

Abstract
BACKGROUND AND PURPOSE
Opioid antagonists, such as naloxone and naltrexone, exhibit agonistic properties at the mutated μ receptor, MOR-S196ACSTA. In our previous study, systemic naloxone (10 mg·kg−1, s.c.) elicited antinociceptive effect without the induction of tolerance, dependence or rewarding effect in mice 2 weeks after intrathecal administration of double-stranded adeno-associated virus-MOR-S196ACSTA-eGFP. Here, we have investigated if this antinociceptive paradigm would be effective in a mouse model of neuropathic pain.
EXPERIMENTAL APPROACH
Spinal nerves were ligated in male C57BL/6 mice 3 or 4 weeks after intrathecal injection of the lentivirus encoding the construct of MOR-S196ACSTA-eGFP (LV-MOR-S196ACSTA). Anti-allodynic effects of daily s.c.injections of saline, naltrexone (10 mg·kg−1) or morphine (10 mg·kg−1) were assessed by the von Frey test. After 14 days of treatment with saline, naltrexone or morphine, signs of natural withdrawal were measured at 22 and 46 h after the last injection. To determine the rewarding effects induced by morphine or naltrexone, the conditioned place preference test was carried out.
KEY RESULTS
Anti-allodynic effects, as measured by von Frey test, increased after naltrexone or morphine treatment in mice transfected with LV-MOR-S196ACSTA in the spinal cord. Cessation of treatment with morphine, but not naltrexone, induced natural withdrawal and rewarding effects.
CONCLUSIONS AND IMPLICATIONS
Systemic injection of naltrexone after the expression of a mutant μ opioid receptor, MOR-S196ACSTA, in the spinal cord may have therapeutic potential for chronic neuropathic pain, without the development of dependence or addiction.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Tables of Links
| TARGETS |
|---|
| GPCRa |
| MOP receptor, μ opioid receptor |
| Catalytic receptorb |
| TLR4, Toll-like receptor 4 |
| LIGANDS |
|---|
| Morphine |
| Naloxone |
| Naltrexone |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
The opioid agonists such as morphine activate μ opioid (MOP) receptors (Cox et al., 2015), resulting in strong antinociceptive responses. These agents have been used in the control of moderate and severe postoperative pain for decades. However, they also induce many adverse effects, such as respiratory depression, nausea, vomiting, constipation, tolerance and dependence. Although non-opioid pharmacological strategies (such as the use of laxative or bulking agents that counteract opioid constipation, and dopaminergic or 5-HT receptor antagonists that prevent the nausea and vomiting commonly brought on by opioid analgesics) are used together with opioid administration to alleviate such side effects while maintaining the analgesic potency of the opioids, these drugs do not prevent the development of tolerance and addiction to opioid analgesics during long-term administration.
Opioid receptors are members of the rhodopsin subfamily of GPCRs (Dhawan et al., 1996). There are three pharmacologically distinct types of opioid receptor: μ, δ and κ. The opioid receptors share 73–78% amino acid identity within the transmembrane domain (TM) regions, as compared with a 60% overall amino acid identity (Minami and Satoh, 1995). In the chimeric receptor construct μδ2 – in which the nucleotide sequence of the δ opioid receptor-1 from the N-terminus up to the first extracellular loop is replaced with a similar sequence from the MOP receptor-1 – classical alkaloid antagonists, such as naloxone or naltrexone, behave as full agonists. Subsequently it was shown that antagonist activation at the μδ2 chimeric opioid receptor was due to the mutation of a conserved serine to leucine in TM4 (Claude et al., 1996). However, when the analogous serine to leucine or alanine substitution was made in the wild-type MOP receptor (i.e. MOR-S196L/A), opioid antagonists had only partial agonist activity (Claude et al., 1996; Yang et al., 2003). On the other hand, opioid receptor antagonists behaved as full agonists when the amino acids in TM7, Thr327 and Cys330 of the MOR-S196L were converted to Ala and Ser respectively (Claude-Geppert et al., 2005).
To develop the antagonist-mediated activation of mutant MOP receptors (MOR-S196A or MOR-S196ACSTA) as a pain-management paradigm, the double-stranded adeno-associated virus type 2 (dsAAV2) has been used as a vector to deliver the constructs of MOR-S196A or MOR-S196ACSTA into various sites within the pain pathway of mice. Naloxone elicited antinociceptive effect in mice injected with dsAAV2-MOR-S196A or dsAAV2-MOR-S196ACSTA directly into the dorsal horn of the spinal cord at the level of S2/S3 (Chen et al., 2007) or inot the periaqueductal grey (Chou et al., 2012). After subchronic naloxone treatment, no naloxone tolerance or dependence was observed in the dsAAV2-MOR-S196A- or MOR-S196ACSTA-injected mice (Chen et al., 2007; Chou et al., 2012). However, the exposure of the spinal cord segment after partial dorsal laminectomy during dsAAV2 injection greatly reduces the utility of this approach. A less invasive delivery method [i.e. intrathecal (i.t.) injection of the virus into the subarachnoid space of the spine] was used to express the transgene at the lumbar region of the spinal cord, with the retrograde transport into the dorsal root ganglion (DRG) neurons (Kao et al., 2010). Naloxone was found to elicit antinociceptive effect without the induction of tolerance or dependence in these mice also (Kao et al., 2010).
Although antagonist-mediated antinociceptive effects without tolerance or dependence have been observed in mice with the mutant MOP receptors (MOR-S196A or MOR-S196ACSTA) by tail-flick test or hot plate test, their effects on chronic pain have not been tested. In the current study, we used a chronic pain model in which neuropathic pain was induced by spinal nerve ligation (SNL), to investigate the anti-allodynia effect of chronic naltrexone treatment in mice injected i.t. with the lentivirus encoding the mutant MOP receptor construct. Our data suggest that systemic injection of naltrexone after the expression of MOR-S196ACSTA in the spinal cord may have therapeutic potential for chronic neuropathic pain without the development of dependence.
Methods
Construction of the lentiviral vector
The mutant receptor MOR-S196ACSTA with eGFP as reporter was cloned into pCDH plasmid containing a cytomegalovirus promoter (System Biosciences, Mountain View, CA, USA). Lentivirus was produced in HEK293TN cells (System Biosciences) by co-transfecting the pPACK packaging plasmid mix (System Biosciences) and the pCDH-MORS196ACSTA-eGFP. The culture medium containing viral particles were collected 48 or 72 h after transfection, and concentrated by PEG-it virus concentration solution (System Biosciences). The virus was titrated using Lenti-X qRT-PCR titration kit (Clontech Laboratories Inc., Mountain View, CA, USA). The titer of virus was determined to be 1.2 × 1012 copies per mL.
Animals
All animal care and experimental protocols complied with institutional and international standards (Principles of Laboratory Animal Care, National Institutes of Health), and were approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center (Taiwan, ROC). Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 80 animals were used in this study.
Adult male C57BL/6 mice (25–30 g; from BioLASCO Taiwan Co., Ltd, Yilan, Taiwan) were kept in an animal room with a 12 h light/dark cycle at a temperature of 25 ± 2°C and humidity of 55%. A standard diet and water were provided ad libitum. Each experimental group had at least eight mice.
Experimental design
Experiment 1
As shown in Figure 1A, mice were injected with LV-MOR-S196ACSTA-eGFP i.t. 4–5 weeks before SNL surgery. Before i.t. injection of virus, the antinociceptive effect of naltrexone (10 mg·kg−1, s.c.) was assessed by the tail-flick test as pretest data. The antinociceptive effects of naltrexone (10 mg·kg−1, s.c.) were tested once a week until 4–5 weeks after i.t. injection of virus. These tests were designed to determine whether the MOR-S196ACSTA-eGFP gene had been expressed enough to show its characteristics to convert the antagonistic property of naltrexone to an agonist. The animals that showed significant antinociceptive effects of naltrexone were used in subsequent experiments and divided into four groups of eight animals each as follow: sham-operated mice treated with saline; SNL-operated mice treated with saline; SNL-operated mice treated with naltrexone; SNL-operated mice treated with morphine. The basal paw withdrawal pressure was recorded by using the von Frey test before surgery or sham operation. The mice were then given a daily s.c. injection of saline (1 mL·kg−1), naltrexone (10 mg·kg−1), or morphine (10 mg·kg−1) for 14 days – days 6–19 after sham or SNL surgery. The basal paw withdrawal pressure was also recorded once a day on days 1, 3, and 5, and again on days 7, 9, 11, 13, 15, 17, and 19 before and after saline or drug injection to determine the anti-allodynia effects of the treatments. The withdrawal symptoms were assessed at 22 and 46 h after the last drug treatment and the mice were then killed humanely, with an overdose of chloral hydrate (350 mg/kg, i.p.).
Figure 1.
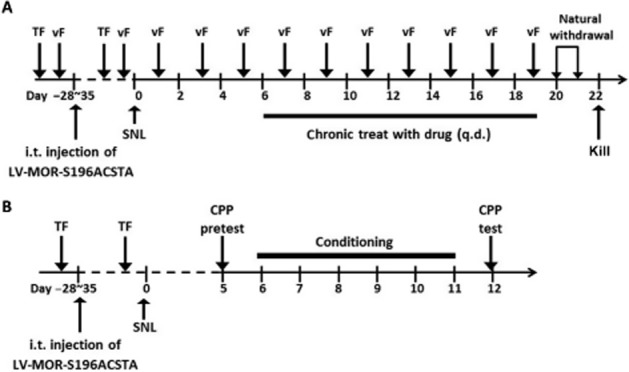
Experimental design. (A) Tests for anti-allodynia effects and physical dependence of drugs (B) Tests for rewarding effect of drugs. TF, tail-flick test; vF, von Frey test.
Experiment 2
To determine the rewarding effects induced by morphine or naltrexone in mice that had been transfected with MOR-S196ACSTA in the spinal cord, the conditioned place preference (CPP) test was carried out (Figure 1B). The tail-flick tests were first carried out before and after i.t. injection of LV-MOR-S196A-CSTA in mice to select those that responded to naltrexone and showed agonist antinociceptive effects after 3–4 weeks gene transfection. The selected mice were divided into three groups of eight animals each as follow: SNL-operated mice treated with saline; SNL-operated mice treated with naltrexone; and SNL-operated mice treated with morphine. SNL surgery was then performed on those selected mice. The mice were allowed to recover from the surgery for 4 days. Then, the CPP pretest was performed in the afternoon of day 5. After 6 days of conditioning (days 6–11), the CPP test was performed again in the afternoon of day 12 to determine the rewarding effects induced by the chronic drug treatment.
Injection of the lentivector into the subarachnoid space of the spinal cord
The direct lumbar puncture method (Wigdor and Wilcox, 1987; Fairbanks, 2003) was applied in conscious mice. Mice were covered with a soft cloth over the head and upper body, and held firmly by the pelvic girdle (iliac crest). Five μl of the virus was injected with a 50 μL Hamilton syringe (Hamilton, Reno, NV, USA) attached to a 30-G, 0.5 in. sterile disposable needle, which was inserted into the i.t. space at the cauda equina region according to the method described by Wigdor and Wilcox (1987). Puncture of the dura was indicated by a flick of the tail. In preliminary experiments with intrathecal injections using the substance P nociceptive assay (Fairbanks, 2003), the success rate of our intrathecal injections was higher than 90%. There was no paralysis caused by intrathecal injection in this study.
Determination of the antinociceptive effect of drugs
Drug-induced antinociception was evaluated by using the tail-flick test (D'Amour and Smith, 1941). Using a tail-flick apparatus (model: Ugo Basile Tail Flick Unit 37360; Ugo Basile S.R.L., Monvalle Varese, Italy), the intensity of the heat source was set at 85, which resulted in a basal tail-flick latency of between 2.0 and 3.0 s for most of the animals. The tail-flick latency was recorded at 30, 60, 90, 120 and 180 min after drug administration. The pre-drug latency was determined from an average of three pre-drug determinations, and cut-off latency was selected at 10 s. The areas under the latency-tme curve (AUCs) were calculated as (tail flick latency at each time point – baseline tail flick latency) × time, according to the linear trapezoidal rule (Figure 3B) without extrapolation to infinite time.
Figure 3.
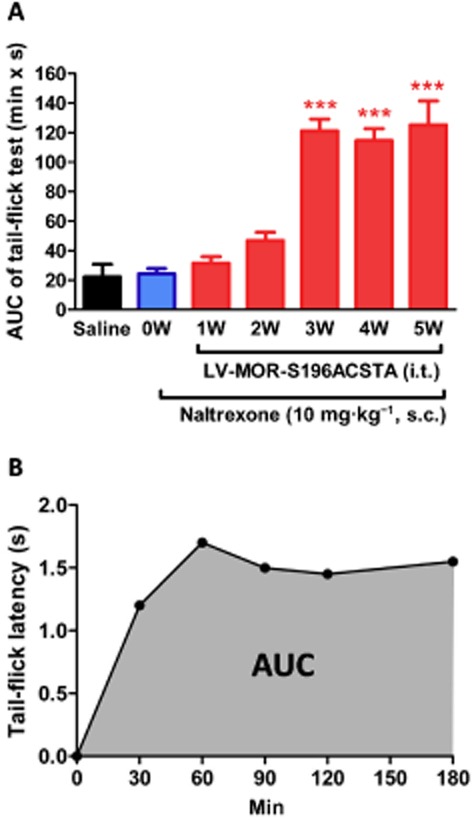
The development of the antinociceptive effects of naltrexone (A) The time course of the developed antinociceptive effects of naltrexone before and after gene transfection (1–5 weeks). (B) A representative time-effect curve of naltrexone (10 mg·kg−1, s.c.) determined by the tail-flick test after transfection of the MOR-S196ACSTA-eGFP gene into the spinal cord of C57BL/6J mice for 3 weeks (3 W). Data are presented as means ± SEM. One-way anova and Newman–Keuls test were used to analyse the data. ***P < 0.001, significantly different from pretest data of naltrexone, before gene transfection (0 W).
Detection of the expression of mutated MOP receptors
Mice were anaesthetized with chloral hydrate (350 mg·kg−1, i.p.) and perfused transcardially with Tyrode calcium-free buffer (NaCl 116 mM, KCl 5.36 mM, MgCl2 1.57 mM, MgSO4 0.405 mM, NaH2PO4 1.23 mM, glucose 5.55 mM, NaHCO3 26.2 mM, pH 7.4), followed by 4% paraformaldehyde in 0.1 M phosphate buffer (KH2PO4 187.5 mM, Na2HPO4 212.5 mM). The L3–L5 DRGs were dissected and then put in 10% sucrose solution followed by 20% and 30% sucrose solution at 4°C. The samples were then embedded in OCT compound and frozen immediately in a −80°C freezer. Serial DRG slices (10 μm) were sectioned with a cryostat at −20°C. The slices were mounted on SuperFrost Plus slides (Menzel-Glaser, Braunschweig, Germany), and the eGFP expression of the mutated MOP receptors was visualized with a fluorescence microscope. In order to investigate whether these mutated MOP receptors were co-localized with calcitonin gene-related peptide (CGRP), a well-characterized marker of the DRG sensory neurons, immunohistochemistry was carried out on the DRG at L3–L5 level. Antibodies were diluted in blocking buffer (0.01 M PBS containing 1% goat serum and 0.1% Triton X-100). Anti-CGRP antibody (AB5920; Chemicon, Temecula, CA, USA) was used at a dilution of 1:2000. Secondary antibody Alexa Fluuor® 568 (Invitrogen, Carlsbad, CA, USA) was diluted 1:400 to against rabbit or mouse IgG for immunofluorescence detection. Sections were then washed with PBS and mounted onto cover slips using mounting medium (HIS002B, Serotec, Oxford, UK). The processed sections were examined with Olympus fluorescence instruments (model: BH2-RFL-T3; Olympus, Tokyo, Japan) on an upright microscope and an Olympus BX50 camera with SPOT software (version 4.6; Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
SNL surgery
SNL was conducted based on a method previously described for rats (Kim and Chung, 1992). The mice were anaesthetized with pentobarbital (80 mg·kg−1, i.p.), and the hairs on their back were clipped. A midline incision above the lumbar spine exposed the left L6 transverse process. The transverse process was removed carefully with a small scraper. The left L5 spinal nerves were tightly ligated with 8-0 nylon thread, and the wound was sutured with 3-0 thread. In sham-operated mice, the surgical procedure was identical to that of the SNL group, except that the spinal nerves were not ligated. Mice were monitored until recovery from the anaesthetic after the surgery and a further 5 days were allowed for recovery before any tests were given. In this study, we did not use analgesics to relieve the pain induced by surgery because they would affect our assessment of experimental treatments on pain (allodynia) relief.
Measurement of mechanical allodynia
The mice were individually placed in suspended cages with wire mesh bottoms and allowed to acclimatise to their environment for at least 30 min. The mechanical stimulus was applied from underneath to the plantar aspect of the hind paw, with a gradual increase in pressure by means of an Electronic von Frey device (IITC Inc., Woodland Hills, CA, USA). The end point was characterized by the removal of the paw, followed by clear flinching movements. After paw withdrawal, the intensity of the pressure was automatically recorded. The test was carried out before (pretest) and 30 min after saline or morphine injection, or 1 h after naltrexone injection. The area under the time-paw pressure curve (AUC) was calculated as (|paw withdrawal pressure at each time point – baseline withdrawal pressure at day -1)| × time, according to the linear trapezoidal rule (Figure 4C).
Figure 4.
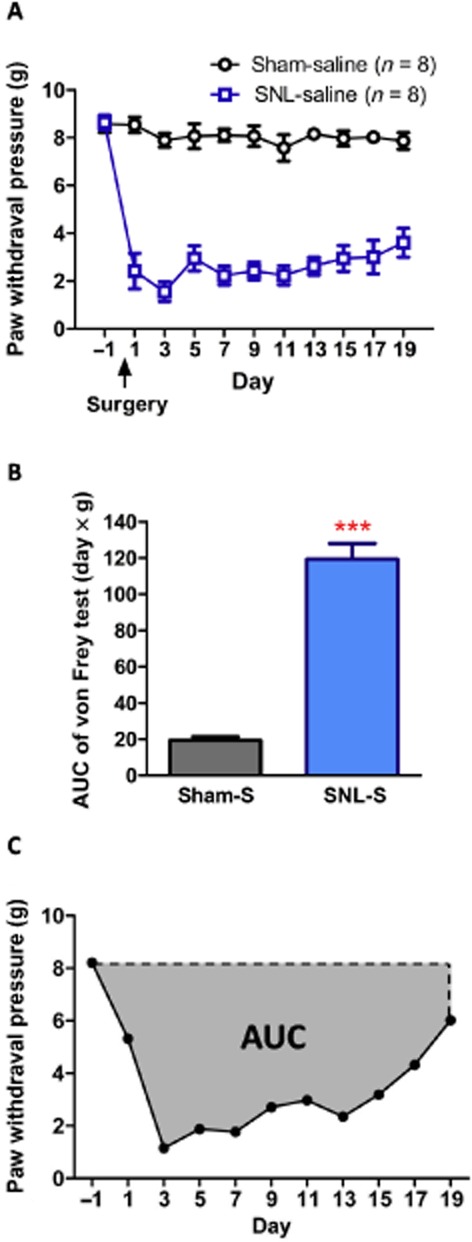
SNL induced mechanical allodynia, but sham operation did not alter mechanical paw withdrawal pressure at any time points tested from days 1 to 19. (A) Time course of paw withdrawal pressure and (B) corresponding AUC from days −1 to 19. (C) An example of AUC determination. Data shown are means ± SEM. ***P < 0.001, significantly different from sham-saline group; Student's t-test.
Assessment of the withdrawal symptoms
To assess reactions to the withdrawal of the treatments, each mouse was placed in a test chamber consisting of a transparent round plastic box (20 cm diameter, 33 cm height) for 30 min at 22 and 46 h after the last drug treatment. The withdrawal symptoms (jump, paw tremor, wet dog shaking, teeth chattering, diarrhoea, ptosis and piloerection) were counted. Jump and paw tremor frequencies were recorded during the test time, and a score of 1 was assigned to every three jumps or five paw tremors. Diarrhoea and ptosis events were recorded for every 5 min interval in which they occurred (maximal score = 6). The presence or absence of piloerection was noted. A global opiate withdrawal score was calculated by summing the values for each sign (Papaleo and Contarino, 2006).
Assessment of the rewarding effect of drugs
To determine the rewarding effects induced by morphine or naltrexone, the CPP test was carried out as reported previously (Chen et al., 2007; Kao et al., 2010; Chou et al., 2012). In this study, a distinctive environment was paired repeatedly with administration of a drug, and a different environment was associated with non-drugged state. The CPP test apparatus, made from an acrylic plastic box (33 × 15 × 15 cm), was divided into three compartments. Two identically sized compartments (15 × 15 × 15 cm) were constructed at both sides, separated by guillotine doors (7 × 3.5 cm) in the central unit. To provide a visual cue, one of the large compartments was covered by paper in a checkerboard pattern (3 × 3 cm black and white squares) on the three walls, while the other large compartment had its three walls covered by purely white paper. To give more visual cues, a blue and a red light bulb were hung separately above the two large compartments. During the experiments, the CPP apparatus was kept in an isolated dark room, which was free from noise. For CPP conditionings, the SNL mice (day 6 after SNL surgery) were given saline in the morning and saline (control group) or morphine (10 mg·kg−1, s.c.) or naltrexone (10 mg·kg−1, s.c.) in the afternoon for 6 days. A distinctive environment was paired repeatedly with administration of saline, and a different environment was associated with drug injection. The animals were kept for 40 min in the corresponding compartment with the guillotine doors closed. The CPP tests were performed on the day before conditioning (day 5 after the SNL surgery) and the day after conditioning (day 12 after the SNL surgery). The CPP test was done by placing the mice in the central compartment of the apparatus with the guillotine doors opened for 15 min. The time that the mice stayed in each compartment was recorded to determine the place preference. The measurement of drug rewarding effect was determined by an increase in the time spent in the compartment previously paired with drug injection over the time spent in the saline-paired compartment.
Data analysis
The data were expressed as means ± SEM. Student's t-test, one-way anova, and Newman–Keuls test were used to analyse the data.
Drugs
Naltrexone (Sigma, St. Louis, MO, USA), morphine (Pharmaceutical Plant of Controlled Drugs, Food and Drug Administration, Ministry of Health and Welfare, Taiwan), chloral hydrate (Sigma), pentobarbital (Sigma).
Results
Expression of the mutated MOP receptor in mice after i.t. injection of LV-MOR-S196ACSTA-eGFP
The expression of the transgene was monitored by the eGFP fluorescence after all the behavioural tests (about 8 weeks after i.t. injection of virus). The green fluorescence was observed in all sizes of cells in L3–L5 DRGs (Figure 2A). When the DRGs were stained with CGRP (a marker for nociceptive sensory neurons), [red colour, (Figure 2B) ], co-localizations of CGRP with the eGFP were seen in some of these DRG cells (Figure 2C).
Figure 2.
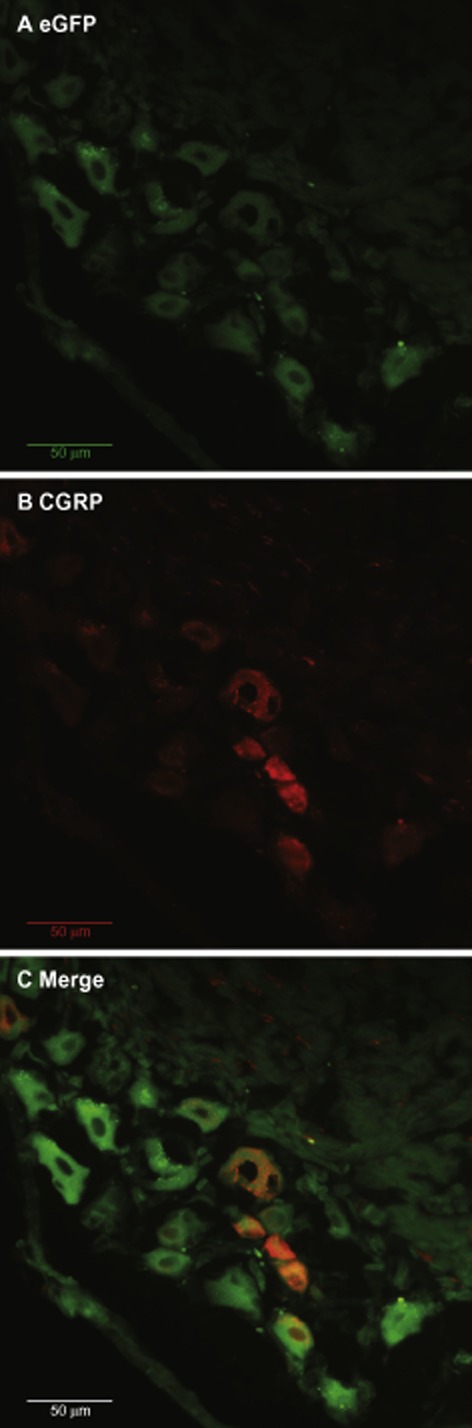
Representative fluorescence micrographs of the DRG after local i.t. injection of LV-MOR-S196ACSTA-eGFP into the spinal cord. (A) eGFP fluorescence (B) CGRP immunofluorescence (C) The merging of eGFP (green) with CGRP immunofluorescence (red) revealed co-localization (orange) of MOR-S196ACSTA and CGRP in the L5 DRG; magnification 40 ×. The scale bars represent 50 μm.
Naltrexone elicited antinociceptive responses in mice injected with LV-MOR-S196ACSTA i.t
As shown in Figure 3A, naltrexone (10 mg·kg−1, s.c.) did not elicit any antinociceptive effect before lentivirus injection. The AUC value after treatment with naltrexone alone was similar to that after treatment with saline. In contrast, starting from 3 weeks after i.t. administration of LV-MOR-S196ACSTA, 68% (19/28) of the mice responded to naltrexone (10 mg·kg−1, s.c.) with a markedly increased AUC value. The naltrexone-induced antinociceptive responses were unchanged when measured 4 or 5 weeks after the i.t. injection of the virus (Figure 3A). A time–effect curve of naltrexone-induced antinociceptive responses, 3 weeks after the virus injection is shown in Figure 3B.
Naltrexone elicited anti-allodynia effects in mice transfected with LV-MOR-S196ACSTA-eGFP i.t. and spinal nerve ligated
As shown in Figure 4, the ipsilateral paw withdrawal pressure was decreased 1 day after SNL surgery (P < 0.001). The SNL-induced allodynia persisted at least for 19 days. On the other hand, the sham operation did not alter the paw withdrawal pressure. The AUC of the time–response curve of the von Frey test from days −1 to 19 was calculated to represent the allodynia or anti-allodynia effect (Figure 4C). As shown in Figure 4B, the AUC of SNL-S group was much greater than that of the sham surgery group (P < 0.001). These results indicated that the allodynia of the ipsilateral hind paw was induced by the ligation of the L5 spinal nerve.
As shown in Figure 5A-1 and A-2, sham operation did not induce allodynia in either ipsilateral or contralateral hind paws; and saline had no effect in mice with sham operation in either ipsilateral or contralateral hind paws. SNL surgery induced significant allodynia on ipsilateral hind paws and persisted at least for 19 days (Figure 5B-1 and B-2). Saline treatment did not affect or improve the allodynia in mice that received SNL surgery (Figure 5B-1 and B-2). On the other hand, naltrexone (10 mg·kg−1, s.c.) (Figure 5C) and morphine (10 mg·kg−1, s.c.) (Figure 5D) both elicited significant anti-allodynia effects on ipsilateral hind paw in mice, which had been transfected with MOR-S196ACSTA in the spinal cord. The AUC was consequently decreased after naltrexone treatment (P < 0.01, Figure 5C-2), and after morphine treatment (P < 0.001, Figure 5D-2).
Figure 5.
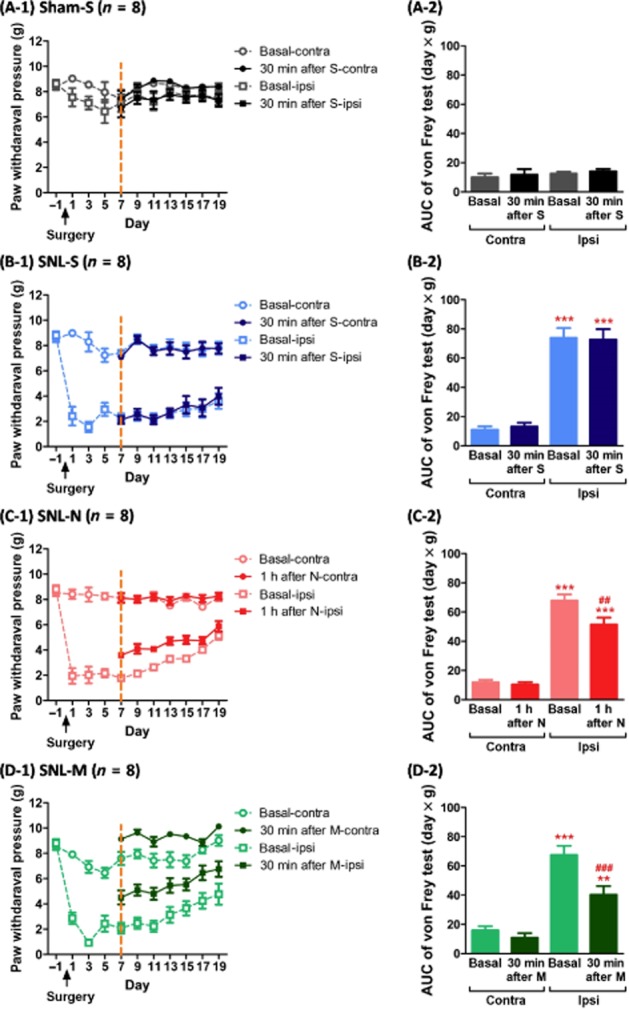
The anti-allodynia effects of naltrexone (10 mg·kg−1, s.c.) or morphine (10 mg·kg−1) on the ipsilateral (injured) or contralateral (uninjured) hind paw, determined by the von Frey test after sham or SNL surgery in mice which had been transfected with MOR-S196ACSTA in the spinal cord. Time course of (A-1) sham operation with the chronic saline treatment group, (B-1) SNL surgery with the chronic saline treatment group, (C-1) SNL surgery with the chronic naltrexone treatment group, and (D-1) SNL surgery with the chronic morphine treatment group. (A-2), (B-2), (C-2) and (D-2) are the corresponding AUC values (days × g) from days 7 to 19 of each group. Data shown are means ± SEM. **P < 0.01, ***P < 0.001, significantly different from basal level of contralateral site (basal-contra); ##P < 0.01, ###P < 0.001, significant effect of treatment; one-way anova and Newman–Keuls test.
When we compared the basal paw withdrawal pressure of the four groups (Figure 6A-1), we found there was no difference among the three SNL groups if we tested just before daily drug treatment in the morning and the corresponding AUC values were similar (Figure 6A-2). If we tested 30 min after daily drug treatment, the AUC of naltrexone group (SNL-N) was less than that of the saline-treated group (P < 0.01; Figure 6B-1 and B-2). The AUC of the morphine-treated group was also less than that of the saline-treated group (P < 0.001; Figure 6B-1 and B-2). In summary, both naltrexone (10 mg·kg−1, s.c.) and morphine (10 mg·kg−1, s.c.) had significant acute anti-allodynia effects in mice expressing MORS196ACSTA in the spinal cord.
Figure 6.
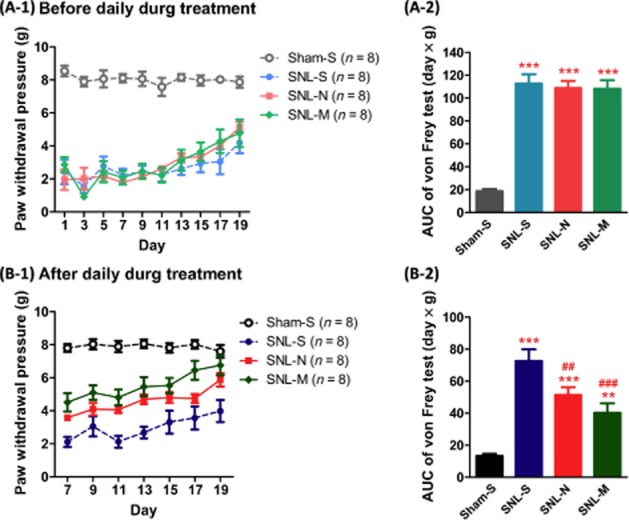
Effects of naltrexone or morphine on SNL-induced allodynia in mice which had been transfected with MOR-S196ACSTA in the spinal cord. The von Frey tests were recorded before (A-1) or after (B-1) the administration of saline, naltrexone (10 mg·kg−1, s.c.) or morphine (10 mg·kg−1, s.c.). (A-2), the corresponding AUC from days 1 to 19 of (A-1); (B-2), the corresponding AUC from days 7 to 19 of (B-1). Data shown are means ± SEM. **P < 0.01, ***P < 0.001, significantly different from sham-saline group; ##P < 0.01, ###P < 0.001, significantly different from SNL-saline group; one-way anova and Newman–Keuls test.
Chronic treatment with naltrexone did not elicit physical dependence or rewarding effect
The natural withdrawal symptoms were assessed in this study at 22 and 46 h after the last drug treatment (Figure 7). The individual scores for each withdrawal symptom are shown in the Supporting information Table S1. Significant withdrawal signs were shown in the morphine-treated group (SNL-M) but chronic naltrexone treatment did not induce signs of withdrawal at either 22 or 46 h after the last naltrexone treatment (Figure 7).
Figure 7.
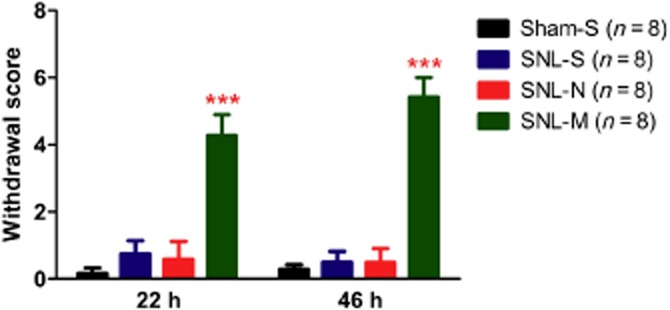
Morphine, but not naltrexone induced withdrawal symptoms in mice that had been transfected with MOR-S196ACSTA in the spinal cord and with SNL. Withdrawal symptoms were assessed for 30 min at 22 and 46 h after the last drug treatment (S, saline; N, naltrexone, M, morphine). Data shown are means ± SEM. ***P < 0.001, significantly different from SNL-saline; one-way anova and Newman–Keuls test.
The rewarding effects of saline, morphine and naltrexone in mice, which had been transfected with MOR-S196ACSTA in the spinal cord and spinal nerve ligated were determined by the CPP test (Figure 8). The time spent in the morphine-paired compartment was increased after conditioning for 6 days (post-test on day 12 vs. pretest on day 5, P < 0.001) in these mice. However, neither saline nor naltrexone (10 mg·kg−1, s.c.) treatment induced any rewarding effects in these mice (Figure 8).
Figure 8.
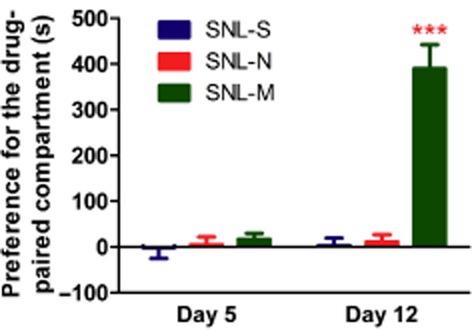
Morphine, but not naltrexone induced rewarding effects in mice that had been transfected with MOR-S196ACSTA in the spinal cord and with SNL. Data are expressed as preference for the drug-paired compartment as determined by time spent in the drug-paired compartment minus time spent in the saline-paired compartment. Data are presented as mean ± SEM (n = 8). ***P < 0.001, significantly different from preconditioning test on day 5; Student's paired t-test.
Discussion
Our current study showed that systemic administration of naltrexone could have anti-allodynia effects in a neuropathic pain model, without inducing physical dependence and rewarding effects in mice that had been transfected with MOR-S196ACSTA in the spinal cord.
In the current study, lentivirus rather than dsAAV2 was used as a vector to deliver the mutant MOP receptor construct into the spinal cord of mice. The results of our present study indicated that lentivirus was as effective in delivering the construct as the dsAAV2 (Kao et al., 2010). The expression of the transgene at DRG sensory neurons resulted in the opioid receptor antagonist naltrexone exerting agonist effects. The antinociceptive effects of naloxone in mice expressing mutant MOP receptors in lumbar spinal cord has been established in our lab (Kao et al., 2010). However, whether such a pain-control strategy can be applied to chronic pain was unknown. In the current study, we used a neuropathic pain model. Neuropathic pain is generally defined as a chronic pain state resulting from peripheral or central nerve injury because of acute events or systemic disease (Woolf and Mannion, 1999). Patients with neuropathic pain often suffer from spontaneous pain, allodynia (pain response to normal innocuous stimuli), and hyperalgesia (aggravated pain evoked by noxious stimuli) (Woolf and Mannion, 1999; Zimmermann, 2001). In addition, neuropathic pain may spread beyond the cutaneous distribution of the injured nerves and be expressed bilaterally in mirror image sites, suggesting the involvement of a central mechanism (Woolf, 1996). Commonly used neuropathic pain models in mice include partial sciatic nerve ligation, tight ligation of one-third or one-half of the sciatic nerve (Malmberg and Basbaum, 1998), and L5 SNL, tight ligation or transection of L5 spinal nerve (Ramer et al., 1998; Zwick et al., 2003). In this study, the L5 SNL was used to induce neuropathic pain. Marked mechanical allodynia was observed in mice after SNL surgery, but not after the sham surgery. This indicated that the allodynia is a direct result of the L5 spinal nerve injury.
In our preliminary study, we found that naltrexone treatment from day 1 reduced the survival rate to about 50%. On one hand, half of the mice died before day 6; the other half survived until the end of the study. On the other hand, all the mice treated with saline or morphine after sham or SNL surgery survived more than 17 days. Many studies have reported that β-endorphin in plasma and brain was increased when the experimental animals were exposed to nociceptive stimulus or when patients suffer from pain (Atkinson et al., 1983; Aloisi et al., 1995; Zangen et al., 1998). β-endorphin is spontaneously released in the brain in response to pain, thereby producing a natural analgesia (Zangen et al., 1998). Moreover, the endogenous opioid system is also a key factor in the stress response. Activation of the stress system by corticotropin-releasing hormone stimulates the secretion of hypothalamic β-endorphin and other pro-opiomelanocortin-derived peptides, which reciprocally inhibit the activity of the stress system (Merenlender-Wagner et al., 2009). To test whether the deaths of mice after naltrexone treatment started from day 1 after SNL surgery might have been the result of the inhibition of β-endorphin-activated opioid receptors by naltrexone, we analysed the plasma β-endorphin concentration of mice that underwent sham or SNL surgery and were treated with saline or naltrexone (10 mg·kg−1, s.c.). We found that the plasma β-endorphin concentration of the SNL-N group was significantly higher than that of the other three groups on days 3 and 5, as shown in the Supporting Information Fig. S1. These data indicate that the lower survival rate might be partly due to the blockade by naltrexone of the activation of opioid receptors, following release of endogenous β-endorphin in the brain. Therefore, in the present study, we initiated the treatment of the SNL-mice with saline, naltrexone, or morphine from day 6 instead of day 1 and found that there was no mortality during this experimental protocol.
Opioids possess strong antinociceptive effects on various types of pain related to abnormal physical condition; but certain types of pain are difficult to control with an opioid. For example, neuropathic pain, caused by nerve injury, does not respond effectively to morphine (Arner and Meyerson, 1988) at the normal dose. Possible reasons for the decreased effectiveness of morphine in neuropathic pain include decreased expression of MOP receptors in the spinal cord (Porreca et al., 1998) and DRG (Rashid et al., 2004), enhanced spinal release of dynorphin A (Nichols et al., 1997) and cholecystokinin (Nichols et al., 1996), increased expression of spinal metabotropic glutamate receptor 1 (Fundytus et al., 2001), and activation of tonic descending facilitation pathways from the brain (Vanderah et al., 2001). In this study, both morphine and naltrexone treatment at a high dose (10 mg·kg−1, s.c.) elicited anti-allodynia effects on ipsilateral paws of mice that had been transfected with MOR-S196ACSTA in the spinal cord and then with SNL. In addition, morphine also increased the mechanical pain threshold of the contralateral paw of mice (Figure 5D-1). Although morphine (10 mg·kg−1, s.c.) had a better antinociceptive effect than naltrexone (10 mg·kg−1, s.c.) for acute pain, as determined by tail-flick test in mice that had been transfected with MOR-S196ACSTA in the spinal cord (Kao et al., 2010), it showed no improvement over naltrexone (10 mg·kg−1, s.c.) for anti-allodynia effects in these mice with SNL, as a model of chronic pain.
Activation of microglia plays an important role in the pathogenesis of pain hypersensitivity following nerve injury (Inoue and Tsuda, 2009; McMahon and Malcangio, 2009; Milligan and Watkins, 2009). Naloxone and naltrexone have been reported to have a neuroprotective effect by inhibiting the activation of microglia (Das et al., 1995; Chang et al., 2000; Hutchinson et al., 2008). Chang and co-workers demonstrated that (−)- and (+)-isomers of the opioid antagonists, naloxone and naltrexone, also blocked TLR4 signalling, which is involved in activation of microglia, and reversed neuropathic pain (Chang et al., 2000; Hutchinson et al., 2008). Blockade of TLR4 signalling by naloxone and naltrexone does not involve the opioid receptors. Therefore in our current study, the effect of naltrexone to suppress SNL-induced allodynia might be not only through the activation of mutant MOP receptors, but also through the blockade of TLR4 signalling in microglia.
The development of tolerance and dependence limits the use of opioids for pain control. In this study, we also investigated this issue in our model of SNL in mice transfected with the MOR-S196ACSTA gene in the spinal cord. We found that significant natural withdrawal symptoms were observed in these mice after chronic morphine treatment (10 mg·kg−1, s.c., daily for 14 days) but not after similar chronic naltrexone treatment. This is because naltrexone can activate the mutated opioid receptors in the spinal cord only, but not the endogenous MOP receptors at the supraspinal level, that are known to be involved in opioid physical dependence. The numerical values of the withdrawal symptom score in the present study are much lower than those in an earlier study (Papaleo et al., 2006). In this earlier study, mice were treated with a schedule of increasing doses of morphine, from 10 mg kg−1 to 50 mg kg−1 or from 20 mg kg−1 to 100 mg kg−1, i.p., twice daily, for 6 days). However, in our study, mice were treated with morphine (10 mg kg−1, s.c.) once a day for 14 days. This may explain why the withdrawal symptom scores in our study are much lower.
Although many studies have indicated that opioid-induced rewarding effects are suppressed under neuropathic pain-like states in rodents (Ozaki et al., 2002; 2003; Oe et al., 2004; Niikura et al., 2008), we found that morphine (10 mg·kg−1, s.c., conditioning for 6 days) still elicited rewarding effects in mice with both SNL and MOR-S196ACSTA expressed in the spinal cord. However, naltrexone (10 mg·kg−1, s.c., conditioning for 6 days) did not elicit any rewarding effect in these mice. Several studies have indicated that relief of pain is rewarding (Navratilova et al., 2013). Analgesic agents that are not rewarding in the absence of pain, such as lidocaine, should become rewarding when there is ongoing or spontaneous pain (King et al., 2009; Navratilova et al., 2013). Relief of pain produces negative reinforcement through activation of the mesolimbic dopamine pathway (Navratilova et al., 2012). In the present study, naltrexone (10 mg·kg−1, s.c.) had anti-allodynia effects, but did not elicit rewarding effects in mice with both SNL and MOR-S196ACSTA expression in the spinal cord. This might be due to the fact that naltrexone behaved as an agonist only in the mutant MOP receptors that were locally expressed in lumbar spinal cord and therefore relieved pain. However, naltrexone still worked as an antagonist in wild-type opioid receptors in the brain. It is known that opioid antagonists elicit aversion in rodents with wild-type receptors (Douglas et al., 1976; Grevert and Goldstein, 1977; Mucha and Iversen, 1984; Mucha and Herz, 1985; Mucha and Walker, 1987). In our present study, naltrexone (10 mg·kg−1, s.c.) did not elicit aversion in the CPP test. That might be due to the combination of antinociceptive/allodynia effects of naltrexone on the mutant MOP receptors in the spinal cord and the aversive effect of naltrexone on wild-type opioid receptors in the brain. Furthermore, the animal must associate the treatment effects with the context of the CPP chamber to induce chamber preference. Only rapid-onset treatments that produce rapid pain relief can elicit CPP (Navratilova et al., 2013). In our study, the onset time of naltrexone was about 30 min to 1 h by tail-flick test. Failure to associate the treatment effects with the context of the CPP chamber might be another reason for the failure to observe CPP responses after naltrexone treatment.
Our current study indicated that i.t. administration of a lentivirus, encoding the construct of a mutant MOP receptor, to express MOR-S196ACSTA in the spinal cord, and systemic administration of an opioid antagonist naltrexone had anti-allodynia effects in a model of neuropathic pain, without induction of dependence or addiction. This approach could be a new strategy for the treatment of neuropathic pain.
Acknowledgments
We thank Mr M. Swofford for the English editing of the paper. This study was supported by grants from the National Science Council (NSC97-2320-B-016-003), Taiwan, Republic of China, and from the National Institutes of Health/National Institute on Drug Abuse (1 RO1-DA023905), USA.
Glossary
Abbreviations
- AAV
adeno-associated virus
- CPP
conditioned place preference
- DRG
dorsal root ganglion
- dsAAV
double-stranded adeno-associated virus
- MOP
μ opioid
- SNL
spinal nerve ligation
- TM
transmembrane domain
Author contributions
J.-H. K. was in-charge of virus production; elisa test for β-endorphin; immunohistochemistry tests; data handling and manuscript draft writing. M.-J. G. performed the behavioural tests for pain and allodynia. P.-P. Y. set up neuropathic pain animal model. P.-Y. L. and H. H. L. performed project discussions; supplied the mutant MOR containing plasmid; and revised the paper. P.-L. T. was responsible for the project; for designing the experiments; data examination; project discussion; and revising the paper.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12790
Figure S1 Plasma concentration of β-endorphin in mice that had been transfected with MOR-S196ACSTA in the spinal cord and with saline or naltrexone treatment started from day 1 after sham or SNL surgery. *P < 0.05, **P < 0.01 when compared with sham-S group at each time point.
Table S1 Physical dependence induced by chronic morphine but not naltrexone in mice which had been transfected with MOR-S196ACSTA in the spinal cord and with SNL.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. British Journal of Pharmacology. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloisi AM, Albonetti ME, Muscettola M, Facchinetti F, Tanganelli C, Carli G. Effects of formalin-induced pain on ACTH, beta-endorphin, corticosterone and interleukin-6 plasma levels in rats. Neuroendocrinology. 1995;62:13–18. doi: 10.1159/000126983. [DOI] [PubMed] [Google Scholar]
- Arner S, Meyerson BA. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23. doi: 10.1016/0304-3959(88)90198-4. [DOI] [PubMed] [Google Scholar]
- Atkinson JH, Kremer EF, Risch SC, Morgan CD, Azad RF, Ehlers CL, et al. Plasma measures of beta-endorphin/beta-lipotropin-like immunoreactivity in chronic pain syndrome and psychiatric subjects. Psychiatry Res. 1983;9:319–327. doi: 10.1016/0165-1781(83)90005-7. [DOI] [PubMed] [Google Scholar]
- Chang RC, Rota C, Glover RE, Mason RP, Hong JS. A novel effect of an opioid receptor antagonist, naloxone, on the production of reactive oxygen species by microglia: a study by electron paramagnetic resonance spectroscopy. Brain Res. 2000;854:224–229. doi: 10.1016/s0006-8993(99)02267-2. [DOI] [PubMed] [Google Scholar]
- Chen SL, Ma HI, Han JM, Tao PL, Law PY, Loh HH. dsAAV type 2-mediated gene transfer of MORS196A-eGFP into spinal cord as a pain management paradigm. Proc Natl Acad Sci U S A. 2007;104:20096–20101. doi: 10.1073/pnas.0703409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SH, Kao JH, Tao PL, Law PY, Loh HH. Naloxone can act as an analgesic agent without measurable chronic side effects in mice with a mutant mu-opioid receptor expressed in different sites of pain pathway. Synapse. 2012;66:694–704. doi: 10.1002/syn.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude PA, Wotta DR, Zhang XH, Prather PL, McGinn TM, Erickson LJ, et al. Mutation of a conserved serine in TM4 of opioid receptors confers full agonistic properties to classical antagonists. Proc Natl Acad Sci U S A. 1996;93:5715–5719. doi: 10.1073/pnas.93.12.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude-Geppert PA, Liu J, Solberg J, Erickson-Herbrandson LJ, Loh HH, Law PY. Antagonist efficacy in MORS196L mutant is affected by the interaction between transmembrane domains of the opioid receptor. J Pharmacol Exp Ther. 2005;313:216–226. doi: 10.1124/jpet.104.076505. [DOI] [PubMed] [Google Scholar]
- Cox BM, Christie MJ, Devi L, Toll L, Traynor JR. Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br J Pharmacol. 2015;172:317–323. doi: 10.1111/bph.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, McMillian MK, Bing G, Hong JS. Modulatory effects of [Met5]-enkephalin on interleukin-1 beta secretion from microglia in mixed brain cell cultures. J Neuroimmunol. 1995;62:9–17. doi: 10.1016/0165-5728(95)00083-e. [DOI] [PubMed] [Google Scholar]
- D'Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, et al. International union of pharmacology. XII. Classification of opioid receptors. Pharmacol Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- Douglas ME, Downs JB, Dannemiller FJ, Hodges MR, Munson ES. Change in pulmonary venous admixture with varying inspired oxygen. Anesth Analg. 1976;55:688–695. doi: 10.1213/00000539-197609000-00016. [DOI] [PubMed] [Google Scholar]
- Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Deliv Rev. 2003;55:1007–1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Yashpal K, Chabot JG, Osborne MG, Lefebvre CD, Dray A, et al. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR(1)) alleviates pain and restores opioid efficacy after nerve injury in rats. Br J Pharmacol. 2001;132:354–367. doi: 10.1038/sj.bjp.0703810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevert P, Goldstein A. Some effects of naloxone on behavior in the mouse. Psychopharmacology (Berl) 1977;53:111–113. doi: 10.1007/BF00426478. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Tsuda M. Microglia and neuropathic pain. Glia. 2009;57:1469–1479. doi: 10.1002/glia.20871. [DOI] [PubMed] [Google Scholar]
- Kao JH, Chen SL, Ma HI, Law PY, Tao PL, Loh HH. Intrathecal delivery of a mutant micro-opioid receptor activated by naloxone as a possible antinociceptive paradigm. J Pharmacol Exp Ther. 2010;334:739–745. doi: 10.1124/jpet.109.165399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, et al. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain. 1998;76:215–222. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Merenlender-Wagner A, Dikshtein Y, Yadid G. The beta-endorphin role in stress-related psychiatric disorders. Curr Drug Targets. 2009;10:1096–1108. doi: 10.2174/138945009789735147. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Satoh M. Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci Res. 1995;23:121–145. doi: 10.1016/0168-0102(95)00933-k. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology (Berl) 1985;86:274–280. doi: 10.1007/BF00432213. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Iversen SD. Reinforcing properties of morphine and naloxone revealed by conditioned place preferences: a procedural examination. Psychopharmacology (Berl) 1984;82:241–247. doi: 10.1007/BF00427782. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Walker MJ. Aversive property of opioid receptor blockade in drug-naive mice. Psychopharmacology (Berl) 1987;93:483–488. doi: 10.1007/BF00207239. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, et al. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci U S A. 2012;109:20709–20713. doi: 10.1073/pnas.1214605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, King T, Porreca F. Evaluation of reward from pain relief. Ann N Y Acad Sci. 2013;1282:1–11. doi: 10.1111/nyas.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols ML, Bian D, Ossipov MH, Malan TP, Jr, Porreca F. Antiallodynic effects of a CCKB antagonist in rats with nerve ligation injury: role of endogenous enkephalins. Neurosci Lett. 1996;215:161–164. doi: 10.1016/0304-3940(96)12964-5. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F. Enhancement of the antiallodynic and antinociceptive efficacy of spinal morphine by antisera to dynorphin A (1-13) or MK-801 in a nerve-ligation model of peripheral neuropathy. Pain. 1997;69:317–322. doi: 10.1016/S0304-3959(96)03282-4. [DOI] [PubMed] [Google Scholar]
- Niikura K, Narita M, Nakamura A, Okutsu D, Ozeki A, Kurahashi K, et al. Direct evidence for the involvement of endogenous beta-endorphin in the suppression of the morphine-induced rewarding effect under a neuropathic pain-like state. Neurosci Lett. 2008;435:257–262. doi: 10.1016/j.neulet.2008.02.059. [DOI] [PubMed] [Google Scholar]
- Oe K, Narita M, Imai S, Shibasaki M, Kubota C, Kasukawa A, et al. Inhibition of the morphine-induced rewarding effect by direct activation of spinal protein kinase C in mice. Psychopharmacology (Berl) 2004;177:55–60. doi: 10.1007/s00213-004-1929-0. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Narita M, Iino M, Sugita J, Matsumura Y, Suzuki T. Suppression of the morphine-induced rewarding effect in the rat with neuropathic pain: implication of the reduction in mu-opioid receptor functions in the ventral tegmental area. J Neurochem. 2002;82:1192–1198. doi: 10.1046/j.1471-4159.2002.01071.x. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Narita M, Iino M, Miyoshi K, Suzuki T. Suppression of the morphine-induced rewarding effect and G-protein activation in the lower midbrain following nerve injury in the mouse: involvement of G-protein-coupled receptor kinase 2. Neuroscience. 2003;116:89–97. doi: 10.1016/s0306-4522(02)00699-1. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Contarino A. Gender- and morphine dose-linked expression of spontaneous somatic opiate withdrawal in mice. Behav Brain Res. 2006;170:110–118. doi: 10.1016/j.bbr.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Tang QB, Bian D, Riedl M, Elde R, Lai J. Spinal opioid mu receptor expression in lumbar spinal cord of rats following nerve injury. Brain Res. 1998;795:197–203. doi: 10.1016/s0006-8993(98)00292-3. [DOI] [PubMed] [Google Scholar]
- Ramer MS, Murphy PG, Richardson PM, Bisby MA. Spinal nerve lesion-induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain. 1998;78:115–121. doi: 10.1016/S0304-3959(98)00121-3. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Inoue M, Toda K, Ueda H. Loss of peripheral morphine analgesia contributes to the reduced effectiveness of systemic morphine in neuropathic pain. J Pharmacol Exp Ther. 2004;309:380–387. doi: 10.1124/jpet.103.060582. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigdor S, Wilcox GL. Central and systemic morphine-induced antinociception in mice: contribution of descending serotonergic and noradrenergic pathways. J Pharmacol Exp Ther. 1987;242:90–95. [PubMed] [Google Scholar]
- Woolf CJ. Windup and central sensitization are not equivalent. Pain. 1996;66:105–108. [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Yang W, Law PY, Guo X, Loh HH. In vivo activation of a mutant mu-opioid receptor by antagonist: future direction for opiate pain treatment paradigm that lacks undesirable side effects. Proc Natl Acad Sci U S A. 2003;100:2117–2121. doi: 10.1073/pnas.0334906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangen A, Herzberg U, Vogel Z, Yadid G. Nociceptive stimulus induces release of endogenous beta-endorphin in the rat brain. Neuroscience. 1998;85:659–662. doi: 10.1016/s0306-4522(98)00050-5. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]
- Zwick M, Molliver DC, Lindsay J, Fairbanks CA, Sengoku T, Albers KM, et al. Transgenic mice possessing increased numbers of nociceptors do not exhibit increased behavioral sensitivity in models of inflammatory and neuropathic pain. Pain. 2003;106:491–500. doi: 10.1016/j.pain.2003.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Plasma concentration of β-endorphin in mice that had been transfected with MOR-S196ACSTA in the spinal cord and with saline or naltrexone treatment started from day 1 after sham or SNL surgery. *P < 0.05, **P < 0.01 when compared with sham-S group at each time point.
Table S1 Physical dependence induced by chronic morphine but not naltrexone in mice which had been transfected with MOR-S196ACSTA in the spinal cord and with SNL.