al., 2009; Afilalo et al., 2010; Lange et al., 2010; Pergolizzi et al., 2012).
The mechanisms of action of tapentadol have been studied in vitro (Tzschentke et al., 2007), as well as in neurochemical and behavioural studies in animals in vivo (Schröder et al., 2010; 2011; Bee et al., 2011; Schiene et al., 2011; Tzschentke et al., 2012), but little is known about the mechanism of action in individual neurons. The locus coeruleus (LC) is the major source of noradrenergic innervation of the brain and spinal cord (Foote et al., 1983). LC neurons express both μ receptors and α2-adrenoceptors that converge via G-protein βγ-subunits to activate a single population of inwardly rectifying K+ (KIR3.x) channels (North and Williams, 1985; nomenclature follows Alexander et al., 2013b). The LC also expresses a high density of NAT (Schroeter et al., 2000). As such, the LC is also a useful model to study inhibition of NAT by assaying enhancement of actions of α2-adrenoceptors on KIR3.x channels (Surprenant and Williams, 1987).
A previous study of electrical activity of LC neurons in vivo (Torres-Sanchez et al., 2013) reported that systemic administration of tapentadol inhibits spontaneous action potential activity. The authors suggested that the actions of tapentadol on these neurons are mediated predominantly by activation of α2-adrenoceptors but the direct membrane actions and relative potencies for μ receptors and NAT could not be determined using this experimental approach. The aim of the present study was to investigate the relative potency and efficacy of tapentadol in functioning LC neurons using patch-clamp recording in brain slices. The results establish that tapentadol is an agonist at μ receptors with lower intrinsic activity than morphine and oxycodone, but greater than that of buprenorphine. Tapentadol has similar potency in single LC neurons as an activator of µ receptors and inhibitor of NAT. At the cellular level, the interaction between these mechanisms on activation of KIR3.x channels is additive.
Methods
Preparation of brain slices
The Animals Ethics Committee of the University of Sydney, Sydney, NSW, Australia, which complies with the National Health and Medical Research Council ‘Australian code of practice for the care and use of animals for scientific purposes’, approved all experiments. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of N = 114 animals were used in the experiments described here.
Male Sprague Dawley rats (3 to 5 weeks old, n = 114) were used in this study. Animals were housed in groups of two to four under a 12 h/12 h light–dark cycle at 22 ± 2°C with environmental enrichment and free access to food and water. LC slices were prepared as described previously (Dang et al., 2011; 2012). Briefly, rats were anaesthetized with isoflurane (4% in air), decapitated and the brains rapidly removed and blocked. Horizontal brain slices (260–300 μm thick) containing LC were obtained from the pons, using a vibratome (Leica biosystem, VT100, Wetzlar, Germany) in cooled artificial cerebrospinal fluid (ACSF) containing the following (in mM): 126 NaCl, 2.5 KCl, 2.4 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, 11.1 glucose, 95%O2-5%CO2. Slices were incubated in warm (37°C) oxygenated ACSF for at least 30 min before recording.
Electrophysiological recordings
Whole-cell voltage-clamp recordings of visualized LC neurons (infrared Dopt optics, Multiclamp 700B amplifier from Molecular Devices, CA, USA) were acquired at holding potential of −60 mV using Axograph X (Axograph Scientific, Sydney, Australia) as previously described (Dang et al., 2012). Data were sampled at 10 kHz and filtered at 20–50 Hz. Series resistance (≤15 ΜΩ) was compensated by 75% and monitored throughout experiments. Patch pipettes with resistance of 2–4 MΩ were filled with a solution containing the following (in mM): 115 K MES (2-[morpholino]- ethane-sulfonic acid), 20 NaCl, 1.5 MgCl2, 10 BAPTA, 5 HEPES, 4 Mg- ATP and 0.4 Na-GTP and pH 7.3–7.4 was adjusted using KOH. All drugs were applied by superfusion of the recording chamber (∼2 mL min−1, 35°C). Bestatin (10 μM) and thiorphan (1 μM) were included in all experiments using [Met]enkephalin to limit degradation of the peptide. In all experiments involving application of noradrenaline, prazosin (1 μM) was included for at least 5 min before superfusing noradrenaline to prevent potential effects of α1-adrenoceptor activation on KIR3.x channels in these neurons (Osborne et al., 2002). Solutions of noradrenaline were freshly prepared when reapplication time exceeded 5 min to avoid oxidative decomposition. A single concentration of tapentadol was applied per slice.
Data analyses
Data were analysed using Graph-Pad Prism (Graph-Pad V5, San Diego, CA, USA), except for isobolographic analysis. All data are presented as mean ± SEM. Significant differences were analysed by Student's t-test (paired or unpaired two-tailed Student's t-tests) or, where appropriate, one-way anova followed by Dunnett's test, or two-way anova followed by Bonferroni post-test. Concentration-response curves were calculated for each group and fitted to a logistic equation using Graph-Pad Prism, with minimal constrained to zero. Isobolographic analysis was performed using a modification of the method of Tallarida (2007). By this method, additivity is based on the concept of concentration equivalence for drug A and B (or dual mechanisms) follows from individual concentration-effect curves (Grabovsky and Tallarida, 2004), that is a concentration α of drug A is equal to a concentration of drug B denoted beq (α). So combinations of these two concentrations (a,b) would be beq (α) + b. The theoretical additive EC50 value was compared with the observed EC50 for the combination by t-test using FlashCalc 4.5.3 (Dr Michael Ossipov, University of Arizona, Tucson, AZ, USA).
Materials
Stock solutions of tapentadol HCl supplied by Grünenthal GmbH (Aachen, Germany), [Met5]-enkephalin (Sigma-Aldrich, Sydney, NSW, Australia), bestatin (Sigma-Aldrich), morphine HCl (GSK, Sydney, NSW, Australia), idazoxan HCl (Tocris, Bristol, UK), naloxone HCl (Sigma-Aldrich), NA (Sigma-Aldrich), cocaine (GSK) and oxycodone HCl (National Institute on Drug Abuse Drug Supply Program, Chapel Hill, NC, USA), were dissolved in distilled water. Other drugs included UK14304 (Tocris), prazosin HCl (Sigma-Aldrich), dissolved in 20% and 10% dimethyl sulfoxide respectively. Thiorphan (Sigma-Aldrich) was dissolved in 50% ethanol. (+)-Pentazocine from Research Biochemical (Natick, MA, USA) was dissolved in distilled water with one to two drops (almost 50 μL) of glacial acetic acid. Buprenorphine (National Institute on Drug Abuse Drug Supply Program) was dissolved in distilled water using HCI (1 M, one to two drops). The maximum concentration of dimethyl sulfoxide used in the superfusion solution was <0.01% and for ethanol was <0.0001%.
Results
Relative potency and efficacy of tapentadol at μ-opioid receptors
To determine the relative potencies and efficacies of tapentadol and several other opioids at μ receptors, whole-cell patch-clamp of KIR3.x currents were recorded at a VH of −60 mV in the presence of idazoxan (1 μM) and prazosin (300 nM) to block any action of increased noradrenergic tone that might be caused by the inhibition of NAT by tapentadol. Concentration-response curves for [Met]enkephalin were constructed using two to three concentrations per cell. For morphine and tapentadol concentration-response curves, a single concentration of tapentadol or morphine was applied per slice to minimize any possibility that desensitization would confound potency or maximal responses. As shown in Figure 1A, a supramaximal concentration of tapentadol (30 μM) produced an outward current that was consistently smaller than the current produced by a supramaximal concentration of the full α2-adrenoceptor agonist UK14304 (3 μM), in the same cells (Table 1). Naloxone (1 μM) fully reversed the effects of tapentadol (n = 12).
Figure 1.
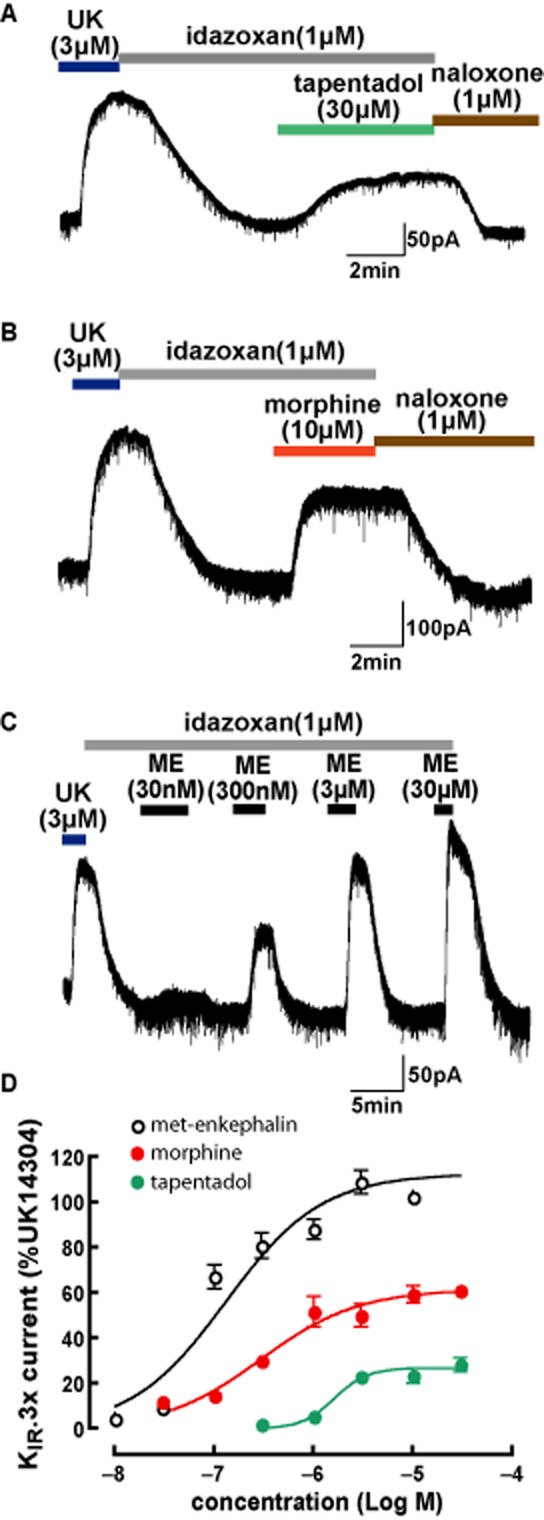
KIR3.x currents induced by activation of μ receptors in LC neurons. Superfusion (shown by bars) of tapentadol (A), morphine (B) and [Met]enkephalin (ME; C) activate KIR3.x currents at holding potential (−60 mV) in the presence of the α2-adrenoceptor antagonist, idazoxan. All actions are reversed by the μ receptor antagonist, naloxone. (D) Concentration-response curves for [Met]enkephalin, morphine and tapentadol. The amplitude of the hyperpolarization plotted as a percentage of the amplitude of a supramaximal concentration of UK14304 (UK).
Table 1.
Membrane actions, MOPr agonism and potency of tapentadol and other opioids in LC neurons
| Imax (pA) | Imax/IUK14304% | pEC50 (-log[μM])a | EC50 (μM) | |
|---|---|---|---|---|
| Tapentadol | 91 ± 15 (n = 12) | 29 ± 4*† | 5.8 ± 0.2 | 1.8 |
| Morphine | 151 ± 25 (n = 8) | 59 ± 4* | 6.5 ± 0.3 | 0.3 |
| [Met]enkephalin | 222 ± 13 (n = 5) | 126 ± 7 | 6.9 ± 0.1 | 0.13 |
Data are expressed as mean ± SEM and the number of experiments is shown in parentheses.
SEM of fitted curve.
Significantly different from [Met]enkephalin (unpaired t-tests, all P < 0.001).
Significantly different from morphine (unpaired t-test, all P < 0.001).
As expected from previous studies (Osborne et al., 2000; Dang and Williams, 2005; Virk and Williams, 2008; Bailey et al., 2009), the effect of morphine was lower than the maximal response produced by [Met]enkephalin in LC neurons (Figure 1B, Table 1), that is morphine acted as a partial agonist for activation of KIR3.x currents in rat LC neurons using whole-cell recording in brain slices. The maximum response to tapentadol was smaller than that produced by morphine (Table 1) suggesting that tapentadol has lower intrinsic efficacy than morphine at the μ receptors.
[Met]enkephalin was a potent and efficacious agonist in LC (Figure 1D, Table 1), with a response amplitude similar to that produced by UK14304 (3 μM). The maximum responses relative to UK14304 (3 μM) for both morphine (30 μM) and tapentadol (30 μM) were considerably smaller than that produced by [Met]enkephalin (Table 1). As summarized in Table 1, tapentadol is approximately sixfold less potent than morphine. The EC50 for morphine was similar to that previously reported in LC neurons using patch-clamp recordings (Osborne et al., 2000).
Intrinsic activity of tapentadol relative to other μ receptor agonists using partial antagonism
If tapentadol does indeed have lower intrinsic agonist activity than [Met]enkephalin and morphine, it should partially antagonize both agonists. As shown in Figure 2, both morphine (Figure 2A) and tapentadol (Figure 2B) partially reduced the actions of a supramaximal concentration of [Met]enkephalin. Moreover, tapentadol partially reduced the effect of a supramaximal concentration of morphine (Figure 2C).
Figure 2.
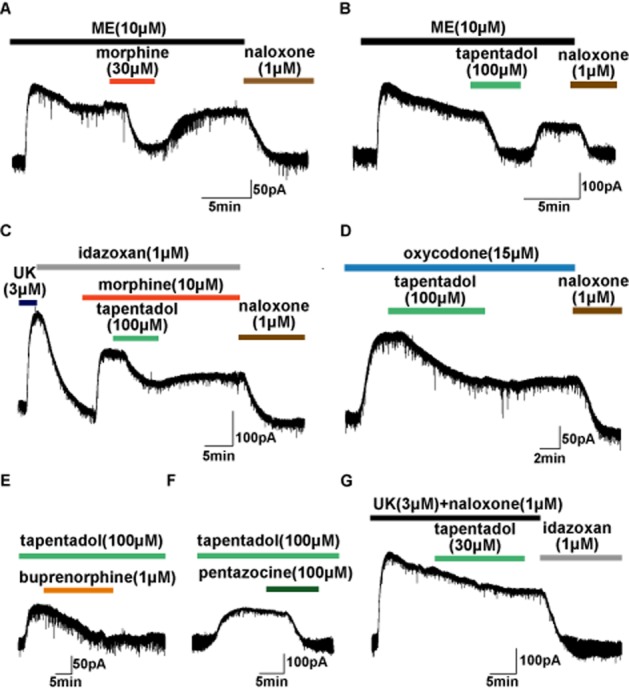
Intrinsic activity of tapentadol at μ receptors (adrenoceptors were blocked from A to F with idazoxan, 1 μM and prazosin 300 nM). (A) Morphine (30 μM) reversibly antagonized the current induced by a supramaximal (and desensitized) concentration of [Met]enkephalin (ME) confirming the lower intrinsic activity of morphine. (B) Tapentadol had lower intrinsic activity than [Met]enkephalin and reversibly antagonized the current induced by a supramaximal (desensitized) concentration of [Met]enkephalin (C) Tapentadol partially and reversibly reduced the current induced by a supramaximal concentration of morphine. (D) Tapentadol partially reduced the current induced by a supramaximal concentration of oxycodone. (E) Buprenorphine and (F) pentazocine almost completely antagonized currents induced by a supramaximal concentration of tapentadol. (G) Tapentadol did not inhibit the current induced by supramaximal concentration of UK14304 (UK) in the presence of naloxone.
The relative intrinsic activity of tapentadol was further compared with several other clinically used μ receptor agonists including oxycodone, buprenorphine and pentazocine. Tapentadol partially reduced the effect of a supramaximal concentration of oxycodone (Figure 2D). Because buprenorphine and pentazocine are weak partial agonists at μ receptors in LC neurons (Virk et al., 2009), producing very small outward currents, they were both tested as partial antagonists, with tapentadol applied first. Buprenorphine (Figure 2E) and pentazocine (Figure 2F) both antagonized the actions of a supramaximal concentration of tapentadol, suggesting that tapentadol has greater intrinsic activity than these two partial agonists.
To test the possibility that tapentadol might inhibit KIR3.x channels in LC neurons, independently of its action on μ receptors [as does thadone; (Rodriguez-Martin et al., 2008)], slices were superfused with naloxone (1 μM), prior to and during application of tapentadol in the presence of a supramaximal concentration of UK14304 (Figure 2G) to fully activate KIR3.x currents via α2-adrenoceptors. Prazosin (300 nM) was included in these experiments to prevent any effects of the activation of α1-adrenoceptors (Osborne et al., 2002). Application of a saturating concentration of UK14304 (3 μM) induced a slowly desensitizing outward current, as previously reported (Dang et al., 2012) but tapentadol had no apparent effect on the current (Figure 2G). To account for the decline in the UK14304-induced current because of desensitization, the slope of the declining current was fitted with a linear function prior to application of tapentadol and the increased slope produced by tapentadol was calculated. Superfusion of tapentadol in the presence of UK14304 did not have any effect on the steady decay of the outward current (t = 0.054, P > 0.96 paired t-test, d.f. = 6). These results establish that partial antagonism of [Met]enkephalin, morphine and oxycodone by tapentadol is due to its lower intrinsic activity rather than direct block of KIR3.x channels.
The actions of tapentadol at the μ receptor are summarized in Table 2. The results suggest a relative order of intrinsic activity of [Met]enkephalin > morphine ≈ oxycodone > tapentadol > buprenorphine ≈ pentazocine.
Table 2.
Relative intrinsic activity of tapentadol and other opioids in LC neurons
| Opioid (n) | Iopioid (pA) | Iopioid+tapent (pA) | Itapent/opioid (%) |
|---|---|---|---|
| [Met]enkephalin (4) | 177 ± 6 | 26 ± 9 | 14 ± 5 |
| Morphine (4) | 153 ± 18 | 73 ± 9* | 48 ± 3 |
| Oxycodone (7) | 209 ± 37 | 93 ± 27* | 41 ± 5 |
| Buprenorphine (5) | 69 ± 6a | 3.4 ± 1.7* | n.d. |
| Pentazocine (4) | 117 ± 17a | 1.3 ± 2.0* | n.d. |
Data are expressed as mean ± SEM.
Itapentadol applied first then combined with listed opioid.
Significantly different from [Met]enkephalin (unpaired t-tests, all P < 0.01).
n.d., not determined.
Tapentadol enhances the action of exogenously applied noradrenaline in LC neurons
The potency of tapentadol to inhibit NAT and thereby potentiate the actions of noradrenaline on LC neurons was studied by blocking μ receptors, then superfusing a low concentration of noradrenaline with tapentadol as previously established for the actions of cocaine in LC neurons (Surprenant and Williams, 1987). In the presence of naloxone (1 μM) and prazosin (300 nM), without addition of noradrenaline, tapentadol (100 μM) produced little or no outward current (<5 pA, n = 3, data not shown), suggesting little basal noradrenergic tone in slices under the present recording conditions. However, as shown in Figure 3, tapentadol potentiated α2-adrenoceptor responses when noradrenaline was exogenously applied. Responses to noradrenaline (3 or 10 μM) were variable from cell to cell and slice to slice, presumably because of variation in the diffusion path through slices to relevant α2-adrenoceptors, which produced a large variation in subsequent NAT inhibition. Indeed, there appeared to be an inverse correlation between the amplitude of response to noradrenaline and extent of potentiation produced by tapentadol (not shown). To control for these differences, the extent of potentiation produced by different concentrations of tapentadol was determined only if the initial response to 3 or 10 μM noradrenaline was between 5 (less than 5% was considered too small to calculate a reliable percentage of enhancement) and 20% of the maximum response produced by [Met]enkephalin. Tapentadol produced a concentration-dependent enhancement of noradrenaline-induced KIR3.x currents in the concentration range of 0.1–30 μM. In the presence of high concentrations of tapentadol (30–100 μM), no further potentiation of the response to noradrenaline could be achieved by further addition of a supramaximal concentration of cocaine [10 μM; (Surprenant and Williams, 1987) (101 ± 1% of tapentadol response, n = 3)], indicating that NAT inhibition was maximal at these concentrations. All subsequent data were normalized to the enhancement of outward current produced by a supramaximal concentration of cocaine (10 μM). The concentration-response curve for tapentadol is presented in Figure 4. The IC50 for tapentadol at NAT in LC slices was 2.3 μM (pIC50 = 5.6 ± 0.1).
Figure 3.
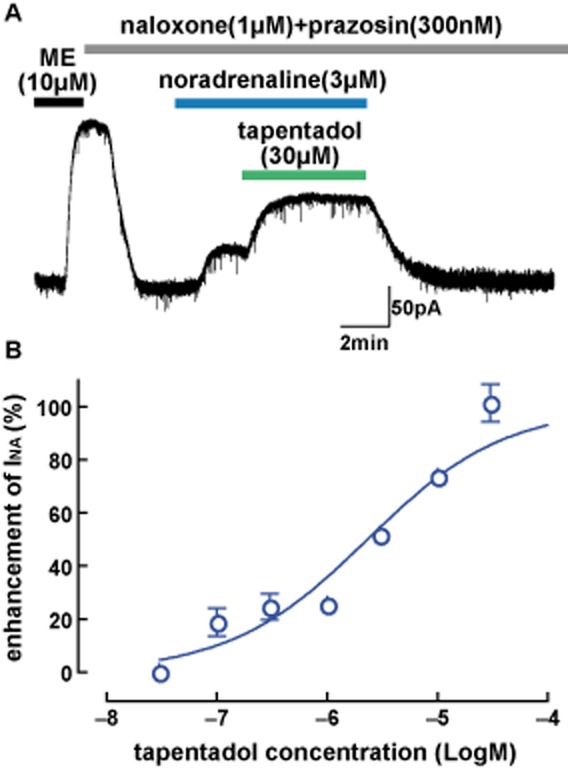
Tapentadol potentiates responses of LC neurons to noradrenaline. (A). Typical recording showing that tapentadol in the continuous presence of noradrenaline enhances the outward current induced by noradrenaline (INA) via inhibiting the noradrenaline reuptake transporter (NAT). In these experiments, the μ receptors were blocked by naloxone (1 μM). (B). Concentration-response curve for enhancement of the noradrenaline current (INA) by tapentadol (IC50 = 2.3 μM).
Figure 4.
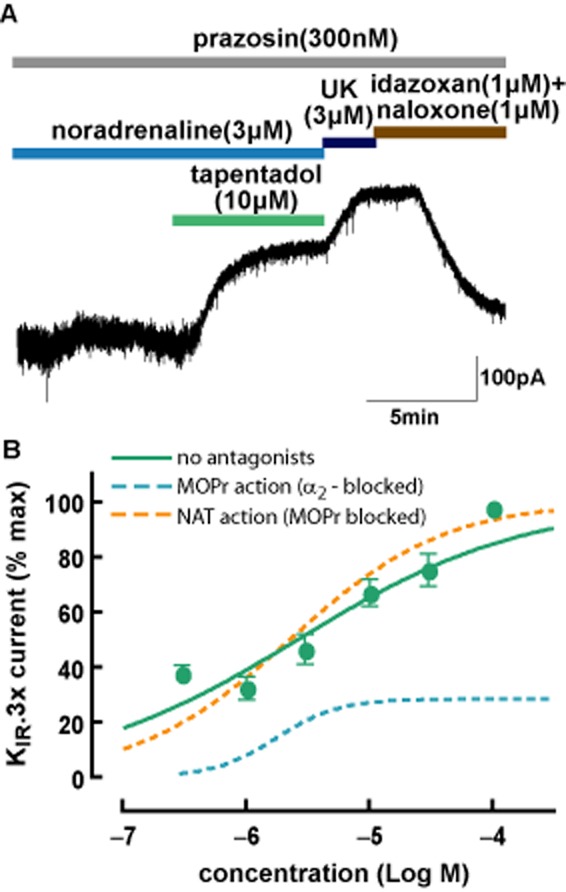
(A) Tapentadol with combined mode of action enhanced the effects of noradrenaline in single LC neurons. All data were normalized by the maximum effect induced by UK14304 (UK; 3 μM). (B) Comparison between the concentration-response curve of tapentadol as a μ receptor (MOPr) agonist alone (with adrenoceptors blocked; data from Figure 1), tapentadol as a NAT inhibitor alone (with μ receptors blocked; data from Figure 3) and tapentadol acting on both mechanisms in the absence of both μ receptor and α2-adrenoceptor antagonists.
Interaction between the two mechanisms of action of tapentadol in single LC neurons
To test how the combined actions of tapentadol on μ receptors and α2-adrenoceptors, via NAT inhibition, interact with respect to KIR3.x currents, tapentadol was applied to LC neurons in the presence of a low concentration of noradrenaline (3 μM) without addition of antagonists of either receptor. To construct the concentration-response curve, a single concentration of tapentadol was applied per slice. Figure 4A shows the enhancement of the effect of noradrenaline (3 μM) under these conditions. Data were normalized to the maximum KIR3.x current induced by a saturating concentration of UK14303 (3 μM) after subtraction of the small current induced by noradrenaline (3 μM). As shown in Figure 4B, the potency of tapentadol acting on both μ receptors and NAT (EC50 = 2.6 μM, pEC50 = 5.6 ± 0.1, n = 5 for each concentration of tapentadol) was not significantly different from the potency of tapentadol as a NAT inhibitor (EC50 = 2.3 μM, P > 0.05, two-way anova, Bonferroni post-test). As confirmed by isobolographic analysis (Grabovsky and Tallarida, 2004; Tallarida, 2006), the interaction between these two mechanisms of action on a single neuron level was additive (P > 0.15), as the theoretical additive EC50 for independent actions on μ receptors and NAT (3.8 ± 1.2 μM) was not significantly different from EC50 for tapentadol with its combined mechanism (2.53 ± 1.4 μM).
Discussion
The present study demonstrates that tapentadol is a μ receptor agonist and NAT inhibitor in single LC neurons in brain slices. Tapentadol exhibited sixfold lower potency than morphine for activation of KIR3.x currents in LC neurons. A previous study (Tzschentke et al., 2007) reported that the binding affinity of tapentadol for rat μ receptors was more than 40-fold lower than that of morphine (Ki = 96 nM for tapentadol vs. Ki = 2.2 nM for morphine). Similarly, in a human recombinant μ receptor [35S]GTPγS assay, tapentadol (EC50 = 670 nM) exhibited approximately 30-fold lower potency than morphine [EC50 = 22 nM; (Tzschentke et al., 2007)]. From this, tapentadol would have been expected to exhibit substantially lower potency than morphine in LC neurons. A likely explanation for the smaller differences between the agonist potencies of tapentadol and morphine observed here compared with the previous study is that the μ receptor reserve is substantially lower in LC neurons than the heterologous expression system used previously (Tzschentke et al., 2007). This interpretation is consistent with the overall potency difference for morphine (14-fold) between the two assay systems. The differences in relative intrinsic activities are also consistent with a large difference in receptor reserve. The maximal effect of tapentadol was 88% of that of morphine in the previous study (Tzschentke et al., 2007) but less than 50% in the LC. Agonists with relatively high intrinsic efficacy (morphine) are predicted to exhibit greater reduction in EC50 than lower intrinsic efficacy agonists when receptor reserve increases (Strange, 2008). It is therefore likely that the relative potency and intrinsic activity differences between morphine and tapentadol reported here more closely reflect differences between μ receptor interactions in brain neurons and those in previous studies using heterologous expression systems, as discussed elsewhere (Law et al., 2000; Morgan and Christie, 2011).
Tapentadol exhibited a relative intrinsic activity at μ receptors with respect to activation of KIR3.x currents that was less than [Met]enkephalin, morphine and oxycodone but greater than the weak partial agonists buprenorphine and pentazocine. This was confirmed, where possible, both by examining the relative maximal response to each agonist and partial antagonism. Furthermore, the present study confirmed that measurement of relative intrinsic activity of tapentadol was not confounded by direct block of KIR3.x channels, as was observed for other opioids such as methadone (Rodriguez-Martin et al., 2008).
Many studies have sought to separate the analgesic effect of μ receptor activation from their side effects. It is well established that the μ receptor mediates both analgesic activity and adverse effects of μ receptor agonists (Matthes et al., 1996). Agonists with a relatively low intrinsic activity at μ receptors could overcome some of the limitations of very high intrinsic efficacy μ receptor agonists in terms of therapeutic window and side effects. Indeed, buprenorphine is a very low intrinsic efficacy μ receptor agonist, but produces strong analgesia, and shows an improved tolerability profile compared with morphine (Cowan et al., 1977; Jasinski et al., 1978). In opioid tolerant individuals, buprenorphine also shows a greatly reduced overdose liability (Fatseas and Auriacombe, 2007). It is tempting to speculate that favourable tolerability of tapentadol in humans, particularly with respect to gastrointestinal side effects (Hartrick et al., 2009; Tzschentke et al., 2009; Afilalo et al., 2010; Lange et al., 2010; Pergolizzi et al., 2012), could in part be due to its relative intrinsic activity at μ receptors which is intermediate between morphine and buprenorphine.
The present study has also established that tapentadol enhances the outward current induced by application of noradrenaline in LC neurons by effectively increasing the concentration of noradrenaline reaching postsynaptic α2-adrenoceptors, as do other NAT inhibitors (Surprenant and Williams, 1987). Tapentadol exhibited potency for NAT inhibition similar to that for activation of the μ receptor.
The finding that the interaction between the two mechanisms of action of tapentadol is additive with respect to activation of KIR3.x currents in LC neurons is not surprising because both mechanisms are known to converge via a common transduction system through G-protein βγ-subunits to activate a single population of KIR3.x channels (North and Williams, 1985; Stone and Wilcox, 2004). However, previous studies have demonstrated that the interaction between the two mechanisms of action of tapentadol in vivo, which is predominantly mediated at the spinal level, is synergistic rather than additive (Schröder et al., 2011). Possible explanations for the synergistic interaction of tapentadol acting on both μ receptor and NAT in vivo could be due to receptor activation at multiple sites along the physiological pain-modulatory pathways, rather than being mediated at the single cell level (Stone and Wilcox, 2004). The synergistic interaction observed in vivo could include a network effect activation of the descending inhibitory pathways at supraspinal level via μ receptor activation together with potentiation of this effect via NAT inhibition at the spinal level, as well as inhibition of nociceptive transmission between primary afferent and spinal second order neurons via pre- and postsynaptic μ receptors and α2-adrenoceptors.
Although the LC is useful to study the outcome of μ receptor pharmacology and NAT inhibition in neurons, the influence of neuronal activity in this nucleus in pain modulation is not fully understood. Electrical or chemical stimulation of the LC produces analgesia in acute and inflammatory pain states (Jones and Gebhart, 1986; Jones, 1991; West et al., 1993) as would be expected from enhancement of spinal noradrenaline release. However, direct inhibition of LC activity by microinjection of μ receptor agonists in vivo is also antinociceptive (Jongeling et al., 2009). The predicted overall effect of suppression of LC neuronal activity by tapentadol on pain modulation in vivo is therefore uncertain.
In conclusion, the present study has established that tapentadol has almost the same potency for NAT inhibition and μ receptor activation in single functioning neurons. Its reduced intrinsic activity at μ receptor compared with morphine, together with its inhibition of NAT may both contribute to improved tolerability profile.
Acknowledgments
This study was supported by a grant from Grünenthal GmbH (Germany) and the National Health and Medical Research Council of Australia (project grant1 1011979, fellowship 1045964 to MJC). The authors are grateful to Dr Michael Ossipov for providing FlashCalc 4.5.3.
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- KIR3.x
G-protein-coupled, inwardly rectifying K+ channels
- LC
locus coeruleus
- NAT
noradrenaline reuptake transporter
Conflict of interest
The current work was financially supported in part by a grant from Grünenthal GmbH to MJC. TMT is an employee of Grünenthal GmbH.
References
- Afilalo M, Etropolski MS, Kuperwasser B, Kelly K, Okamoto A, Van Hove I, et al. Efficacy and safety of Tapentadol extended release compared with oxycodone controlled release for the management of moderate to severe chronic pain related to osteoarthritis of the knee: a randomized, double-blind, placebo- and active-controlled phase III study. Clin Drug Investig. 2010;30:489–505. doi: 10.2165/11533440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, et al. Role of protein kinase C and mu-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bee LA, Bannister K, Rahman W, Dickenson AH. Mu-opioid and noradrenergic alpha(2)-adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve-injured rats. Pain. 2011;152:131–139. doi: 10.1016/j.pain.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Lewis JW, Macfarlane IR. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol. 1977;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT. Morphine-induced mu-opioid receptor desensitization. Mol Pharmacol. 2005;68:1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- Dang VC, Chieng B, Azriel Y, Christie MJ. Cellular morphine tolerance produced by betaarrestin-2-dependent impairment of mu-opioid receptor resensitization. J Neurosci. 2011;31:7122–7130. doi: 10.1523/JNEUROSCI.5999-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Chieng BC, Christie MJ. Prolonged stimulation of mu-opioid receptors produces beta-arrestin-2-mediated heterologous desensitization of alpha(2)-adrenoceptor function in locus ceruleus neurons. Mol Pharmacol. 2012;82:473–480. doi: 10.1124/mol.112.079350. [DOI] [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL. Spinal antinociceptive synergism between morphine and clonidine persists in mice made acutely or chronically tolerant to morphine. J Pharmacol Exp Ther. 1999;288:1107–1116. [PubMed] [Google Scholar]
- Fatseas M, Auriacombe M. Why buprenorphine is so successful in treating opiate addiction in France. Curr Psychiatry Rep. 2007;9:358–364. doi: 10.1007/s11920-007-0046-2. [DOI] [PubMed] [Google Scholar]
- Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- Grabovsky Y, Tallarida RJ. Isobolographic analysis for combinations of a full and partial agonist: curved isoboles. J Pharmacol Exp Ther. 2004;310:981–986. doi: 10.1124/jpet.104.067264. [DOI] [PubMed] [Google Scholar]
- Hartrick C, Van Hove I, Stegmann JU, Oh C, Upmalis D. Efficacy and tolerability of tapentadol immediate release and oxycodone HCl immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther. 2009;31:260–271. doi: 10.1016/j.clinthera.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Pevnick JS, Griffith JD. Human pharmacology and abuse potential of the analgesic buprenorphine: a potential agent for treating narcotic addiction. Arch Gen Psychiatry. 1978;35:501–516. doi: 10.1001/archpsyc.1978.01770280111012. [DOI] [PubMed] [Google Scholar]
- Jones SL. Descending noradrenergic influences on pain. Prog Brain Res. 1991;88:381–394. doi: 10.1016/s0079-6123(08)63824-8. [DOI] [PubMed] [Google Scholar]
- Jones SL, Gebhart GF. Quantitative characterization of ceruleospinal inhibition of nociceptive transmission in the rat. J Neurophysiol. 1986;56:1397–1410. doi: 10.1152/jn.1986.56.5.1397. [DOI] [PubMed] [Google Scholar]
- Jongeling AC, Johns ME, Murphy AZ, Hammond DL. Persistent inflammatory pain decreases the antinociceptive effects of the mu opioid receptor agonist DAMGO in the locus coeruleus of male rats. Neuropharmacol. 2009;56:1017–1026. doi: 10.1016/j.neuropharm.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange B, Kuperwasser B, Okamoto A, Steup A, Haufel T, Ashworth J, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010;27:381–399. doi: 10.1007/s12325-010-0036-3. [DOI] [PubMed] [Google Scholar]
- Law PY, Erickson LJ, El-Kouhen R, Dicker L, Solberg J, Wang W, et al. Receptor density and recycling affect the rate of agonist-induced desensitization of mu-opioid receptor. Mol Pharmacol. 2000;58:388–398. doi: 10.1124/mol.58.2.388. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Christie MJ. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br J Pharmacol. 2011;164:1322–1334. doi: 10.1111/j.1476-5381.2011.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Williams JT. On the potassium conductance increased by opioids in rat locus coeruleus neurones. J Physiol. 1985;364:265–280. doi: 10.1113/jphysiol.1985.sp015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne PB, Chieng B, Christie MJ. Morphine-6 beta-glucuronide has a higher efficacy than morphine as a mu-opioid receptor agonist in the rat locus coeruleus. Br J Pharmacol. 2000;131:1422–1428. doi: 10.1038/sj.bjp.0703697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne PB, Vidovic M, Chieng B, Hill CE, Christie MJ. Expression of mRNA and functional alpha(1)-adrenoceptors that suppress the GIRK conductance in adult rat locus coeruleus neurons. Br J Pharmacol. 2002;135:226–232. doi: 10.1038/sj.bjp.0704453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Malseed RT, Goldstein FJ. Augmentation of central and peripheral morphine analgesia by desipramine. Arch Int Pharmacodyn Ther. 1982;259:222–229. [PubMed] [Google Scholar]
- Pergolizzi J, Alegre C, Blake D, Alen JC, Caporali R, Casser HR, et al. Current considerations for the treatment of severe chronic pain: the potential for tapentadol. Pain Pract. 2012;12:290–306. doi: 10.1111/j.1533-2500.2011.00487.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martin I, Braksator E, Bailey CP, Goodchild S, Marrion NV, Kelly E, et al. Methadone: does it really have low efficacy at micro-opioid receptors? Neuroreport. 2008;19:589–593. doi: 10.1097/WNR.0b013e3282f97b64. [DOI] [PubMed] [Google Scholar]
- Schiene K, De Vry J, Tzschentke TM. Antinociceptive and antihyperalgesic effects of tapentadol in animal models of inflammatory pain. J Pharmacol Exp Ther. 2011;339:537–544. doi: 10.1124/jpet.111.181263. [DOI] [PubMed] [Google Scholar]
- Schröder W, Vry JD, Tzschentke TM, Jahnel U, Christoph T. Differential contribution of opioid and noradrenergic mechanisms of tapentadol in rat models of nociceptive and neuropathic pain. Eur J Pain. 2010;14:814–821. doi: 10.1016/j.ejpain.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Schröder W, Tzschentke TM, Terlinden R, De Vry J, Jahnel U, Christoph T, et al. Synergistic interaction between the two mechanisms of action of tapentadol in analgesia. J Pharmacol Exp Ther. 2011;337:312–320. doi: 10.1124/jpet.110.175042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD. Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol. 2000;420:211–232. [PubMed] [Google Scholar]
- Stone LS, Wilcox GL. Alpha-2-adrenergic and opioid receptor additivity in rat locus coeruleus neurons. Neurosci Lett. 2004;361:265–268. doi: 10.1016/j.neulet.2003.12.065. [DOI] [PubMed] [Google Scholar]
- Strange PG. Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br J Pharmacol. 2008;153:1353–1363. doi: 10.1038/sj.bjp.0707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A, Williams JT. Inhibitory synaptic potentials recorded from mammalian neurones prolonged by blockade of noradrenaline uptake. J Physiol. 1987;382:87–103. doi: 10.1113/jphysiol.1987.sp016357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther. 2006;319:1–7. doi: 10.1124/jpet.106.104117. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. Interactions between drugs and occupied receptors. Pharmacol Ther. 2007;113:197–209. doi: 10.1016/j.pharmthera.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Sanchez S, Alba-Delgado C, Llorca-Torralba M, Mico JA, Berrocoso E. Effect of tapentadol on neurons in the locus coeruleus. Neuropharmacology. 2013;72:250–258. doi: 10.1016/j.neuropharm.2013.04.053. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Christoph T, Kogel B, Schiene K, Hennies HH, Englberger W, et al. (-)- (1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel mu-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323:265–276. doi: 10.1124/jpet.107.126052. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Jahnel U, Kogel B, Christoph T, Englberger W, De Vry J, et al. Tapentadol hydrochloride: a next-generation, centrally acting analgesic with two mechanisms of action in a single molecule. Drugs Today (Barc) 2009;45:483–496. doi: 10.1358/dot.2009.45.7.1395291. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Folgering JH, Flik G, De Vry J. Tapentadol increases levels of noradrenaline in the rat spinal cord as measured by in vivo microdialysis. Neurosci Lett. 2012;507:151–155. doi: 10.1016/j.neulet.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Virk MS, Williams JT. Agonist-specific regulation of mu-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol. 2008;73:1301–1308. doi: 10.1124/mol.107.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Arttamangkul S, Birdsong WT, Williams JT. Buprenorphine is a weak partial agonist that inhibits opioid receptor desensitization. J Neurosci. 2009;29:7341–7348. doi: 10.1523/JNEUROSCI.3723-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West WL, Yeomans DC, Proudfit HK. The function of noradrenergic neurons in mediating antinociception induced by electrical stimulation of the locus coeruleus in two different sources of Sprague-Dawley rats. Brain Res. 1993;626:127–135. doi: 10.1016/0006-8993(93)90571-4. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, et al. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]