

Abstract
BACKGROUND AND PURPOSE
Acute activation of κ opioid (KOP) receptors results in anticocaine-like effects, but adverse effects, such as dysphoria, aversion, sedation and depression, limit their clinical development. Salvinorin A, isolated from the plant Salvia divinorum, and its semi-synthetic analogues have been shown to have potent KOP receptor agonist activity and may induce a unique response with similar anticocaine addiction effects as the classic KOP receptor agonists, but with a different side effect profile.
EXPERIMENTAL APPROACH
We evaluated the duration of effects of Mesyl Sal B in vivo utilizing antinociception assays and screened for cocaine-prime induced cocaine-seeking behaviour in self-administering rats to predict anti-addiction effects. Cellular transporter uptake assays and in vitro voltammetry were used to assess modulation of dopamine transporter (DAT) function and to investigate transporter trafficking and kinase signalling pathways modulated by KOP receptor agonists.
KEY RESULTS
Mesyl Sal B had a longer duration of action than SalA, had anti-addiction properties and increased DAT function in vitro in a KOP receptor-dependent and Pertussis toxin-sensitive manner. These effects on DAT function required ERK1/2 activation. We identified differences between Mesyl Sal B and SalA, with Mesyl Sal B increasing the Vmax of dopamine uptake without altering cell-surface expression of DAT.
CONCLUSIONS AND IMPLICATIONS
SalA analogues, such as Mesyl Sal B, have potential for development as anticocaine agents. Further tests are warranted to elucidate the mechanisms by which the novel salvinorin-based neoclerodane diterpene KOP receptor ligands produce both anti-addiction and adverse side effects.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Keywords: salvinorin A, Salvia divinorum, cocaine self-administration, drug seeking, κ opioid receptor, addiction, nociception, dopamine transporter
Introduction
Activation of κ opioid (KOP) receptors by traditional agonists such as U50,488H, U69,593 and the novel neoclerodane diterpene salvinorin A (SalA) have been shown previously to have anti-addiction effects in preclinical models of addiction (Negus et al., 1997; Mello and Negus, 1998; Gray et al., 1999; Schenk and Partridge, 1999; 2000; Schenk et al., 1999; 2000; Morani et al., 2009; Wee et al., 2009; receptor nomenclature follows Alexander et al., 2013). Unfortunately, side effects including sedation, aversion, dysphoria and depression limit their therapeutic use. SalA has been shown to have reduced side effects compared with the traditional KOP receptor agonists; however, pro-depressive effects were still observed (Morani et al., 2012). The short duration of effects of SalA also make it undesirable as a potential therapeutic agent (Prisinzano, 2005; Butelman et al., 2009; Teksin et al., 2009; Ranganathan et al., 2012). Utilizing the unique structure of SalA, several investigators have synthesized novel derivatives with varying opioid receptor selectivities, potencies and efficacies (Harding et al., 2005; Prisinzano and Rothman, 2008; Lovell et al., 2011). One SalA analogue, β tetrahydropyran Sal B, was shown to have anti-addiction effects in preclinical studies similar to those reported for SalA, although side effects were not reported (Prevatt-Smith et al., 2011). The development of longer acting KOP receptor agonists possessing desirable anti-addiction effects and reduced side effects remains to be investigated along with the mechanism of action of such effects (Kivell et al., 2014).
Acute administration of cocaine leads to increases in dopamine levels in the dorsal striatum (dStr) and nucleus accumbens (NAcb), leading to rewarding effects. It has been proposed that KOP receptor agonists oppose the effects of drugs, such as cocaine, by modulating the dopamine system (Spangler et al., 1997; Shippenberg et al., 2007; Wee and Koob, 2010). Acute administration of KOP receptor agonists such as: U50,488H, U69,593 and SalA have all been shown to cause a decrease in dopamine concentrations in the NAcb and dStr (Spanagel et al., 1990; Shippenberg et al., 2007). This decrease has been linked to reduced release of dopamine from the presynaptic neuron. KOP receptors are also localized in the same neurons as the dopamine transporter (DAT) in the NAcb (Svingos et al., 2001), suggesting that KOP receptors may also play a role in regulating DAT function. DAT clears dopamine from the synapse and its activity is rapidly regulated by kinases.
Identification of functional agonists that are able to differentially activate signalling pathways may yield analogues with desirable anti-addiction effects without the unwanted side effects limiting their therapeutic use. There are currently no reported potent SalA analogues with partial KOP receptor agonist or antagonist activities. Activation of p38-MAPK, β-arrestin recruitment and late activation of ERK1/2 have all been identified as candidate pathways for the undesirable effects of KOP receptor agonists (Bruchas et al., 2007; 2011; Land et al., 2009; Muschamp et al., 2011). ERK1/2 activation is responsible for the discriminative stimulus effects induced by U50,488H (Yoshizawa et al., 2011). Repeated stress has also been shown to activate ERK1/2 in the mouse striatum in a KOP receptor -dependent manner (Bruchas et al., 2008). Mice, following the acute administration of the KOP receptor agonist U50,488H display potentiated ethanol-conditioned place preference (CPP) and forced swim stress potentiated ethanol-CPP. These effects were shown to be KOP receptor-dependent (Sperling et al., 2010). Stress-induced potentiation of cocaine CPP was prevented by the KOP receptor antagonist norbinaltorphimine (norBNI) and was absent in mice lacking the prodynorphin gene (McLaughlin et al., 2003), suggesting an important role for the KOP receptor system in modulating stress-induced behaviours.
Many experimental models are used to predict addiction behaviours in humans, including various measures of natural reward, CPP and intracranial self-stimulation (ICSS). No single model is able to successfully model such complex behaviours. However, drug self-administration models are considered to be the gold standard (Kmiotek et al., 2012). Within self-administration models, different paradigms such as long and short access models, progressive ratio models and extinction models are commonly used to model various aspects of addiction and drug-reward properties (Dworkin and Stairs, 2002). All models have various strengths and weaknesses and model different aspects of addiction in humans. In this study, we used a drug-prime model of relapse in rats trained to self-administer cocaine following a short period of extinction. This model has been extensively used to measure the anticocaine-like effects of traditional KOP receptor agonists (Schenk and Partridge, 1999; 2000; Schenk et al., 1999; 2000; Morani et al., 2009); SalA (Morani et al., 2009) and novel SalA analogues (Prevatt-Smith et al., 2011; Morani et al., 2013) allowing for direct comparisons between these compounds. Future work comparing acute and chronic administration will identify if differences between novel SalA analogues and traditional compounds are present. It is generally accepted that acute administration of KOP receptor agonists attenuate the rewarding effects of drugs of abuse, possibly by inducing punishing/aversive-like effects; whereas, chronic exposure to KOP receptor agonists may potentiate drug-seeking and/or drug taking, via stress mediated mechanisms and modulation of withdrawal (Wee and Koob, 2010; Graziane et al., 2013).
In this study, the novel SalA analogue, Mesyl Sal B, was investigated. Mesyl Sal B is a selective KOP receptor agonist with a similar affinity for the receptor (Ki 2.3 nM for Mesyl Sal B, compared with 1.9 nM for SalA) and potency (EC50 of 30 ± 5 nM compared with 40 ± 10 nM for SalA) (Harding et al., 2005). In this study, we showed that Mesyl Sal B was a longer acting KOP receptor agonist than SalA, and that it attenuated cocaine-prime induced drug seeking or taking following acute administration. We also showed that Mesyl Sal B increased DAT function and activated early, but not late, phase ERK1/2 in the dStr, NAcb and prefrontal cortex. Unlike SalA and U69,593, the increases in DAT function seen with Mesyl Sal B were not due to increases in cell-surface DAT expression.
Methods
Animals
All animal care and experimental procedures conform to the UK laws governing animal experimentation and were approved by the Animal Ethics Committee (VUW), New Zealand. The authors have consulted the ARRIVE guidelines (Kilkenny et al., 2010) and reporting of experiments (McGrath et al., 2010). A total of 128 male Sprague Dawley rats (250–400 g) were used for self-administration experiments and for brain tissue, and 27 male mice (B6-SJL, 23–27 g) were used for tail-withdrawal assays.
All rats were bred and housed in the Victoria University of Wellington, New Zealand (VUW) animal facility. Juvenile mice were purchased from the Malaghan Institute of Medical Research (Wellington, New Zealand), prior to housing within the VUW animal facility. All animals were maintained on a 12-h light/dark cycle and given free access to food and water except during experimental sessions. All procedures were conducted during the light cycle.
Hot water tail-withdrawal assay
To determine the duration of action of Mesyl Sal B, the antinociception hot water tail-withdrawal assay was carried out in mice (n = 9) following the method of Simonin et al. (1998). Briefly, mice were restrained in a Plexiglas tube (internal diameter of 24 mm) and allowed to acclimatize for 15 min. One-third of each mouse tail was marked and immersed in water (50 ± 0.5°C) with a maximum cut-off latency set at 10 s (Simonin et al., 1998; Valjent et al., 2015). Repeated measurements were carried out at 1, 5, 10, 15, 30, 45 60, 90, 120, 150 min, and the time for the animal to show a tail-withdrawal response was recorded following i.p. administration of vehicle (80% propylene glycol, 20% DMSO diluted with the same volume PBS) or KOP receptor agonist dissolved in vehicle by an observer, unaware of the treatments given to the experimental groups. The maximum possible effect (MPE) of analgesia was then calculated using the following formula: % MPE = 100 × (test latency − control latency)/(10 − control latency).
Cocaine self-administration
A total of 16 rats were used for self-administration experiments. Under deep anaesthesia [ketamine/xylazine (90/9 mg·kg−1, i.p.)], a silastic catheter was inserted into the right external jugular vein. The distal end (22 gauge stainless steel tubing) was passed subcutaneously to an exposed portion of the skull where it was fixed to embedded jeweller's screws with dental acrylic. Post-surgery catheters were infused daily with sterile saline solution (containing 30 U·mL−1 heparin, 250 000 U·mL−1 penicillin G potassium and 8000 U·mL−1 streptokinase), and rats were rested for 5 days. Self-administration training and reinstatement tests were conducted in standard operant chambers equipped with two retractable levers (Med Associates, ENV-001, St. Albans, VT, USA). Self-administration procedures were as described previously (Schenk and Partridge, 1999; 2000; Morani et al., 2009). Briefly, depression of the active lever led to a programmed intravenous (i.v.) infusion (0.1 mL) of cocaine-HCl dissolved in physiological saline containing heparin (3.0 U·mL−1). Infusions lasted 12 s and were accompanied by illumination of a light above the active lever. Rats were trained to self-administer cocaine daily for 2 h sessions, and trained on a fixed response-1 (FR-1) schedule of reinforcement followed by FR-5. The reinstatement test was conducted in a single day and consisted of three phases. During the first phase, the animals were allowed to self-administer cocaine (0.5 mg·kg−1·infusion−1, FR-5) for 1 h, followed by a 3 h extinction phase when cocaine was replaced with heparinized saline (FR-5). The extinction period (3 h) was chosen because this was the time taken for responses to decline to less than 10% of baseline responding. Only animals that reached this criterion were used in experiments. At the beginning of the third phase (reinstatement), rats (n = 5–6 per group) received Mesyl Sal B or vehicle. These injections were administered 45 min prior to the cocaine-prime (20 mg·kg−1, i.p.), and responding under extinction was measured for 1 h. The 45 min pre-incubation for Mesyl Sal B was based on in vivo pharmacological studies (Figure 2). Some rats were also treated with the KOP receptor antagonist norBNI (2 mg·kg−1, i.p.) 30 min prior to Mesyl Sal B (0.3 mg·kg−1, i.p.) prior to reinstatement testing to evaluate KOP receptor-mediated effects.
Figure 2.
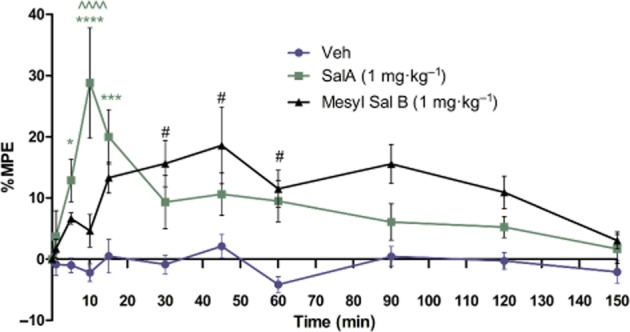
Duration of action of Mesyl Sal B in vivo in the hot water tail immersion assay in mice. Tail-withdrawal latency is expressed as percent maximal possible effect (% MPE ± SEM) versus time. SalA (1 mg·kg–1) has rapid analgesic effects compared with vehicle at 5, 10 and 15 min. Mesyl Sal B (1 mg·kg−1) has a slower onset and longer duration of action with increased tail-withdrawal latencies at 30, 45 and 60 min. SalA and Mesyl Sal B showed significant differences at 10 min (SalA compared to veh *P < 0.05, ***P < 0.001 ****P < 0.0001, Mesyl Sal B compared to veh #P < 0.05, SalA compared with Mesyl Sal B ∧∧∧∧P < 0.0001). Repeated measures anova revealed a significant effect of time [F(3603) = 4.952; P < 0.0001], drug [F(6625) = 19.88; P < 0.0001] and a drug and time interaction [F(3580) = 3.69; P < 0.001]. Two-way anova followed by Bonferroni post test (n = 9).
Spontaneous locomotion activity
A total of 14 drug-naïve male rats were used to determine locomotor activity by previously described methods (Morani et al., 2013). Briefly, following a 30 min habituation phase in the open field chamber (Med Associates ENV-520), drug-naïve rats were injected with either vehicle (75% DMSO, i.p.) or Mesyl Sal B (0.3 mg·kg−1, i.p.), and ambulation counts were measured at 5 min intervals for 60 min.
Cell culture
HEK-293, green monkey kidney (COS7) and mouse neuroblastoma cells (N2A) (ATCC, Manassas, VA, USA) were transfected 48 h prior to experiments with 0.6 μg yellow fluorescent protein-tagged human DAT (YFP-hDAT) (J. Javitch, New York, NY, USA) and 0.4 μg myc-rKOP receptor (L. Devi, New York, NY, USA) using Lipofectamine 2000. HEK-293 and COS7 cells were maintained in DMEM containing 10% FBS, 1% penstrep antibiotic and 1% L-glutamine, and N2A cells were maintained in RPMI-1640 medium containing 10% FBS and 1% penstrep antibiotic.
Imaging experiments
ASP+ uptake
DAT function was measured using 4-(4-(dimethylamino)-styrl)-N-methylpyridinium (ASP+). Methods were carried out as previously described (Schwartz et al., 2003; Bolan et al., 2007; Zapata et al., 2007). In brief, transfected cells were placed into a temperature controlled Olympus Fluoview FV1000 confocal microscope (Sydney, NSW, Australia) in KREBs buffer (130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4.6H20, 1.2 mM KH2PO4, 10 mM HEPES, 10 mM D-glucose, pH 7.4), medium was aspirated and cells imaged (120 s, image every 5 s, YFP filter: 485 nm excitation; 545 nm emission, ASP+ filter: 570 nm excitation; 670 nm emission). ASP+ (10 μM) was added after capture of the first image followed by agonist (1–10 μM) or DMSO vehicle at 250 s. For cellular experiments KOP receptor specificity (norBNI, 1 μM, 30 min), Pertussis toxin (PTX 100 ng·mL−1 16–24 h) and ERK1/2 signalling pathways (U0126, 20 μM 30 min) were investigated, cells were incubated with the appropriate drug before ASP+ addition. For binding experiments, cells were incubated with agonist or DMSO vehicle for 250 s prior to ASP+ addition with images taken at maximum speed for 100 s.
Analysis for ASP+ uptake and binding studies
YFP-hDAT expressing cells were manually outlined on the image, and the accumulation of ASP+ over time determined. The rates of uptake for a period of 60 s before and 60 s following drug were calculated at the time of maximal effect. The change in slope was calculated as a percentage change [(slope after drug/slope before drug) × 100]. For binding studies, data from 3 to 7 s after ASP+ addition were analysed.
Total internal reflective fluorescence microscopy (TIRFM)
Methods followed previously published protocols (Furman et al., 2009; Kivell et al., 2010). Cells were placed onto the stage of an Olympus FV1000 microscope in a cell chamber for 20 min in aerated culture medium (100X objective, 1.45 NA; Olympus, PLAPO, Sydney, NSW, Australia). A region of cells was selected using the TIRFM camera (QImaging Digital Camera 6.2.0.6; Olympus) and Image Pro Plus 7.0 acquisition software (Media Cybernetics, Rockville, MD, USA). A penetration depth of 100 nM and a refractive index of 1.52 were set. Images (473 nm laser, YFP filter: 485 nm excitation; 545 nm emission) were captured every minute for 40 min from time 0 with an exposure time of 3 s. After 10 min, agonists (10 μM) were added. Pixel intensities of individual cells at each time point were analysed using ImageJ (National Institutes of Health, Bethesda, MD, USA).
ERK phosphorylation
Transfected HEK-293 cells were treated with 10 μM KOP receptor agonist or vehicle (0.28% DMSO in KREBS buffer) following serum starvation (30 min), lysed in RIPA buffer (10 mM Tris-HCL, pH 7.5, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS and 1% sodium deoxycholate; pH 7.5) and run on SDS-PAGE gels, followed by overnight transfer to PVDF membranes (Millipore Corporation, Billerica, MA, USA). Polyacrylamide gels were imaged on a Fuji FLA-5100 laser scanner before transferring to PVDF to confirm YFP-hDAT expression. Once transferred, membranes were probed with mouse monoclonal P-ERK 1/2 followed by anti-mouse Cy5, and then striped and reprobed with rabbit monoclonal ERK 1/2, followed by anti-rabbit Cy3. ERK 1/2 signalling was inhibited using the MEK inhibitor U0126 (20 μM). To analyse data, band intensities were measured using ImageJ, and P-ERK1/2 was normalized to total ERK1/2 expression. To determine ERK phosphorylation ex vivo, vehicle (75% DMSO) or KOP receptor agonist (2.0 mg·kg−1, i.p.) was administered at selected times (0, 10–15, 20, 60 and 120 min) and rats killed following CO2 asphyxiation. A total of 40 rats were utilized to determine P-ERK1/2 levels. The brain was then rapidly removed and the dStr, NAcb and prefrontal cortex dissected. All brain regions were placed into microcentrifuge tubes. Tissue was lysed in 300 μL RIPA buffer containing protease and phosphatase inhibitors, on ice. Lysates were centrifuged at 16 000 g for 15 min, and the DNA pellet removed and processed as above.
Cell surface biotinylation
Protocols were followed as previously published (Kivell et al., 2010). Transfected cells were treated with agonist or vehicle 30 min prior to biotin incubation (1 mg·mL−1, EZ-link-NHS-Sulfo-SS-biotin). Cells were lysed in 400 μL of RIPA buffer for 1 h in the dark at 4°C with shaking. Lysates were centrifuged at 16 000 g for 30 min at 4°C and 50 μL removed (total fraction). Supernatant was incubated with 100 μL streptavidin beads for 1 h at room temperature, centrifuged (4°C for 1 min at 500 g) and 50 μL of supernatant removed (internal fraction). Bound proteins were eluted from washed beads with 50 μL of SDS-PAGE loading buffer (cell-surface fraction). Fractions were run on an SDS-PAGE gel, the gel was scanned to visualize YFP-hDAT, transferred overnight onto PVDF and blotted for GAPDH for verification of fraction identities.
Rotating disk electrode voltammetry (RDEV)
Methods were carried out as previously described (Povlock and Schenk, 1997; Danielson et al., 2015). A total of 56 rats were used for these RDEV studies. DStr, NAcb and prefrontal cortex tissue was dissected from freshly killed drug-naïve rat brains, weighed, finely chopped in ice-cold PBS and transferred to microcentrifuge tubes. Tissue was washed eight times in oxygenated, 37°C Krebs buffer, allowing to tissue to settle between each wash. Tissue was resuspended in 300 μL of buffer and transferred to the RDEV chamber where the glassy carbon rotating electrode (Pine Instruments, AFMDO3GC, Durham, NC, USA) was lowered and rotated at 2000 r.p.m. A potential of +450 mV was applied and the resulting current recorded (eDAQ, Denistone, NSW, Australia). Tissue was left to reach baseline prior to KOP receptor agonist (500 nM) or DMSO vehicle addition. For the low to infinite trans model (Meiergerd and Schenk, 1994; Povlock and Schenk, 1997), increasing concentrations of dopamine were added, and the current allowed to reach baseline between each addition. For single additions, once the current had reached baseline, a single addition of dopamine was added (2 μM). For medial prefrontal cortex experiments, once baseline had been reached, the noradrenaline transporter inhibitor desipramine-HCl (1 μM) was added 5 min prior to dopamine addition. For the norBNI experiments, tissue was pre-incubated at 37°C with 1 μM norBNI in oxygenated, 37°C Krebs buffer for 30 min. To determine whether ERK activation was required for KOP receptor mediated increases in DAT function, dStr tissue was pre-incubated with U0126 (10 μM) or vehicle (0.02% DMSO in Krebs buffer) prior to KOP receptor agonist addition and determination of DAT function. Uptake data were collected for 10 s beginning 1 s after addition of dopamine, and a linear regression calculated. Uptake was expressed as pmol·s−1·g−1 of tissue. For the low to infinite trans-model, uptake values were entered into GraphPad Prism, and a nonlinear regression (one-site binding hyperbole) was fitted to each repeat and the Vmax and Km values obtained.
Data analysis
Data are expressed as mean ± SEM. Phase 3 of the reinstatement tests were analysed using one-way anova, followed by Tukey's post hoc test. All imaging experiments were carried out over three transfections with at least two dishes per transfection analysed; n equals the number of animals studied. GraphPad Prism (San Diego, CA, USA) was used to determine statistical significance, with Student t-tests carried out for comparing a single treatment to control, one-way anova followed by Bonferroni post hoc tests to compare multiple treatments, and two-way anova used to compare multiple effects. Values of P < 0.05 were considered significant.
Materials
The KOP receptor agonist SalA was isolated and purified from Salvia divinorum leaves (>98% pure by HPLC) (Butelman et al., 2007), and Mesyl Sal B was synthesized from SalA as described previously (Harding et al., 2005). Desipramine HCl, dopamine HCl, U0126, U69,593, U50, 488H and norBNI were purchased from Sigma (Auckland, New Zealand), ASP+, and PTX from Tocris (Bristol, UK). Other chemicals were purchased as indicated: streptavidin beads and EZ-link NHS-Sulfo-SS-biotin (Pierce, Rockford, IL, USA), mouse monoclonal anti-GAPDH (Abcam, Cambridge, MA, USA), mouse monoclonal anti P-ERK1/2 (1:500; Santa Cruz Biotechnology, Paso Robles, CA, USA), rabbit monoclonal anti ERK1/2 (Cell Signalling Technologies, Danvers, MA, USA), anti-rabbit Cy3 and anti-mouse Cy5 (1:1000; GE Healthcare, Fairfield, CT, USA), YFP-hDAT (J. Javitch) and myc-rKOP receptor (L. Devi). Addition of these fluorescent tags does not alter the trafficking, localization or function of the proteins (Jordan and Devi, 1999; Zapata et al., 2007).
Results
Mesyl Sal B is a neoclerodane diterpene prepared from SalA. Mesyl Sal B has a mesylate group at the C2 position (Figure 1) and has previously been reported to be a selective and potent full KOP receptor agonist with similar binding affinity to SalA at the KOP receptor (Table 1) and similar potency in GTPγS assays [SalA: 40 ± 10 nM EC50; Mesyl Sal B: 30 ± 5 nM (means±SD)] (Harding et al., 2005).
Figure 1.

Chemical structures and in vitro functional potencies (EC50) for SalA (A) and Mesyl Sal B (B). Reported previously in Harding et al. (2005). [35S]GTPγS assays in CHO cells stably expressing human KOP receptors. EC50 SalA 40 ± 10 nM (EC50 ± SD), Emax 120 ± 2% compared with U50, 488H [% stimulation of binding compared with U50, 488H (500 nM)]. Mesyl Sal B: EC50 30 ± 5; Emax 112%.
Table 1.
Binding affinities of salvinorin A and Mesyl Sal B at the KOP receptor using [125I]IOXY as radioligand
| Ki ± SD, nM | |||
|---|---|---|---|
| Compound | μ | δ | κ |
| SalA | >1000 | 5 790 ± 980 | 1.9 ± 0.2 |
| Mesyl Sal B | 6820 ± 660 | >10 000 | 2.3 ± 0.1 |
Reported previously in Harding et al. (2005).
In vivo pharmacology
No previous in vivo work for Mesyl Sal B has been reported. Therefore, we determined its duration of action in vivo. Mesyl Sal B significantly increases the latency of tail-withdrawal at 30, 45 and 60 min compared with vehicle controls (P < 0.05); whereas, SalA had significant effects at 5 (P < 0.05), 10 (P < 0.0001) and 15 min (P < 0.001; n = 9 for all groups). Significant differences between SalA and Mesyl Sal B were observed at 10 min (P < 0.0001), with SalA showing significantly higher latencies (Figure 2).
Traditional KOP receptor agonists and SalA have demonstrated anti-addiction effects. The effects of the longer acting SalA derivative, Mesyl Sal B, on addiction behaviours have not been tested. The effects of acute Mesyl Sal B administration on cocaine-prime induced reinstatement of drug seeking (Figure 3A) shows Mesyl Sal B (0.3, 1.0 mg·kg−1) dose-dependently attenuated this effect [F (3,23) = 36.82, P < 0.0001]. This effect was KOP receptor-dependent, as attenuation of reinstatement was prevented in rats pretreated with the KOP receptor antagonist norBNI (p = 0.0002) (Figure 3B). No effects on spontaneous locomotor activity were observed with Mesyl Sal B (0.3 mg·kg−1, i.p.) (Figure 3C).
Figure 3.

Mesyl Sal B attenuates cocaine-prime induced cocaine-seeking behaviour in rats. (A) Active lever press responses following Mesyl Sal B 45 min prior to a priming injection of cocaine (20 mg·kg−1) ***P < 0.001, data compared with vehicle treated controls (0 mg·kg−1): one-way anova followed by Tukey's post hoc test: n = 5–6 for each group. (B) norBNI (30 min pretreatment, 2 mg·kg−1, s.c.) followed by Mesyl Sal B (0.3 mg·kg−1, ip) during phase 3 of reinstatement tests (mean ± SEM). (C) Spontaneous locomotor activity in vehicle and Mesyl Sal B treated rats over 60 min (mean ± SEM) (Student's t-test) (n = 7 for each group).
Using ASP+ uptake methods, changes in DAT function in cells expressing YFP-hDAT and myc-rKOP receptor were measured. Mesyl Sal B dose-dependently (1–10 μM) increased the rate of ASP+ uptake (Figure 4A) (P < 0.001). The increase in DAT function observed was similar to U50,488H with a 21% (±5%) increase (Figure 4B) (P < 0.001) and was approximately half that of SalA (45 ± 7%) (P < 0.001) (Figure 4C). The KOP receptor antagonist norBNI (1 μM, 30 min) completely inhibited the increase caused by 10 μM Mesyl Sal B (Figure 4A) (P < 0.01), U50,488H (Figure 4B) (P < 0.001) and SalA (Figure 4C) (P < 0.001).
Figure 4.

Mesyl Sal B modulates DAT function in vitro via a KOP receptor-dependent and PTX-sensitive mechanism in cells transiently expressing YFP-hDAT and Myc-rKOP receptor (A) Mesyl Sal B, (B) U50,488H and (C) SalA cause concentration-dependent increases in ASP+ uptake. These effects (at 10 μM Mesyl Sal B) were norBNI reversible (30 min pretreatment, 1 μM compared to 10 μM drug, #P < 0.05, ##P < 0.01, ###P < 0.001). Mesyl Sal B (10 μM) also caused an increase in ASP+ uptake in (D) N2A and (E) COS7 cells. (F) ASP+ binding following Mesyl Sal B (10 μM). The addition of DMSO (at time 0) shows that the vehicle control did not lead to a significant decrease in uptake from vehicle for any of the agonists (one sample Student's t-test). Experiments were carried out over three transfections with at least two dishes used per transfection (n = 46–64 cells). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the vehicle control without KOP receptor agonist (mean ± SEM).
The KOP receptor is a G0/i protein-coupled receptor. Pretreatment of cells with PTX (100 ng·mL−1 for 16–24 h) blocked the effects of 10 μM Mesyl Sal B (Figure 4A) (P < 0.01), U50,488H (Figure 4B) (P < 0.001) and SalA (Figure 4C) (P < 0.001). The ability of Mesyl Sal B to increase ASP+ uptake was not restricted to HEK-293 cells, as this effect at 10 μM was also seen in N2A cells (Figure 4D) (P < 0.01) and COS7 cells (Figure 4E) (P < 0.01). In addition to increasing ASP+ uptake, Mesyl Sal B also significantly increased ASP+ binding to DAT in HEK-293 cells (Figure 4F). Neither norBNI, PTX, nor vehicle (DMSO) had effects on ASP+ uptake on their own (data not shown).
Previous studies have shown KOP receptor agonists activate ERK1/2 (Belcheva et al., 2005), and ERK1/2 activation is known to increase DAT function and cell-surface expression (Moron et al., 2003). Therefore, ERK1/2 is a possible link between KOP receptor and DAT. We determined the effects of Mesyl Sal B on phosphorylation of ERK1/2 with a time-course experiment in HEK-293 cells transiently expressing YFP-hDAT and myc-rKOP receptor. Mesyl Sal B rapidly phosphorylated ERK1/2 with significant increases seen at 3 and 5 min (Figure 5A,B) (P < 0.05). ERK1/2 is activated by direct phosphorylation of MEK. The selective MEK inhibitor U0126 (20 μM for 30 min) attenuated ERK1/2 activation (Figure 5C,D) (P < 0.001). To determine if the increase in ASP+ uptake following KOP receptor activation by Mesyl Sal B (10 μM) was dependent on ERK1/2 activation, ASP+ uptake experiments were performed on cells pre-incubated with the MEK inhibitor U0126 (20 μM, 30 min). U0126 inhibited the increase in ASP+ uptake by Mesyl Sal B (10 μM) (Figure 5E) (P < 0.001). Although high doses of U0126 (50 μM) altered DAT surface expression and may modulate ASP+ uptake, control experiments showed no effects on ASP+ uptake compared with controls (U0126, 20 μM, 30 min) (data not shown).
Figure 5.

Mesyl Sal B modulates DAT function through an ERK1/2-dependent pathway. (A) Fluorescent scan of a representative gel showing YFP-hDAT expression (top), P-ERK1/2 expression (middle) and total ERK1/2 expression (bottom) after Mesyl Sal B treatment (10 μM). (B) P-ERK1/2 expression was normalized to total ERK1/2 and expressed graphically as a fold change compared with time 0 (one-way anova followed by Bonferroni post test, *P < 0.01, n = 4). (C & E) Inhibition of ERK1/2 activation by U0126 pretreatment (20 μM, 30 min) one-way anova followed by Bonferroni post test, *P < 0.05, **P < 0.01 compared with appropriate control, ###P < 0.001 compared with appropriate Mesyl Sal B treated control, n = 6. (D) A representative Western blot of the effect of U0126 on the Mesyl Sal B-induced phosphorylation of ERK1/2. (E) The increase in uptake caused by Mesyl Sal B was inhibited by U0126; one-way anova followed by Bonferroni post test, ***P < 0.001 compared with DMSO control, ###P < 0.001 compared with Mesyl Sal B treated control respectively (n = 32–50). The addition of DMSO as a vehicle control did not lead to a significant decrease in uptake from 0 (one sample Student's t-test).
To determine if the effects on ERK1/2 activation in cell-culture models translated to animal models, we also investigated the ability of Mesyl Sal B (2 mg·kg−1, i.p.) to phosphorylate ERK1/2 in brain tissue taken from the rat dStr, NAcb and prefrontal cortex. Mesyl Sal B significantly increased ERK1/2 phosphorylation at 10–15 min in the dStr, NAcb and prefrontal cortex (Figure 6) (P < 0.05).
Figure 6.

Mesyl Sal B modulates ERK phosphorylation in vivo. (A) P-ERK1/2 expression in the dStr, (C) NAcb and (E) prefrontal cortex was normalized to total ERK1/2 and expressed as fold change (mean ± SEM) compared with time 0. ERK1/2 phosphorylation following Mesyl Sal B administration (Student's t-test, *P < 0.05, n = 6–17). Representative Western blot showing P-ERK1/2 expression (top) and ERK1/2 expression (bottom) after Mesyl Sal B administration in (B) dStr, (D) NAcb and (F) prefrontal cortex. MM = molecular weight marker.
To determine whether Mesyl Sal B modulates DAT function in the rat brain, we performed RDEV on tissue from the rat NAcb that was pre-incubated with the KOP receptor agonist (500 nM, 4 min) prior to RDEV experiments. Mesyl Sal B caused a significant increase in Vmax (1509 ± 115, pmol·s−1·g−1) (P < 0.05) with no change in Km (1.5 ± 0.5 μM) compared with vehicle control (Vmax:1026 ± 181) (Km: 1.5 ± 0.6 μM) similar to the effects seen with the positive control U50,488H compared with vehicle [Vmax, 1979 ± 335 pmol·s−1·g−1 (P < 0.05)] with no change in Km (2.3 ± 0.8 μM) (Figure 7A; Table 2). To investigate the effects of Mesyl Sal B on different brain regions, a single-addition model (dopamine, 2 μM) was used. Mesyl Sal B (500 nM) (P < 0.05), like control U50,488H (P < 0.01), showed a significant increase in dopamine clearance in the NAcb, dStr and prefrontal cortex (P < 0.01) that was prevented by norBNI (1 μM, 30 min) (Figure 7C) (P < 0.05). SalA showed a faster onset of action of 1 min compared with 4 min for U50,488H and Mesyl Sal B (data not shown), and significantly increased dopamine uptake in the dStr, NAcb and prefrontal cortex, respectively (P < 0.001, P < 0.001 and P < 0.05) at this time point. The increase in dopamine uptake was dependent on ERK1/2 activation as pre-incubation with MEK inhibitor U0126 prevented the Mesyl Sal B increase in dopamine uptake in dStr tissue (Figure 7D). Incubation with norBNI alone had no effect on dopamine uptake (Figure 7C).
Figure 7.

Mesyl Sal B modulates DAT function ex vivo via a KOP receptor dependent mechanism. (A) NAcb tissue was pre-incubated with vehicle (DMSO matched), or KOP receptor agonist U50,488H or Mesyl Sal B before sequential addition of dopamine. Data points shown are the mean ± SEM (n = 6–8 tissue homogenates). Mesyl Sal B significantly increased Vmax (1567 ± 228, pmol·s–1·g–1) (**P < 0.01) with no change in Km (1.9 ± 0.6 μM) compared with vehicle control. U50,488H also showed an increase in Vmax [1837 ± 406 pmol·s−1·g−1 (*P < 0.05)] with no change in Km (1.9 ± 0.9 μM). (B) Mesyl Sal B (500 nM) and U50,488H showed a significant increase in dopamine clearance in the NAcb (P < 0.05 and P < 0.01, respectively), dorsal striatum (P < 0.01 and P < 0.01, respectively) and prefrontal cortex (P < 0.01 and P < 0.01, respectively) following a single addition of dopamine (2 μM). (C) This increase in dopamine clearance was inhibited by prior incubation of dStr tissue with norBNI (1 μM, 30 min) (#P < 0.05). Incubation with norBNI alone had no effect on dopamine uptake. Pre-incubation of dStr tissue with U0126 (10 μM) prevented Mesyl Sal B from increasing DAT function (Mesyl Sal B + U0126 compared with DMSO + U0126 (P = 0.9368); Mesyl Sal B/vehicle compared with Mesyl Sal B + U0126 (***P < 0.001); one-way anova followed by Bonferroni post test, (n = 6–7). DA = dopamine.
Table 2.
Kinetics of dopamine uptake in rat nucleus accumbens tissue
| Treatment | Vmax (pmol·s–1·g–1) | Km (μM) |
|---|---|---|
| Vehicle | 740 (73) | 0.71 (0.24) |
| U50,488H | 1837 (406)* | 1.95 (0.93) |
| Mesyl Sal B | 1567 (228)** | 1.89 (0.61) |
Values in the Table are means (SEM). *P < 0.05, **P < 0.01, significantly different from vehicle.
An increase in Vmax is often associated with an increase in cell-surface expression of transporters. To investigate whether the increase in Vmax of DAT with Mesyl Sal B (Figure 7) was due to changes in cell-surface expression of DAT, we utilized an in vitro cell-culture model and performed cell-surface biotinylation and TIRFM experiments to determine if there were any changes in cell-surface DAT. In biotinylation experiments, a 30 min incubation with the KOP receptor agonists U69,593 (Figure 8B) and SalA (Figure 8C) (10 μM) caused a significant increase in cell-surface DAT levels 37 ± 15% (P < 0.05), and 25 ± 12% (P < 0.01) respectively. Mesyl Sal B had no effect on cell-surface DAT levels (P = 0.2063) (Figure 8A). Results from TIRFM studies show that at 30 min following U69,593 (Figure 8E) and SalA [(Figure 8F) exposure, HEK-293 cells increased cell-surface expression of DAT (P < 0.05), an effect that is not seen with Mesyl Sal B (Figure 8D). No significant changes in cell-surface DAT were seen with short incubations of any of the KOP receptor agonists tested with either biotinylation or TIRFM protocols (Mesyl Sal B, SalA, U69,593; data not shown).
Figure 8.

The increase in DAT function following Mesyl Sal B is not via an increase in DAT cell-surface expression in HEK-293 cells. (A–C) HEK-293 cells were incubated with KOP receptor agonists (10 μM, 30 min) before biotinylation studies. (D–F) TIRFM studies were carried out on HEK-293 cells expressing YFP-hDAT and myc-rKOP receptor with drug addiction occurring at 10 min. (A) Mesyl Sal B biotinylation (one-way anova followed by Bonferroni post test; n = 12–13), (D) Mesyl Sal B TIRFM experiments (two-way anova followed by Bonferroni post test; n = 9–10). (B) U69,593 biotinylation (one-way anova followed by Bonferroni post test, *P < 0.05; n = 6–8), (E) TIRFM studies with U69,593 (two-way anova followed by Bonferroni post test, *P < 0.05, **P < 0.01; n = 12). (C) SalA biotinylation experiments (one-way anova followed by Bonferroni post test, **P < 0.01; n = 12–16) and (F) TIRFM studies with Sal A (two-way anova followed by Bonferroni post test, *P < 0.05; n = 7–9). T = total, I = internal, CS = cell surface.
Discussion
SalA is a full agonist at KOP receptors (Roth et al., 2002) and has similar efficacy to the traditional KOP receptor agonists U50,488H and U69,593 and the endogenous ligand dynorphin in GTPγS assays (Chavkin et al., 2004; Prevatt-Smith et al., 2011). Acutely administered SalA has anti-addiction effects in rats (Morani et al., 2009), similar to the effects seen with traditional KOP receptor agonists (Schenk and Partridge, 1999; Schenk et al., 1999; Shippenberg et al., 2007). SalA attenuated cocaine-induced drug-seeking behaviour without suppressing sucrose reinforcements (Morani et al., 2009). Also, SalA suppressed cocaine-induced hyperactivity (Chartoff et al., 2008) and cocaine behavioural sensitization without inducing sedation and aversion in rats at the dose required to attenuate drug seeking (Morani et al., 2012). In the present study, Mesyl Sal B, like SalA, showed no sedative effects in spontaneous locomotion experiments. Although SalA has a better toxicity profile (Mowry et al., 2003; Johnson et al., 2011) and fewer undesirable side effects (Morani et al., 2012) than traditional KOP receptor agonists, its poor pharmacokinetic properties make it undesirable for therapeutic development (Beguin et al., 2006; Prisinzano and Rothman, 2008). With this in mind, the development of new SalA analogues with improved pharmacokinetics and reduced side effects has the potential to yield useful KOP receptor ligands for the treatment of addiction (Kivell et al., 2014), pain (McCurdy et al., 2006), neuroprotection (Su et al., 2012) and for development of non-addictive analgesics (Groer et al., 2007).
The C-2 position of SalA is an important position for its binding to KOP receptors and metabolic stability (Chavkin et al., 2004; Prisinzano, 2005; Kane et al., 2008). Mesyl Sal B has a mesylate group (-SO2-CH3) substitution at the C-2 position and retains selective KOP receptor binding affinity and potency (Harding et al., 2005). No previous in vivo work has been reported on Mesyl Sal B; therefore, we determined its duration of action in mice relative to the known antinociceptive properties of KOP receptor agonists (Simonin et al., 1998; John et al., 2006; McCurdy et al., 2006). Results show that Mesyl Sal B has a slower, more prolonged onset of action in vivo compared with SalA. The effects we show here for SalA are consistent with the literature with antinociceptive effects in the tail-flick assay also reported at 10 min with 1 mg·kg−1 (i.p.) SalA but not at 20 or 30 min (McCurdy et al., 2006). Tail-flick assays performed in mice intrathecally administered SalA also showed significant antinociceptive effects between 5 and 15 min (John et al., 2006). The longer duration of effects seen with Mesyl Sal B in this study are likely to reflect an improvement in its metabolic stability and bioavailability.
Novel neoclerodanes based on the structure of SalA have been suggested as potential targets for the development of anti-addiction pharmacotherapies (Beguin et al., 2006; Prisinzano and Rothman, 2008; Prevatt-Smith and Prisinzano, 2010). Here, we show that Mesyl Sal B retained the ability to dose-dependently attenuate cocaine-prime induced drug-seeking or drug-taking behaviour in a KOP receptor-dependent manner when administered acutely, just like the parent compound SalA (Morani et al., 2009). While this model is limited to cocaine-produced reinstatement following extinction and does not attempt to evaluate cue or stress-induced behaviour, this widely used model does allow us to make direct comparisons with traditional and novel KOP receptor agonists utilizing the same model (Schenk and Partridge, 1999; 2000; Schenk et al., 1999; 2000; Morani et al., 2009; 2013; Prevatt-Smith et al., 2011). Our findings are consistent with ICSS measurements of reward following acute SalA (Potter et al., 2011). In a cocaine challenge following repeated SalA, a decrease in the reward effects of cocaine were also reported. However, the delayed effects of repeated SalA at 24 h were also shown to increase the rewarding impact of ICSS (Potter et al., 2011). Acute activation of the KOP receptor system is widely accepted to have anticocaine effects (Schenk and Partridge, 1999; Schenk et al., 1999; Morani et al., 2009; Potter et al., 2011); in contrast to delayed (Potter et al., 2011) or chronic exposure which shows KOP receptor agonists may potentiate the rewarding effects of drugs by activating stress, anxiety systems or negative emotional states (McLaughlin et al., 2003; Koob and Le Moal, 2008; Wee et al., 2009; Koob et al., 2014). It is worth noting that studies on SalA have shown both pro- (Carlezon et al., 2006; Morani et al., 2009) and antidepressive effects (Braida et al., 2008; 2009; Harden et al., 2012), whereas, traditional KOP receptor agonists are consistently reported to be pro-depressive and aversive (Van't Veer and Carlezon, 2013).
SalA has been shown previously to decrease dopamine levels in ventral (Carlezon et al., 2006) and dStr (Chartoff et al., 2008; Gehrke et al., 2008) and modulate cocaine-induced locomotor behaviours by interacting with dopamine D1 receptor signalling in the dStr (Chartoff et al., 2008). In the present study, SalA (Morani et al., 2009) and Mesyl Sal B attenuated cocaine seeking via KOP receptor activation. Cocaine seeking has been attributed to dopaminergic mechanisms (Schmidt et al., 2005), and KOP receptor activation has previously been shown to modulate dopamine neurotransmission (Thompson et al., 2000). Therefore, the ability of SalA and Mesyl Sal B to modulate dopamine neurotransmission is a possible mechanism underlying their anticocaine effects. It is well known that endogenous KOP receptor activation and administration of KOP receptor agonists attenuate dopamine release (Chefer et al., 1999; 2000; Gray et al., 1999; Thompson et al., 2000; Carlezon et al., 2006). Ultrastructural studies by Svingos et al. (2001) showed that KOP receptors and dopamine transporters are colocalized in the NAcb, identifying an additional mechanism whereby activation of KOP receptors may regulate cocaine-induced behaviours. In the current study, we show that Mesyl Sal B, by activation of KOP receptors, increased DAT function in cells and brain tissue, similar to SalA and the traditional KOP receptor agonist U50,488H.
Many studies have shown differences in dopamine regulation between the NAcb and dStr (Roitman et al., 1999; Wu et al., 2001; Richards and Zahniser, 2009), and there is evidence that the regulation of DAT by KOP receptors is different between these brain regions (Chefer et al., 1999; Thompson et al., 2000). In rats, ex vivo RDEV experiments showed increased dopamine uptake in the ventral but not dStr following acute exposure to U69,593 (Thompson et al., 2000). Here, we are the first to show that Mesyl Sal B rapidly increases DAT function in HEK-293, N2A and COS7 cells and in rat brain tissue taken from the dStr, prefrontal cortex and NAcb. Because these effects are seen in vitro, and in multiple neuronal and non-neuronal cell lines, it is unlikely that intact brain circuitry is required for these effects within individual cells. The differences between the study by Thompson et al. (2000) using U69,593 and the current study with Mesyl Sal B and U50,488H are that we observed increases in DAT function in all tissue coexpressing DAT and KOP receptors. These differences are likely to reflect our use of tissue treated in vitro rather than ex vivo. Quantitative microdialysis experiments in mice have shown no change in dopamine uptake in vivo in the dStr following SalA (Gehrke et al., 2008) nor in the NAcb in rats using fast-scan cyclic voltammetry (Ebner et al., 2010). Further investigations are needed to determine if a direct interaction between KOP receptors and DAT exists in vivo.
It is established that SalA activates ERK1/2 rapidly at 15 min and repeated SalA activates ERK1/2 and CREB in the NAcb (Potter et al., 2011). Here, we show that acute Mesyl Sal B administration rapidly and transiently activates ERK1/2 in vitro at 3 and 5 min and at 10–15 min in the NAcb, dStr and prefrontal cortex of the rat ex vivo. No change in late phase ERK1/2 activation was seen with Mesyl Sal B (Figure 6). Early phase ERK1/2 activation by KOP receptors is G-protein mediated and β-arrestin independent; whereas, late activation (2 h) is β-arrestin dependent (McLennan et al., 2008). In astrocytes, U69,593 has been shown to cause both early- and late-phase ERK1/2 activation in contrast to the SalA analogue Mom Sal B, which only shows early phase ERK1/2 activation (McLennan et al., 2008). The lack of late-phase ERK1/2 activation at 60 and 120 min with Mesyl Sal B (Figure 6) and SalA analogue Mom Sal B is in contrast to U69,593 which shows both early- and late-phase activation (McLennan et al., 2008). Findings presented in this study support the proposal that SalA analogues possess different signalling properties to traditional KOP receptor agonists. Further studies into the recruitment of β-arrestin and p-38 MAPK activation are warranted to evaluate the ligand bias of these novel SalA analogues.
Differential ERK1/2 activation between brain regions may explain reported differences in dopamine regulation seen in previous studies. We show in the current study utilizing ASP+ uptake methods and RDEV that Mesyl Sal B activation of DAT is dependent on rapid ERK1/2 phosphorylation. Pre-incubation with the MEK inhibitor U0126 prevented the Mesyl Sal B induced increase in DAT function in vitro (Figure 5E) and in the dStr ex vivo (Figure 7). The effects seen with Mesyl Sal B are consistent with the effects of SalA and U69,593.
A recent study by Zhou et al. (2013) identified two low MW KOP receptor agonist scaffolds that displayed potent G-protein activation and weak β-arrestin-2 recruitment (Zhou et al., 2013). Recruitment of β-arrestin is associated with the dysphoric effects of KOP receptor agonists and requires late-phase activation of ERK1/2 (McLennan et al., 2008; Al-Hasani and Bruchas, 2011). It has also been shown that p-38 MAPK activation in the dorsal raphe is required for KOP receptor-mediated dysphoria (Land et al., 2009). Another KOP receptor ligand, 6'-guanidinonaltrindole, was shown to activate G-protein coupling to KOP receptors without β-arrestin-2 recruitment in transfected cells and activated Akt but not ERK1/2 in striatal neurons (Schmid et al., 2013). There is growing support for research into the identification of biased ligands for the development of compounds with the desired therapeutic effects and fewer side effects (Melief et al., 2010; Chavkin, 2011; Cox et al., 2015; Kivell et al., 2014; Thompson et al., 2015).
Both Mesyl Sal B and U50,488H increase DAT function in vitro in tissue from the rat NAcb (Figure 7). This effect is due to an increased Vmax with no change in Km. An increase in Vmax is suggestive of an increase in cell-surface DAT or functional DAT on the cell surface. TIRFM and cell-surface biotinylation techniques have been used previously to investigate changes in cell-surface levels of transporters for dopamine (DAT) or for 5-HT (SERT) (Bolan et al., 2007; Zapata et al., 2007; Furman et al., 2009; Kivell et al., 2010; Perry et al., 2010). Utilization of these two complementary techniques has identified differences between Mesyl Sal B, SalA and U69,593. We show that Mesyl Sal B had no effect on cell-surface DAT using biotinylation or TIRFM. SalA and U69,593 both increase cell-surface DAT. This suggests that Mesyl Sal B has a different mechanism of action in its modulation of DAT. It should also be noted that the effects on cell-surface expression of DAT with SalA and U69,593 only occur at around 30 min and were not observed at the earlier rapid time points when increases in DAT function are observed, suggesting other mechanisms are also likely to play a role. Despite this, it is clear that differences between Mesyl Sal B, SalA and classic agonists exist. Differences between SalA and traditional KOP receptor agonists have been reported in the literature previously. For example, SalA caused 40% less KOP receptor internalization compared with U50,488H in CHO cells (Wang et al., 2005).
The interaction between KOP receptor activation and DAT is a new potential target for the development of therapeutic drugs to treat addiction, particularly if differences between classic agonists and novel agonists can be identified. The longer duration of action of Mesyl Sal B and the demonstration of anticocaine effects when administered acutely has identified Mesyl Sal B as a SalA analogue with improved pharmacokinetic properties that warrants further investigation into potential therapeutic utility.
Acknowledgments
This work was supported by grants from the Health Research Council of New Zealand, the Neurological Foundation of New Zealand, Wellington Medical Research Foundation and Lottery Health (to BK) and the National Institutes of Health, USA (NIDA 018151 to T. E. P.). A. W. M. E., A. S. M. and B.S. received studentship awards from Victoria University of Wellington, New Zealand. The authors thank Dr Peter Bosch for technical assistance and Prof. Susan Schenk, Mr Caleb Carati and Mr Alex Crowther for their assistance with self-administration experiments.
Glossary
Abbreviations
- ASP+
4-4(dimethylamino)-styrl)-N-methylpyridinium
- CPP
conditioned place preference
- DAT
dopamine transporter
- dStr
dorsal striatum
- ICSS
intracranial self-stimulation
- Mesyl Sal B
(2S,4aR,6aR,7R,9S,10aS,10bR)-9-(methanesulfonyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naptho[2,1-c]pyran-7-carboxylic acid methyl ester
- MPE
maximum possible effect
- NAcb
nucleus accumbens
- norBNI
norbinaltorphimine
- RDEV
rotating disk electrode voltammetry
- SalA
salvinorin A
- TIRFM
total internal reflective fluorescence microscopy
- U50,488H
(trans-(±)-3,4-dichloro-N-methyl-N(2-[1-pyrrolidinyl]-cyclohexyl) benzenacetamide methanesulfonate
- U69,593
(5α,7α, 8β)-(+)-N-methyl-N(7-(1-pyrrolidinly)-1-oxaspiro(4,5)dec-8-yl)benzeneacetamide
Author contribution
B. M. K. designed the study, contributed to analysis of data and wrote the paper. B. S. performed the experiments, data analysis and wrote the paper. A. W. M. E., L. W., A. S. M. and N. K. performed the research and analysed the data. T. E. P. and D. S. provided salvinorin A and Mesyl Sal B for the study, and T. E. P. provided input into the design of the study and critical review of the manuscript. J. H. M. provided critical input into the writing of the manuscript and analysis of data.
Conflict of interest
The authors have no competing interests to disclose.
References
- Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, et al. Synthesis and in vitro evaluation of salvinorin A analogues: effect of configuration at C(2) and substitution at C(18) Bioorg Med Chem Lett. 2006;16:4679–4685. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, et al. Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. 2005;280:27662–27669. doi: 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, et al. D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol Pharmacol. 2007;71:1222–1232. doi: 10.1124/mol.106.027763. [DOI] [PubMed] [Google Scholar]
- Braida D, Limonta V, Capurro V, Fadda P, Rubino T, Mascia P, et al. Involvement of kappa-opioid and endocannabinoid system on Salvinorin A-induced reward. Biol Psychiatry. 2008;63:286–292. doi: 10.1016/j.biopsych.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Braida D, Capurro V, Zani A, Rubino T, Vigano D, Parolaro D, et al. Potential anxiolytic- and antidepressant-like effects of salvinorin A, the main active ingredient of Salvia divinorum, in rodents. Br J Pharmacol. 2009;157:844–853. doi: 10.1111/j.1476-5381.2009.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, et al. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Xu M, Chavkin C. Repeated swim stress induces kappa opioid-mediated activation of extracellular signal-regulated kinase 1/2. Neuroreport. 2008;19:1417–1422. doi: 10.1097/WNR.0b013e32830dd655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelman ER, Mandau M, Tidgewell K, Prisinzano TE, Yuferov V, Kreek MJ. Effects of salvinorin A, a kappa-opioid hallucinogen, on a neuroendocrine biomarker assay in nonhuman primates with high kappa-receptor homology to humans. J Pharmacol Exp Ther. 2007;320:300–306. doi: 10.1124/jpet.106.112417. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Prisinzano TE, Deng H, Rus S, Kreek MJ. Unconditioned behavioral effects of the powerful kappa-opioid hallucinogen salvinorin A in nonhuman primates: fast onset and entry into cerebrospinal fluid. J Pharmacol Exp Ther. 2009;328:588–597. doi: 10.1124/jpet.108.145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Beguin C, Dinieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Chartoff EH, Potter D, Damez-Werno D, Cohen BM, Carlezon WA., JR Exposure to the selective kappa-opioid receptor agonist salvinorin A modulates the behavioral and molecular effects of cocaine in rats. Neuropsychopharmacology. 2008;33:2676–2687. doi: 10.1038/sj.npp.1301659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C. The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacology. 2011;36:369–370. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, et al. Salvinorin A, an active component of the hallucinogenic sage Salvia divinorum is a highly efficacious kappa-opioid receptor agonist: structural and functional considerations. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- Chefer V, Thompson AC, Shippenberg TS. Modulation of cocaine-induced sensitization by kappa-opioid receptor agonists. Role of the nucleus accumbens and medial prefrontal cortex. Ann N Y Acad Sci. 1999;877:803–806. doi: 10.1111/j.1749-6632.1999.tb09327.x. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Moron JA, Hope B, Rea W, Shippenberg TS. Kappa-opioid receptor activation prevents alterations in mesocortical dopamine neurotransmission that occur during abstinence from cocaine. Neuroscience. 2000;101:619–627. doi: 10.1016/s0306-4522(00)00417-6. [DOI] [PubMed] [Google Scholar]
- Cox BM, Christie MJ, Devi L, Toll L, Traynor JR. Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br J Pharmacol. 2015;172:317–323. doi: 10.1111/bph.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson K, Putt F, Truman P, Kivell BM. The effects of nicotine and tobacco particulate matter on dopamine uptake in the rat brain. Synapse. 2014;68:45–60. doi: 10.1002/syn.21715. [DOI] [PubMed] [Google Scholar]
- Dworkin S, Stairs D. Self-administration of drugs of abuse. In: Waterhouse BD, editor. Methods in Drug Abuse Research: Cellular and Circuit Level Analyses. Boca Raton: CRC Press; 2002. pp. 17–49. [Google Scholar]
- Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology (Berl) 2010;210:241–252. doi: 10.1007/s00213-010-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman CA, Chen R, Guptaroy B, Zhang M, Holz RW, Gnegy M. Dopamine and amphetamine rapidly increase dopamine transporter trafficking to the surface: live-cell imaging using total internal reflection fluorescence microscopy. J Neurosci. 2009;29:3328–3336. doi: 10.1523/JNEUROSCI.5386-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke BJ, Chefer VI, Shippenberg TS. Effects of acute and repeated administration of salvinorin A on dopamine function in the rat dorsal striatum. Psychopharmacology (Berl) 2008;197:509–517. doi: 10.1007/s00213-007-1067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray AM, Rawls SM, Shippenberg TS, McGinty JF. The kappa-opioid agonist, U-69593, decreases acute amphetamine-evoked behaviors and calcium-dependent dialysate levels of dopamine and glutamate in the ventral striatum. J Neurochem. 1999;73:1066–1074. doi: 10.1046/j.1471-4159.1999.0731066.x. [DOI] [PubMed] [Google Scholar]
- Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013;77:942–954. doi: 10.1016/j.neuron.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, et al. An opioid agonist that does not induce mu-opioid receptor – arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden MT, Smith SE, Niehoff JA, McCurdy CR, Taylor GT. Antidepressive effects of the kappa-opioid receptor agonist salvinorin A in a rat model of anhedonia. Behav Pharmacol. 2012;23:710–715. doi: 10.1097/FBP.0b013e3283586189. [DOI] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, et al. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- John TF, French LG, Erlichman JS. The antinociceptive effect of salvinorin A in mice. Eur J Pharmacol. 2006;545:129–133. doi: 10.1016/j.ejphar.2006.06.077. [DOI] [PubMed] [Google Scholar]
- Johnson MW, Maclean KA, Reissig CJ, Prisinzano TE, Griffiths RR. Human psychopharmacology and dose-effects of salvinorin A, a kappa opioid agonist hallucinogen present in the plant Salvia divinorum. Drug Alcohol Depend. 2011;115:150–155. doi: 10.1016/j.drugalcdep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane BE, McCurdy CR, Ferguson DM. Toward a structure-based model of salvinorin A recognition of the kappa-opioid receptor. J Med Chem. 2008;51:1824–1830. doi: 10.1021/jm701040v. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell B, Day D, Bosch P, Schenk S, Miller J. MDMA causes a redistribution of serotonin transporter from the cell surface to the intracellular compartment by a mechanism independent of phospho-p38-mitogen activated protein kinase activation. Neuroscience. 2010;168:82–95. doi: 10.1016/j.neuroscience.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Kivell BM, Ewald AW, Prisinzano TE. Salvinorin A analogs and other kappa-opioid receptor compounds as treatments for cocaine abuse. Adv Pharmacol. 2014;69:481–511. doi: 10.1016/B978-0-12-420118-7.00012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiotek EK, Baimel C, Gill KJ. Methods for intravenous self administration in a mouse model. J Vis Exp. 2012 doi: 10.3791/3739. doi: 10.3791/3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, et al. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76((Pt B)):370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, et al. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell KM, Prevatt-Smith KM, Lozama A, Prisinzano TE. Synthesis of neoclerodane diterpenes and their pharmacological effects. Top Curr Chem. 2011;299:141–185. doi: 10.1007/128_2010_82. [DOI] [PubMed] [Google Scholar]
- McCurdy CR, Sufka KJ, Smith GH, Warnick JE, Nieto MJ. Antinociceptive profile of salvinorin A, a structurally unique kappa opioid receptor agonist. Pharmacol Biochem Behav. 2006;83:109–113. doi: 10.1016/j.pbb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan GP, Kiss A, Miyatake M, Belcheva MM, Chambers KT, Pozek JJ, et al. Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem. 2008;107:1753–1765. doi: 10.1111/j.1471-4159.2008.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiergerd SM, Schenk JO. Striatal transporter for dopamine: catechol structure-activity studies and susceptibility to chemical modification. J Neurochem. 1994;62:998–1008. doi: 10.1046/j.1471-4159.1994.62030998.x. [DOI] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A. 2010;107:11608–11613. doi: 10.1073/pnas.1000751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello NK, Negus SS. Effects of kappa opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther. 1998;286:812–824. [PubMed] [Google Scholar]
- Morani AS, Kivell B, Prisinzano TE, Schenk S. Effect of kappa-opioid receptor agonists U69593, U50488H, spiradoline and salvinorin A on cocaine-induced drug-seeking in rats. Pharmacol Biochem Behav. 2009;94:244–249. doi: 10.1016/j.pbb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani AS, Schenk S, Prisinzano TE, Kivell BM. A single injection of a novel kappa opioid receptor agonist salvinorin A attenuates the expression of cocaine-induced behavioral sensitization in rats. Behav Pharmacol. 2012;23:162–170. doi: 10.1097/FBP.0b013e3283512c1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani AS, Ewald A, Prevatt-Smith KM, Prisinzano TE, Kivell BM. The 2-methoxy methyl analogue of salvinorin A attenuates cocaine-induced drug seeking and sucrose reinforcements in rats. Eur J Pharmacol. 2013;720:69–76. doi: 10.1016/j.ejphar.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moron JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM, et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J Neurosci. 2003;23:8480–8488. doi: 10.1523/JNEUROSCI.23-24-08480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowry M, Mosher M, Briner W. Acute physiologic and chronic histologic changes in rats and mice exposed to the unique hallucinogen salvinorin A. J Psychoactive Drugs. 2003;35:379–382. doi: 10.1080/02791072.2003.10400021. [DOI] [PubMed] [Google Scholar]
- Muschamp JW, Van't Veer A, Carlezon WA., JR Tracking down the molecular substrates of stress: new roles for p38alpha MAPK and kappa-opioid receptors. Neuron. 2011;71:383–385. doi: 10.1016/j.neuron.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of kappa opioids on cocaine self-administration by rhesus monkeys. J Pharmacol Exp Ther. 1997;282:44–55. [PubMed] [Google Scholar]
- Perry SW, Barbieri J, Tong N, Polesskaya O, Pudasaini S, Stout A, et al. Human immunodeficiency virus-1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and Vmax. J Neurosci. 2010;30:14153–14164. doi: 10.1523/JNEUROSCI.1042-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter DN, Damez-Werno D, Carlezon WA, Jr, Cohen BM, Chartoff EH. Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry. 2011;70:744–753. doi: 10.1016/j.biopsych.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlock SL, Schenk JO. A multisubstrate kinetic mechanism of dopamine transport in the nucleus accumbens and its inhibition by cocaine. J Neurochem. 1997;69:1093–1105. doi: 10.1046/j.1471-4159.1997.69031093.x. [DOI] [PubMed] [Google Scholar]
- Prevatt-Smith KM, Prisinzano TE. New therapeutic potential for psychoactive natural products. Nat Prod Rep. 2010;27:23–31. doi: 10.1039/b912196j. [DOI] [PubMed] [Google Scholar]
- Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, et al. Potential drug abuse therapeutics derived from the hallucinogenic natural product salvinorin A. Medchemcomm. 2011;2:1217–1222. doi: 10.1039/C1MD00192B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sci. 2005;78:527–531. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Prisinzano TE, Rothman RB. Salvinorin A analogs as probes in opioid pharmacology. Chem Rev. 2008;108:1732–1743. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, et al. Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist salvinorin A in humans. Biol Psychiatry. 2012;72:871–879. doi: 10.1016/j.biopsych.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TL, Zahniser NR. Rapid substrate-induced down-regulation in function and surface localization of dopamine transporters: rat dorsal striatum versus nucleus accumbens. J Neurochem. 2009;108:1575–1584. doi: 10.1111/j.1471-4159.2009.05910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Patterson TA, Sakai RR, Bernstein IL, Figlewicz DP. Sodium depletion and aldosterone decrease dopamine transporter activity in nucleus accumbens but not striatum. Am J Physiol. 1999;276:R1339–R1345. doi: 10.1152/ajpregu.1999.276.5.R1339. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, et al. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S, Partridge B. Cocaine-seeking produced by experimenter-administered drug injections: dose-effect relationships in rats. Psychopharmacology (Berl) 1999;147:285–290. doi: 10.1007/s002130051169. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B. Sensitization to cocaine's reinforcing effects produced by various cocaine pretreatment regimens in rats. Pharmacol Biochem Behav. 2000;66:765–770. doi: 10.1016/s0091-3057(00)00273-2. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B, Shippenberg TS. U69593, a kappa-opioid agonist, decreases cocaine self-administration and decreases cocaine-produced drug-seeking. Psychopharmacology (Berl) 1999;144:339–346. doi: 10.1007/s002130051016. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B, Shippenberg TS. Reinstatement of extinguished drug-taking behavior in rats: effect of the kappa-opioid receptor agonist, U69593. Psychopharmacology (Berl) 2000;151:85–90. doi: 10.1007/s002130000476. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. Functional selectivity of 6'-guanidinonaltrindole (6'-GNTI) at kappa-opioid receptors in striatal neurons. J Biol Chem. 2013;288:22387–22398. doi: 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Anderson SM, Famous KR, Kumaresan V, Pierce RC. Anatomy and pharmacology of cocaine priming-induced reinstatement of drug seeking. Eur J Pharmacol. 2005;526:65–76. doi: 10.1016/j.ejphar.2005.09.068. [DOI] [PubMed] [Google Scholar]
- Schwartz JW, Blakely RD, Defelice LJ. Binding and transport in norepinephrine transporters. Real-time, spatially resolved analysis in single cells using a fluorescent substrate. J Biol Chem. 2003;278:9768–9777. doi: 10.1074/jbc.M209824200. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A, et al. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 1998;17:886–897. doi: 10.1093/emboj/17.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- Spangler R, Zhou Y, Maggos CE, Schlussman SD, Ho A, Kreek MJ. Prodynorphin, proenkephalin and kappa opioid receptor mRNA responses to acute ‘binge’ cocaine. Brain Res Mol Brain Res. 1997;44:139–142. doi: 10.1016/s0169-328x(96)00249-5. [DOI] [PubMed] [Google Scholar]
- Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology (Berl) 2010;210:199–209. doi: 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- Su D, Riley J, Armstead WM, Liu R. Salvinorin A pretreatment preserves cerebrovascular autoregulation after brain hypoxic/ischemic injury via extracellular signal-regulated kinase/mitogen-activated protein kinase in piglets. Anesth Analg. 2012;114:200–204. doi: 10.1213/ANE.0b013e31823a5d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Chavkin C, Colago EE, Pickel VM. Major coexpression of kappa-opioid receptors and the dopamine transporter in nucleus accumbens axonal profiles. Synapse. 2001;42:185–192. doi: 10.1002/syn.10005. [DOI] [PubMed] [Google Scholar]
- Teksin ZS, Lee IJ, Nemieboka NN, Othman AA, Upreti VV, Hassan HE, et al. Evaluation of the transport, in vitro metabolism and pharmacokinetics of Salvinorin A, a potent hallucinogen. Eur J Pharm Biopharm. 2009;72:471–477. doi: 10.1016/j.ejpb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. Kappa-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GL, Kelly E, Christopoulos A, Canals M. Novel GPCR paradigms at the mu-opioid receptor. Br J Pharmacol. 2015;172:287–296. doi: 10.1111/bph.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Mitchell JM, Besson MJ, Caboche J, Maldonado R. Behavioural and biochemical evidence for interactions between Delta 9-tetrahydrocannabinol and nicotine. Br J Pharmacol. 2002;135:564–578. doi: 10.1038/sj.bjp.0704479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van't Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, et al. Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther. 2005;312:220–230. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Orio L, Ghirmai S, Cashman JR, Koob G. Inhibition of kappa opioid receptors attenuated increased cocaine intake in rats with extended access to cocaine. Psychopharmacology (Berl) 2009;205:565–575. doi: 10.1007/s00213-009-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Kuhar MJ, Carroll FI, Garris PA. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J Neurosci. 2001;21:6338–6347. doi: 10.1523/JNEUROSCI.21-16-06338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa K, Narita M, Saeki M, Narita M, Isotani K, Horiuchi H, et al. Activation of extracellular signal-regulated kinase is critical for the discriminative stimulus effects induced by U-50,488H. Synapse. 2011;65:1052–1061. doi: 10.1002/syn.20937. [DOI] [PubMed] [Google Scholar]
- Zapata A, Kivell B, Han Y, Javitch JA, Bolan EA, Kuraguntla D, et al. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors. J Biol Chem. 2007;282:35842–35854. doi: 10.1074/jbc.M611758200. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, et al. Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem. 2013;288:36703–36716. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]