

Abstract
BACKGROUND AND PURPOSE
The opioid receptor family comprises four structurally homologous but functionally distinct sub-groups, the μ (MOP), δ (DOP), κ (KOP) and nociceptin (NOP) receptors. As most opioid agonists are selective but not specific, a broad spectrum of behaviours due to activation of different opioid receptors is expected. In this study, we examine whether other opioid receptor systems influenced KOP-mediated antinociception.
EXPERIMENTAL APPROACH
We used a tail withdrawal assay in C57Bl/6 mice to assay the antinociceptive effect of systemically administered opioid agonists with varying selectivity at KOP receptors. Pharmacological and genetic approaches were used to analyse the interactions of the other opioid receptors in modulating KOP-mediated antinociception.
KEY RESULTS
Etorphine, a potent agonist at all four opioid receptors, was not anti-nociceptive in MOP knockout (KO) mice, although etorphine is an efficacious KOP receptor agonist and specific KOP receptor agonists remain analgesic in MOP KO mice. As KOP receptor agonists are aversive, we considered KOP-mediated antinociception might be a form of stress-induced analgesia that is blocked by the anxiolytic effects of DOP receptor agonists. In support of this hypothesis, pretreatment with the DOP antagonist, naltrindole (10 mg·kg−1), unmasked etorphine (3 mg·kg−1) antinociception in MOP KO mice. Further, in wild-type mice, KOP-mediated antinociception by systemic U50,488H (10 mg·kg−1) was blocked by pretreatment with the DOP agonist SNC80 (5 mg·kg−1) and diazepam (1 mg·kg−1).
CONCLUSIONS AND IMPLICATIONS
Systemic DOP receptor agonists blocked systemic KOP antinociception, and these results identify DOP receptor agonists as potential agents for reversing stress-driven addictive and depressive behaviours mediated through KOP receptor activation.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Table of Links
| TARGETS | LIGANDS | |
|---|---|---|
| δ receptor | SNC80 | JDTic |
| κ receptor | U50,488H | J113397 |
| μ receptor | naltrindole | diazepam |
| NOP receptor | etorphine | nor BNI |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Three of the four classic members of the opioid receptor family, the μ (MOP), δ (DOP), κ (KOP) receptors, though structurally homologous and capable of activating many of the same second messenger pathways, regulate behaviours very differently (Kieffer and Evans, 2009; Alexander et al., 2013; Cox et al., 2015). MOP receptor agonists, such as fentanyl, are highly effective analgesics for the treatment of moderate to severe pain. These drugs also suppress respiration, induce constipation and stimulate reward circuitry (Gaveriaux-Ruff and Kieffer, 2002). Conversely, KOP receptor agonists are aversive, elicit stress and dysphoria, but do not cause respiratory depression (Wadenberg, 2003). Importantly, systemic KOP receptor agonists, like MOP receptor agonists, are analgesic in assays of acute pain such as the tail-immersion assay (Von Voigtlander and Lewis, 1982; Dortch-Carnes and Potter, 2005). Systemically administered DOP receptor agonists, unlike MOP and KOP receptor agonists, have little or no efficacy in acute pain models but are effective anxiolytics (Saitoh et al., 2004), and are analgesic in rodent models of chronic pain, where the δ agonist responses are up-regulated (Holdridge and Cahill, 2007; Pradhan et al., 2011; van Rijn et al., 2012). Furthermore, DOP receptor agonists have lower abuse liability and produce less euphoric effects than MOP receptor agonists. Accordingly, DOP receptor agonists are not self-administered, nor do they cause dependence (Negus et al., 1998; Brandt et al., 2001; Stevenson et al., 2005).
Some opioid drugs also interact with the fourth member of the opioid receptor family, the nociceptin/orphanin FQ (NOP) receptor. This opioid receptor is distinct from the other three opioid receptors already discussed in that the endogenous opioid peptides derived from pro-dynorphin, pro-enkephalin or pro-opiomelanocortin do not bind to it with high affinity (Zaki and Evans, 1998). Peptides derived from pro-nociceptin/orphanin FQ are considered the primary endogenous ligands for the NOP receptor (Meunier et al., 1995; Reinscheid et al., 1995). Systemic injection of NOP receptor agonists has been shown to effectively block the analgesic responses of MOP and KOP receptor agonists and attenuate the rewarding effects of cocaine and morphine (Mogil et al., 1996; Murphy et al., 1999; Vazquez-DeRose et al., 2013). The oripavines, buprenorphine and etorphine both activate NOP receptors, and in the case of buprenorphine, the MOP receptor-mediated analgesic efficacy in acute pain assays is compromised by co-activation of NOP receptors (Butour et al., 1997; Lutfy et al., 2003).
Opioid receptors are widely distributed throughout the peripheral nervous system and CNS, and the locations of these receptors mediate the diverse effects of opioid agonists (Le Merrer et al., 2009). All four opioid receptors are expressed in the spinal cord on overlapping populations of neurons (Minami and Satoh, 1995b; Minami et al., 1995a). In the brain, MOP, NOP and KOP receptors are expressed throughout the cortex, midbrain and hindbrain (Le Merrer et al., 2009). DOP receptors, in contrast, have a more focal distribution throughout the limbic and prelimbic brain regions (Swanson, 2000; Cahill et al., 2001; Faget et al., 2012); the anxiolytic properties of DOP receptor agonists are attributed to the activation of these receptors. Opioid agonists can produce a range of effects depending on the population of receptors targeted, and therefore route of administration and drug bioavailability are important factors when considering opioid function. Further, given that most opioid drugs are selective but not specific for one or more of the opioid receptors, the potential for complex and opposing activities due to the activation of different members of the opioid receptor family should be anticipated.
Etorphine is a highly efficacious and potent opioid that has been used to immobilize large animals such as elephants (Jainudeen, 1970). Pharmacologically, etorphine is a non-selective opioid receptor agonist with high and comparable affinities of 0.35, 1.5 and 0.1 nM for the MOP, DOP and KOP receptors, respectively (Toll et al., 1998; Li et al., 2000; Clark et al., 2006), and a weaker affinity of 530 nM at NOP receptors (Butour et al., 1997). Etorphine is highly efficacious at the MOP receptor with a maximum stimulation of 115%, and is slightly less efficacious at the DOP and KOP receptors, with maximal stimulations of 60 and 98% respectively (Xu et al., 2008). When given systemically, etorphine is 1000–10 000 times more potent than morphine as an analgesic, and historically this drug has been an important tool in probing opioid receptor pharmacology both in vivo and in vitro (Lewis and Husbands, 2004). The current study was triggered by our observation that etorphine showed no analgesic efficacy in acute pain assays in MOP receptor knockout (KO) mice. This result was completely unexpected given that etorphine is a potent KOP receptor agonist, and selective KOP receptor agonists, such as U50,488H, remain analgesic in MOP KO mice (Kieffer and Gaveriaux-Ruff, 2002). This observation led us to investigate the contribution of other members of the opioid receptor family into the modulation of KOP receptor-mediated antinociception.
Methods
Animals
All animal care and experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the UCLA Institutional Animal Care and Use Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 224 animals were used in the experiments described here.
Male and female MOP KO (exon 2 KO) mice and littermate wild-type (WT) controls (Matthes et al., 1996) were fully backcrossed to the C57Bl/6J line. NOP null mice (NOP-KO) (Nishi et al., 1997) were used to generate the MOP/NOP KO in house; male and female MOP and NOP heterozygotes were bred to form this strain. In experiments where both sexes were used, males and females were evenly distributed between groups, and each experimental group contained 30–60% females. For all experiments, animals were randomly assigned to control and drug treatment groups. For experiments using only WT mice, male C57Bl/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). All experiments were performed with mice between 8 and 12 weeks of age (25–30 g). All animals were housed ventilated plastic cages in groups of four with standard bedding, maintained on a normal 12 h light/dark cycle (temperature at 22°C and 60% humidity), with lights on at 07:00 h, and allowed free access to standard rodent chow (Teklad, Harlan, Indianapolis, IN, USA) and water.
Experiment 1: tail withdrawal thermal nociceptive assay for etorphine and U50,488H antinociception
The dose–response curve for etorphine and U50,488H in WT and MOP KO male and female mice was determined. Antinociception was measured by the tail withdrawal assay in which animals were gently restrained by cupping animals in a soft plastic conical sleeve and 2.5 cm of the tail was immersed in 49°C water. The time to tail withdrawal was measured. After three basal measurements, separate groups of WT and MOP KO animals received escalating doses of U50,488H (0.3–10 mg·kg−1, s.c.). A separate group of WT and MOP KO mice received escalating doses of etorphine (0.1 μg·kg–1 to 10 mg·kg−1, s.c.). The AUC was calculated for the entire dose–response curve of etorphine or U50,488H of WT and MOP KO animals and compared using a Mann–Whitney rank sum test. A separate group of male and female WT and MOP KO animals was pretreated with etorphine (10 mg·kg−1, s.c.) and/or J113397, a NOP antagonist (10 mg·kg−1, s.c.), prior to U50,488H (10 mg·kg−1, s.c.) injection. Etorphine and J11397 were co-administered 10 min prior to U50,488H treatment. The latency to tail withdrawal was measured 40 min after U50,488H injection. U50,488H+etorphine antinociception was also assayed in male and female MOP/NOP KO mice. MOP/NOP KO mice were treated with etorphine (10 mg·kg−1, s.c.) 10 min prior to U50,488H (10 mg·kg−1, s.c.). Tail withdrawal latencies were measured 40 min after U50,488H injection, as described above. A cut-off of 15 s was imposed to avoid tissue damage. Antinociception was reported as per cent maximum possible effect (%MPE) = [test latency − baseline latency/cut-off − baseline latency] × 100. The %MPE of each group 40 min after U50,488H injection was compared using a Kruskal–Wallis test or Mann–Whitney rank sum test. Differences were considered statistically significant when P < 0.05.
Experiment 2: [35S] GTPγS assay
Membrane preparations were carried out as described previously (Befort et al., 2001). Whole brain from MOP/NOP KO was removed, immediately frozen in isopentane or dry ice, and stored at −80°C prior to use. Whole brain membranes were prepared by homogenizing the brain in ice-cold 0.25 M sucrose solution 10 vol (mL·g−1 wet weight of tissue). Samples were then centrifuged at 1100× g for 10 min. Supernatants were collected and diluted five times in buffer containing 50 mM Tris-HCl (pH 7.4) and 1 mM EDTA, following which they were centrifuged at 25 000× g for 30 min. The pellets were homogenized in 2 mL ice-cold sucrose solution (0.32 M), aliquoted and kept at −80°C until further use.
For [35S]GTPγS binding assay, 5 μg of protein was used per well. Samples were incubated with varying concentrations of U50,488H or etorphine (10−5 to 10−12 M) for 1 h at 25°C in assay buffer containing 50 mM TrisHCl (pH 7.4), 3 mM MgCl2, 100 mM NaCl, 0.2 mM EGTA, 30 μM GDP and 0.1 nM [35S]GTPγS. Varying concentrations of etorphine (10−5 to 10−12 M) were also tested in the presence of 1 μM naltrindole and/or 1 μM norbinaltorphimine (norBNI). Incubation was terminated by rapid filtration and washing in ice-cold buffer (50 mM TrisHCl, 5 mM MgCl2, 50 mM NaCl, pH 7.4). Bound radioactivity was quantified using a liquid scintillation counter. Non-specific binding was defined as binding in the presence of 10 μM GTPγS, and basal binding indicates binding in the absence of any agonist. Curve fitting was performed using the statistical program GraphPad Prism (La Jolla, CA, USA). U50,488H was fit with a nonlinear fit, one-site model, whereas a two-site model was performed for the etorphine binding curve. R2 values were used to assess goodness of fit. EC50 values were determined from pooled, fitted data (n = 3, 4 mice per group). Each data point for each mouse was the average of a triplicate, and this average was considered as n = 1. Emax were compared with one-way anova followed by Tukey's post hoc test.
Experiment 3: δ receptor component to etorphine antinociception
The effect of etorphine was examined in the presence of naltrindole. Separate groups of male and female MOP KO mice were pretreated with naltrindole (10 mg·kg−1, s.c.) or saline 20 min prior to etorphine (0.3–10 mg·kg−1, s.c.). Tail withdrawal latencies were measured 40 min after injection (as described in experiment 2). Differences in untransformed withdrawal latencies between naltrindole and saline-treated animals were compared with a two-way repeated measures anova. In addition, %MPE between groups was compared by calculating the AUC for the entire dose-response of etorphine and compared with a Mann–Whitney rank sum test.
Next, male MOP KO mice were injected with etorphine (3 mg·kg−1, s.c.) or vehicle and the time to tail withdrawal was measured 40 min after injection. A second group of male MOP KO mice was pretreated with the DOP antagonist, naltrindole (10 mg·kg−1, s.c.), 20 min prior to etorphine injection and tested as above. A third group was pretreated with the KOP receptor antagonist, JDTic (10 mg·kg−1, s.c.), 24 h before testing and naltrindole (10 mg·kg−1, s.c.) 20 min prior to etorphine injection. %MPE was compared with a Kruskal–Wallis test. Differences were considered significant when P < 0.05. Finally, the dose–response effect of naltrindole in the presence of etorphine (3 mg·kg−1, s.c.) was examined. Separate groups of animals were treated with naltrindole (5–40 mg·kg−1, s.c.) 20 min prior to etorphine injection. Tail withdrawal responses were measured 40 min after injection.
Experiment 4: activation of δ receptor agonists during U50,488H antinociception
Male WT mice were injected with U50,488H (10 mg·kg−1, s.c.) and the time to tail withdrawal (as described in experiment 2) was measured every 20 min for an hour. A separate group of animals was pretreated with SNC80, a DOP receptor antagonist (5 mg·kg−1, s.c.), or saline 20 min prior to U50,488H injection. A third group was pretreated with diazepam (1 mg·kg−1, s.c.) or vehicle (20% EtOH) 20 min prior to U50,488H injection and tested as above. The AUC for both the vehicle and SNC80 or diazepam-treated animals was calculated for the entire time period of testing (0–60 min). and compared using a Mann–Whitney rank sum test.
Experiment 5: forced swim test
For the stress-induced analgesia test, male WT mice were forced to swim in 30°C water for 10 min. Mice were placed in an acrylic glass cylinder (18 cm in diameter, 36 cm high) filled halfway with water kept at a constant temperature by a circulating heater. Ten minutes prior to the swim test, animals were administered with SNC80 (5 mg·kg−1, s.c.) or vehicle. Animals were then forced to swim for 10 min after which they were placed in a standard mouse cage lined with paper towels for 1 min. Tail withdrawal latencies were measured immediately after as described above. Results were compared using a Mann–Whitney rank sum test.
Data analysis
Results are shown as means ± SEM. Means were compared, as shown for each set of results, by a Mann–Whitney Rank sum test, the Kruksal-Wallis test or with one-way anova followed by Tukey's post hoc test. Differences were considered significant when P < 0.05.
Materials
U50,488H hydrochloride, naltrindole hydrochloride, SNC80 and diazepam were purchased from R & D Systems (Minneapolis, MN, USA). Etorphine hydrochloride, JDTic hydrochloride (KOP receptor antagonist) and J113397 hydrochloride (NOP receptor antagonist) were kindly provided by the National Institute on Drug Abuse Drug Supply Program (Research Triangle Institute, Research Triangle Park, NC, USA). All drugs were dissolved in saline (0.9% sodium chloride), with the exception of diazepam, which was dissolved in 20% ethanol in water, and given by s.c. or i.p. injection (0.3 mL per injection).
Results
Etorphine blocks U50,488H antinociception in MOP KO mice
U50,488H and etorphine antinociception was measured using the tail withdrawal assay in WT, MOP KO and MOP/NOP KO mice. Baseline tail withdrawal latencies were not significantly different between genotype or sex (Supporting Information Table S1). No significant differences between U50,488H and etorphine antinociception were observed between WT, MOP KO or MOP/NOP KO mice (Supporting Information Fig. S1). While U50,488H antinociception tended to be higher in the males, no significant differences were found (two-way anova; Fsex(1,73) = 1.91, P > 0.05, Fgenotype(2,73) = 0.78, P > 0.05, Finteraction(2,73) = 0.23, P > 0.05). Etorphine antinociception (0.0001–10 mg·kg−1, s.c.) seen in WT mice was absent in male and female MOP KO mice (Figure 1B, U = 0.0, P < 0.0003). U50,488H (0.3–10 mg·kg−1, s.c.) antinociception was not different between WT and MOP KO mice (Figure 1A, U = 0.52, P > 0.05).
Figure 1.
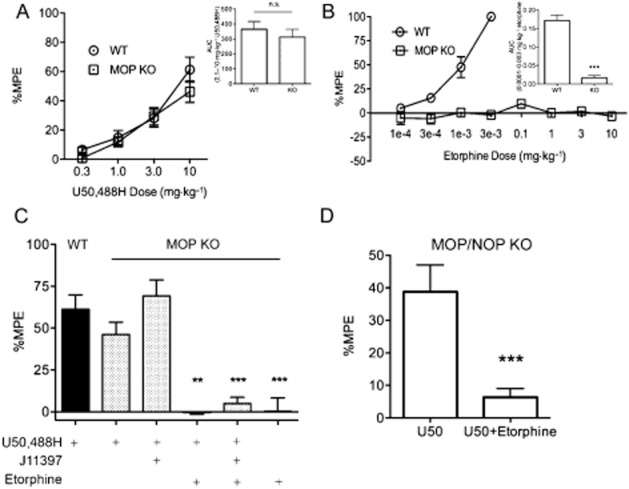
Etorphine interferes with U50,488H-mediated antinociception. (A) Male and female WT and MOP KO mice injected with escalating doses of U50,488H show that KOP receptor-mediated antinociception is not significantly different between genotype. Inset: the AUC was calculated for the entire dose range and was not significantly different between genotypes when compared with a Mann–Whitney rank sum test (n = 21 per group). %MPE = per cent maximum possible effect. n.s. = not significant, error bars = SEM. (B) Male and female WT and MOP KO mice injected with escalating doses of etorphine show that etorphine antinociception was abolished in MOP KO animals. Inset: the AUC was calculated for the entire dose range and was significantly reduced in MOP KO animals when compared with the Mann–Whitney rank sum test (n = 8–9 per group). Error bars = SEM, ***P > 0.0001. (C) Bar graph showing U50,488H (10 mg·kg−1, s.c.) antinociception 40 min after injection in WT and MOP KO mice treated with etorphine (10 mg·kg−1, s.c.) and/or J11397 (10 mg·kg−1, s.c.) 20 min prior to U50,488H injection. Pretreatment with etorphine significantly attenuated U50,488H antinociception response in MOP KO mice when compared with a Kruskal–Wallis multiple comparison test (n = 15–20 per group). Error bars = SEM, ***P < 0.001. (D) Bar graph showing U50,488H (U50; 10 mg·kg−1, s.c.) antinociception 40 min after injection in male and female MOP/NOP KO mice treated with or without etorphine (10 mg·kg−1, s.c.) 20 min prior to U50,488H injection. Pretreatment with etorphine significantly attenuated U50,488H antinociception response in MOP/NOP KO mice when compared with a Mann–Whitney rank sum test (n = 3–15). Error bars = SEM., ***P < 0.001.
We then compared the effect of pretreatment with etorphine (10 mg·kg−1, s.c.) and/or the NOP receptor antagonist J113397 (10 mg·kg−1, s.c.) on U50,488H (10 mg·kg−1, s.c.) antinociception in WT and MOP KO mice 40 min after U50,488H injection (Figure 1C). First, the NOP receptor antagonist J113397 (10 mg·kg−1, s.c.) did not alter U50,488H-mediated antinociception (expressed as %MPE) in MOP KO mice. U50,488H antinociception was significantly attenuated when MOP KO mice were treated with etorphine when compared with a Kruskal–Wallis test (Figure 1C, F(7,118) = 69.84, P < 0.0001). Pretreatment with J11397 had no effect on etorphine inhibition of U50,488H antinociception. These experiments were repeated in male and female MOP/NOP KO mice. Similar to the MOP KO mice, etorphine (10 mg·kg−1, s.c.) blocked U50,488H (10 mg·kg−1, s.c.) antinociception in MOP/NOP KO animals (Figure 1D, U = 0.0, P < 0.001).
Both U50,488H and etorphine are efficacious agonists at the KOP receptor
The efficacy at KOP receptors of two opioid agonists, U50,488H and etorphine, was measured in the GTPγS assay (Figure 2). All experiments were performed on brain tissue from MOP/NOP KO animals. U50,588H binding was best fit with a single curve indicative of a one-site binding model (Figure 2A, R2 = 0.68). Etorphine, a non-specific opioid agonist, showed a bimodal activity profile indicative of two binding sites (DOP and KOP receptor binding) (Figure 2B, R2 = 0.64 for two-site model vs. R2 = 0.56 for one-site model). When etorphine-stimulated GTPγS binding was performed in the presence of the DOP receptor antagonist, naltrindole, a single curve representative of the activity at KOP receptors was found (Figure 2C, R2 = 0.72). Remaining etorphine activity was confirmed to be due to activity at KOP receptors by performing etorphine-stimulated GTPγS binding in the presence of the KOP receptor antagonist, norBNI (Figure 2D; R2 = 0.34). Etorphine showed no detectable GTPγS binding in the presence of both naltrindole and norBNI (data not shown). The EC50 of etorphine at the KOP and DOP receptors was calculated to be 0.07 and 134 nM respectively. Emax values between the specific KOP receptor agonist U50,488H and etorphine in the presence of naltrindole were not significantly different. The Emax value of etorphine at DOP receptors was significantly higher than etorphine activity at KOP receptors (Figure 2E; one-way anova, F(2,12) = 6.43, P < 0.01).
Figure 2.
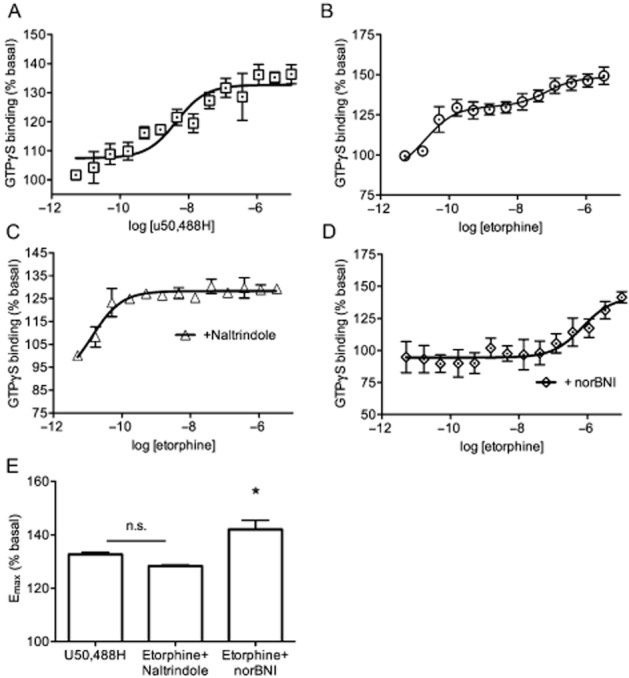
Etorphine and U50,488H show similar maximal activation at KOP receptors. Whole brain membranes from male and female MOP/NOP KO mice were tested in the [35S]GTPγS assay with varying concentrations of (A) U50,488H or (B) etorphine. Etorphine showed a bimodal curve indicated of a two-site binding model. Etorphine binding assays performed in the presence of (C) the DOP receptor antagonist, naltrindole (NTI; 1 μM), or (D) the KOP receptor antagonist, norBNI (1 μM), revealed low and high affinity binding sites relating to etorphine activity at the DOP and KOP receptors. (E) Emax values of the etorphine groups were compared to the specific KOP receptor agonist U50,488H. Emax values of etorphine + norBNI were significantly higher than etorphine + naltrindole when compared using a one-way anova followed by a Tukey's multiple comparison post hoc test (n = 4–7 per group). Error bars = SEM, n.s. = not significant, *P < 0.05.
DOP receptor antagonist reveals etorphine antinociception in MOP KO mice
In order to test whether etorphine activity at DOP receptors blocked KOP receptor-mediated antinociception, the tail withdrawal test was performed with etorphine in the presence or absence of the DOP receptor antagonist, naltrindole. Male and female MOP KO mice treated with etorphine (0.3–10 mg·kg−1, s.c.) showed no antinociception; however, male mice pretreated with naltrindole (10 mg·kg−1, s.c.) revealed a significant etorphine antinociception. The AUC calculated for the etorphine dose–response curve was significantly greater in mice pretreated with naltrindole when compared with a Mann–Whitney rank sum test (Figure 3A; U = 0.0, P < 0.01). Similarly, a significant difference in the etorphine dose–response curves of non-transformed tail withdrawal latencies (in seconds) between naltrindole and saline-pretreated male MOP KO mice was found (Supporting Information Fig. S2; Finteraction(4,32) = 3.53, P < 0.02, Fetorphine(4,32) = 2.67, P = 0.05, Fpretreatment(1,8) = 3.4, P > 0.05). This effect was not observed in female MOP KO mice (Supporting Information Fig. S2). Treatment with naltrindole alone did not have any effect on tail withdrawal thresholds (Figure 3B). When male mice were treated with naltrindole and JDTic (10 mg·kg−1, s.c.), a KOP receptor antagonist, the etorphine antinociception (%MPE) was blocked (Figure 3C; F(3,12) = 8.62, P < 0.002).
Figure 3.
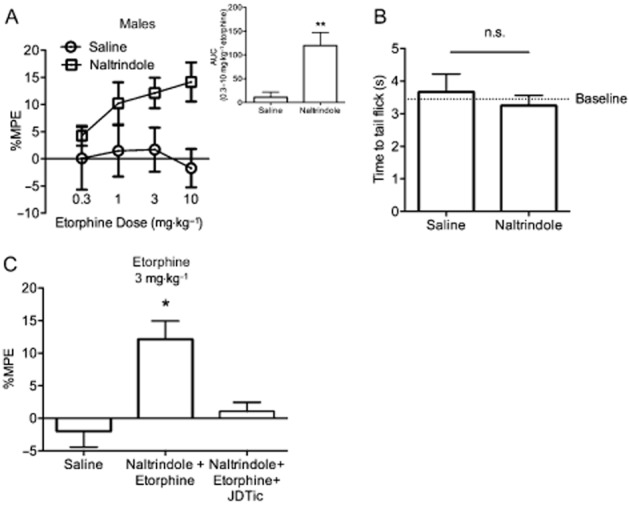
Etorphine antinociception is revealed in the presence of the DOP receptor antagonist, naltrindole. (A) Tail withdrawal latencies were tested 40 min after etorphine (0.3–10 mg·kg−1) in male MOP KO mice. Etorphine alone produced no antinociception. Pretreatment with naltrindole (10 mg·kg−1, s.c.) 20 min prior to etorphine injection revealed significant antinociception. Inset: the AUC was calculated for the entire etorphine dose range and was significantly greater in naltrindole-treated animals compared to saline treated when compared with a Mann–Whitney rank sum test (n = 7–10). %MPE = per cent maximum possible effect, error bars = SEM, **P < 0.01. (B) Naltrindole alone (10 mg·kg−1, s.c.) did not have an antinociceptive effect and was not significantly different from baseline thresholds (pre-drug injection) or saline injection when compared with a one-way anova (n = 10 per group). Error bars = SEM, n.s. = not significant. (C) Male MOP KO mice treated with the KOP receptor antagonist, JDTic (10 mg·kg−1, s.c., injected 24 h prior), blocked the naltrindole-revealed etorphine-mediated antinociception when compared with a Kruskal–Wallis multiple comparison test (n = 3–11). Error bars = SEM., *P < 0.05.
DOP receptor agonist blocks U50,488H-mediated antinociception
In order to further explore whether activity at the DOP receptor can modulate KOP receptor-mediated antinociception, U50,488H antinociception was determined in the presence or absence of the DOP receptor agonist, SNC80. WT male C57Bl/6J mice treated with U50,488H (10 mg·kg−1, s.c.) alone showed a robust analgesic response over 1 h. Pretreatment with SNC80 (5 mg·kg−1, s.c.) significantly attenuated the U50,488H-mediated analgesic response (expressed as %MPE), 40 min after U50,488H injection (Figure 4A, U = 0.0, P < 0.02), lending further credence to the idea that activity at the DOP receptor interferes with KOP receptor-mediated antinociception.
Figure 4.
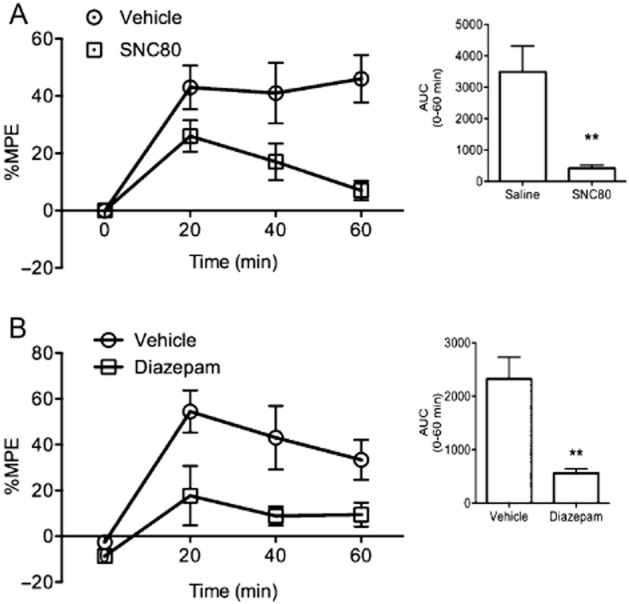
Anxiolytics block systemic KOP receptor-mediated antinociception. (A) Time course of U50,488H (10 mg·kg−1, s.c.) antinociception in male WT mice pretreated with or without the DOP receptor agonist, SNC80 (5 mg·kg−1, s.c.). Inset: bar graph representing AUC for U50,488H antinociception from 0 to 60 min after injection. SNC80 pretreatment significantly attenuated KOP receptor-mediated antinociception when compared with a Mann–Whitney rank sum test (n = 6 per group). (B) Time course of U50,488H antinociception in WT mice treated with or without diazepam (1 mg·kg−1). Inset: bar graph representing the AUC for U50,488H antinociception from 0 to 60 min after injection. Diazepam significantly attenuated the KOP receptor-mediated analgesia when compared with a Mann–Whitney rank sum test (n = 5 per group). %MPE = per cent maximum possible effect, error bars = SEM, ***P < 0.001, **P < 0.01.
Diazepam blocks U50,488H-mediated antinociception
Activity at the DOP receptor is known to have anxiolytic effects and mice lacking DOP receptors have anxiogenic and depressive-like phenotypes (Filliol et al., 2000; Broom et al., 2002; Saitoh et al., 2004; Perrine et al., 2006). In order to test whether the interference of KOP receptor-mediated antinociception by a DOP receptor agonist was due to an anxiolytic effect, the effects of another non-opioid anxiolytic agent, diazepam, on U50,488H antinociception were tested using the tail withdrawal assay. As above, U50,488H (10 mg·kg−1, s.c.) alone induced robust antinociception over 1 h. Pretreatment with diazepam (1 mg·kg−1, i.p.) significantly attenuated U50,488H-mediated antinociception (Figure 4B, U = 0.0, P < 0.001). This provides further support for the hypothesis that etorphine activity at the DOP receptor engages an anxiolytic response that inhibits KOP receptor antinociception.
DOP receptor agonist blocks stress-induced analgesia
Given the hypothesis that activation of the DOP receptor may be inhibiting KOP receptor-mediated antinociception through an anxiolytic mechanism, we confirmed that the dose of SNC80 used in the previous experiments could block classical stress-induced analgesia induced by a forced swim test. Animals forced to swim in 30°C water for 10 min showed a robust stress-induced analgesia (Figure 5); this effect has previously been shown to be mediated by KOP receptor activation in this specific behavioural paradigm (McLaughlin et al., 2003). SNC80 (5 mg·kg−1, s.c.) administered 20 min prior to the stress test significantly attenuated this antinociception (%MPE; U = 7, P < 0.05). These results confirm that SNC80 significantly attenuates KOP receptor-mediated stress-induced analgesia, and provides further evidence that DOP receptor co-activation can block KOP receptor-mediated antinociception by interfering with the stress response.
Figure 5.
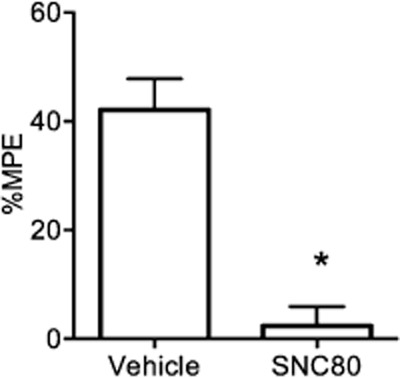
The DOP receptor agonist blocks stress-induced analgesia induced by a 10 min forced swim in 30°C water. The forced swim test produced a robust analgesic response in WT mice and this response was blocked by pretreatment with SNC80 (5 mg·kg−1, s.c.) when compared using the Mann–Whitney rank sum test (n = 6–8 per group). %MPE = per cent maximum possible effect, error bars = SEM, * = P < 0.05.
Discussion
The present study demonstrates that systemic administration of etorphine, a mixed agonist at MOP, DOP, KOP and NOP receptors, loses its analgesic efficacy in the MOP KO mouse. This result is intriguing because KOP receptor-mediated antinociception is intact in MOP KO mice. This observation led us to hypothesize that etorphine activity at NOP receptors may be interfering with the expression of etorphine antinociception in MOP KO mice, based on prior reports that activation of the NOP receptor engages anti-opioid mechanisms (Mogil et al., 1996; Murphy et al., 1999; Vazquez-DeRose et al., 2013). While etorphine affinity for NOP receptors is lower than the other opioid receptor classes, it has been shown to effectively compete with tritiated nociceptin, the endogenous ligand for NOP receptors (Butour et al., 1997). However, etorphine antinociception was not revealed in the presence of a NOP receptor antagonist (Figure 1C) or in MOP/NOP KO animals (Figure 1D). These results argued against the hypothesis that etorphine antinociception is inhibited by activity at the NOP receptors in the MOP KO animals, and led us to examine whether the mixed activity at the DOP and KOP receptors may explain the absence of etorphine antinociception in MOP KO mice.
The possibility that etorphine antinociception was being masked by DOP receptor activity was supported by the finding that MOP KO mice pretreated with the DOP receptor antagonist naltrindole had significant, albeit low, etorphine antinociception that could be blocked by the KOP receptor antagonist JDTic. This result suggested that etorphine activity at both KOP and DOP receptors could have opposing effects on antinociception in the tail withdrawal assay. In support of this premise, pretreatment with a DOP receptor agonist, SNC80, blocked the antinociception of a specific KOP receptor agonist, U50,488H. These data explain the lack of analgesic activity of etorphine in the MOP KO mice and attribute the finding to DOP receptor blockade of KOP receptor-mediated antinociception.
Etorphine is a highly potent, non-selective agonist with nanomolar binding affinities at the KOP, DOP and MOP receptors (Toll et al., 1998). However, based upon the GTPγS performed in the present study, etorphine has considerably greater potency at KOP than DOP receptors in mouse brain tissue. A study using C6 cells expressing DOP receptors found the etorphine EC50 to be around 10 nM (Lee et al., 1999), while the etorphine EC50 in CHO-FLAG-hKOP cells (expressing the human KOP receptor) was found to have a sub-nanomolar EC50 of etorphine (Li et al., 2003). In our study, we found the high affinity site of etorphine (0.07 nM) in MOP/NOP KO brain tissue to be blocked by the KOP receptor antagonist, norBNI, and the low affinity site (134 nM) to be blocked by the DOP receptor antagonist, naltrindole. The pharmacology confirmed the assignment of KOP and DOP receptor contributions to the etorphine-stimulated GTPγS binding but the high potency for etorphine for the KOP receptor site was unanticipated.
Despite using several doses of naltrindole and etorphine, etorphine in the presence of naltrindole never produced an antinociceptive effect above 15% MPE in male MOP KO mice. This is contrast to U50,488H-stimulated antinociception, which was closer to 60%MPE. It is possible that these differences in antinociceptive effects are due to differences in ligand efficacy for KOP receptor signalling resulting in antinociception. Differences in KOP receptor-mediated antinociception between KOP receptor agonists have been reported. For example, U69,593 was found to produce a more robust maximum analgesic response compared to U50,488H (95% vs. 60% MPE) (Smith and French, 2002). Furthermore, there is evidence to suggest that etorphine and U50,488H stimulate separate signalling pathways at the KOP receptor (Li et al., 2003). Therefore, differences in KOP receptor-mediated antinociception between etorphine and U50,488H could be due to a ligand-directed signalling phenomenon, and differences in the magnitude of KOP receptor-mediated antinociception are not without explanation.
Male MOP KO animals pretreated with naltrindole revealed significant etorphine antinociception; however, this effect was not observed in females. This KOP receptor-mediated analgesia is consistent with the observations reported by others that, in rodents, KOP receptor agonists have been found to produce greater antinociceptive responses in males than females (Kavaliers and Innes, 1987; Barrett et al., 2002; Mogil et al., 2003; Sternberg et al., 2004). The sex difference in the KOP receptor component of etorphine analgesia was an interesting finding, given that we did not observe a statistically significant sex difference in U50,488H antinociception, although evoked withdrawal responses tended to be lower in females (Supporting Information Fig. S1). There is ample evidence to support that females are more resistant to stress and stress-induced analgesia, particularly in those paradigms that are mediated by KOP receptors (Menendez et al., 1994; Kastenberger et al., 2012; Russell et al., 2014). The lack of sex difference for the U50,488H response in the present study may be due to testing female animals in different phases of their estrous cycle (proestrus, oestrus, metoestrus and dioestrus), and hence diluting any potential sex difference that may be evident. We did not track oestrus phase in female mice as this greatly complicates experimental design and introduces a female specific stressor (vaginal gavage for cytological examination of circulating ovarian hormones) that may well confound pain. This factor, in addition to the modest antinoceptive effects evident in the present study, made it impractical to detect sex differences with the selective KOP receptor agonist.
Given that all behavioural experiments were performed with systemic drug injection, the specificity of the drugs used is important to consider. U50,488H is a highly selective KOP receptor agonist. It has a binding affinity in the nanomolar range at KOP receptors, with 100- to 1000-fold weaker binding affinity for MOP and DOP receptors respectively (Payza, 2003). Further, U50,488H activity is completely absent in KOP KO animals (Simonin et al., 1998). Given that U50,488H-mediated antinociception was not significantly different between WT and MOP KO animals (Figure 2A) suggests that activity at the MOP receptor is negligible at the doses of U50,488H used in the present study. SNC80 has greater than 1000-fold selectivity for DOP over MOP and KOP receptors in binding assays and is specific when tested in the DOP KO mice (Knapp et al., 1996; Dondio et al., 1997). Likewise, norBNI, JDTic and naltrindole show specificity for KOP and DOP receptors, respectively, based on assessment in receptor KO mice (Payza, 2003; Carroll et al., 2004). So while the systemic administration of these drugs has the risk of off-target effects at other opioid receptors, the specificity of the drugs selected (with the exception of etorphine) lends confidence to the assertion that behavioural effects are due to specific activity at the assigned receptor.
We next considered the locus of purported interaction between the DOP and KOP receptors and possible explanations as to why DOP receptor agonists may block systemic KOP receptor-mediated analgesia. While KOP receptors are found throughout the peripheral nervous system and CNS (Minami et al., 1993), most of the analgesic efficacy of systemically administered KOP receptor agonists is mediated through central targets. Systemic U50,488H (which readily crosses the blood–brain barrier) produces a strong antinociceptive effect, but peripherally restricted KOP receptor agonists do not (Stein et al., 1989; Barber et al., 1994). Therefore, we can assume that the antinociceptive action of systemic U50,488H is mediated by centrally located KOP receptors. Activation of KOP receptors can produce antinociception in the classical opioid sense by inhibiting nociceptive pathways in the spinal cord and brain. They also can produce antinociception indirectly by stimulating the release of corticotrophin releasing factor (CRF). Injection of KOP receptor agonists stimulates the release of CRF and glucocorticoids (Buckingham and Cooper, 1986; Iyengar et al., 1986). Injection of KOP receptor agonists produces anxiety/dysphoria, and activation of the hypothalamic-pituitary-adrenal (HPA) axis and KOP receptor-stimulated release of CRF are thought to mediate these effects. In addition to anxiety, activation of the HPA axis and release of CRF is involved in stress-induced analgesia (Filaretov et al., 1996; Schafer et al., 1996; Butler and Finn, 2009). Injection of exogenous CRF causes analgesic effects in humans and animals (Hargreaves et al., 1987; Mousa et al., 2003). Therefore, a component of KOP receptor-mediated antinociception is mediated by an engagement of non-nociceptive, stress pathways.
DOP receptors are found throughout the peripheral nervous system, spinal cord and brain (Mansour et al., 1995; Cahill et al., 2001; Pradhan and Clarke, 2005; Le Merrer et al., 2009; Pradhan et al., 2011). Intrathecal injection of DOP receptor agonists is analgesic, while systemic injection is not, suggesting different populations of receptors are targeted depending on route of administration. In the spinal cord, KOP and DOP receptor agonists injected intrathecally synergized to produce an elevated antinociceptive response, while a DOP receptor agonist injected into the cerebral ventricles blocked systemic KOP receptor-mediated antinociception (Miaskowski et al., 1990). In the present study, systemically injected δ agonists were found to interfere with KOP receptor-mediated antinociception (Figure 4), and suggest that supraspinal DOP receptors interfere with KOP receptor-mediated antinociception in this study. While systemic administration of DOP receptor agonists has minimal analgesia, they do possess strong anxiolytic and antidepressant effects (Saitoh et al., 2004; Perrine et al., 2006). These properties are thought to be mediated by the limbic and prelimbic brain regions, an area in which DOP receptors are highly expressed. We propose that it is the anxiolytic effects of DOP receptors that interfere with the KOP receptor-mediated stress-induced analgesia, and may be why systemic DOP receptor agonists block U50,488H antinociception (Figure 4). To test this hypothesis, stress-induced analgesia was generated via a forced swim test in 30°C water over 10 min; a paradigm previously shown to induce a KOP receptor-dependent analgesic response (McLaughlin et al., 2003). This is in contrast to other forced swim test paradigms lasting shorter time periods and colder water temperatures that have been shown to be non-KOP receptor-dependent (Kitchen and Pinker, 1990; Contet et al., 2006). We found that pretreatment with SNC80 effectively blocked the stress-induced analgesia in this paradigm. We also found that diazepam, a non-opioid anxiolytic agent, was able to block U50,488H antinociception in a manner similar to SNC80 (Figure 4). This dose of diazepam in mice was shown not to be analgesic on its own (Rosland et al., 1987). This result is supported by previous studies that found diazepam inhibits U50,488H antinociception and conditioned placed aversion (Leri and Franklin, 2000; Nemmani and Ramarao, 2002). It is also in contrast with MOP receptor-mediated antinociception that is reportedly potentiated by benzodiazepines under certain conditions, most likely through a spinal mechanism (Luger et al., 1995). Collectively, these results provide further evidence that activation of the DOP receptors blocks KOP receptor-mediated antinociception via an anxiolytic mechanism. The involvement of KOP receptors in the limbic brain in mediating stress-induced analgesia (Knoll and Carlezon, 2010), and the colocalization of DOP and KOP receptors in this brain region (Le Merrer et al., 2009), identifies it as a likely locus of interaction between these two receptors. The precise location of interaction between DOP and KOP receptors at these supraspinal sites remains to be described.
These results suggest that systemically administered KOP receptor agonists produce antinociception through a stress-induced mechanism, which can be blocked by distinct classes of anxiolytics, including DOP receptor agonists. Given the strong aversive properties of KOP receptor agonists and the anxiolytic effects of DOP agonists mediated by receptors in the limbic and prelimbic brain, we submit that DOP and KOP receptors contribute to stress-induced analgesia and anxiety in an opposing manner. This study further supports DOP receptor agonists as potential therapeutic agents to reverse KOP receptor-mediated behaviours relating to stress.
Acknowledgments
This work was supported by DA005010 (C. J. E.) and DA031243 (A. A. P.), the Shirley and Stefan Hatos Foundation (C. J. E., A. M. T., A. A. P.), and the Canadian Institutes of Health Research (C. M. C., A. M. T). JDTic was kindly donated by the Research Triangle Institute.
Glossary
Abbreviations
- CRF
corticotropin releasing factor
- DOP
δ opioid
- HPA
hypothalamic-pituitary-adrenal axis
- KO
knockout
- KOP
κ opioid
- MOP
μ opioid
- NOP
nociceptin/orphanin FQ
- norBNI
nor-binaltorphimine
- WT
wild type
Author contributions
A. M. T. designed and performed the experiments and wrote the manuscript. K. W. R. and A. A. P. designed and performed the experiments and edited the manuscript. H. A. A. performed the experiments. W. W. maintained the transgenic mouse colony, designed the experiments and edited the manuscript. K. L., C. M. C. and C. J. E. designed the experiments and edited the manuscript. F. I. C. provided JDTic, designed the experiments and edited the manuscript.
Conflict of interest
There is no conflict of interest to disclose.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12810
Figure S1 U50,488H antinociceptive response across sex and genotype. Baseline withdrawal thresholds were taken before drug injection. Male and female WT, MOP KO and MOP/NOP KO mice were treated with U50,488H (10 mg·kg−1, s.c.). Tail withdrawal latencies were measured 40 min after drug injection. While males are tended to have higher U50,488H antinociceptive responses (particularly in MOP/NOP KO), no significant difference between genotype or sex was found when compared with a two-way anova (n = 7–26). Error bars = SEM.
Figure S2 Etorphine antinociception is revealed in the presence of naltrindole in males, but not females. Tail withdrawal latencies were tested 40 min after etorphine (0.3–10 mg·kg−1, s.c.). Etorphine alone did not produce antinociception in either males or females (results presented as raw tail flick response, in seconds). Pretreatment with naltrindole (10 mg·kg−1, s.c.) 20 min prior to etorphine injection revealed significant antinociception in males, but not females when compared with a two-way repeated measures anova (n = 7–10 per group). Error bars = SEM. **P < 0.01.
Table S1 Baseline tail withdrawal latencies in 49°C water.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A, Bartoszyk GD, Bender HM, Gottschlich R, Greiner HE, Harting J, et al. A pharmacological profile of the novel, peripherally-selective kappa-opioid receptor agonist, EMD 61753. Br J Pharmacol. 1994;113:1317–1327. doi: 10.1111/j.1476-5381.1994.tb17142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AC, Smith ES, Picker MJ. Sex-related differences in mechanical nociception and antinociception produced by mu- and kappa-opioid receptor agonists in rats. Eur J Pharmacol. 2002;452:163–173. doi: 10.1016/s0014-2999(02)02274-4. [DOI] [PubMed] [Google Scholar]
- Befort K, Filliol D, Decaillot FM, Gaveriaux-Ruff C, Hoehe MR, Kieffer BL. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J Biol Chem. 2001;276:3130–3137. doi: 10.1074/jbc.M006352200. [Epub 2000 Nov]; 3136. [DOI] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS. Studies of tolerance and dependence with the delta-opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J Pharmacol Exp Ther. 2001;299:629–637. [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH. Nonpeptidic delta-opioid receptor agonists reduce immobility in the forced swim assay in rats. Neuropsychopharmacology. 2002;26:744–755. doi: 10.1016/S0893-133X(01)00413-4. [DOI] [PubMed] [Google Scholar]
- Buckingham JC, Cooper TA. Pharmacological characterization of opioid receptors influencing the secretion of corticotrophin releasing factor in the rat. Neuroendocrinology. 1986;44:36–40. doi: 10.1159/000124618. [DOI] [PubMed] [Google Scholar]
- Butler RK, Finn DP. Stress-induced analgesia. Prog Neurobiol. 2009;88:184–202. doi: 10.1016/j.pneurobio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Butour JL, Moisand C, Mazarguil H, Mollereau C, Meunier JC. Recognition and activation of the opioid receptor-like ORL 1 receptor by nociceptin, nociceptin analogs and opioids. Eur J Pharmacol. 1997;321:97–103. doi: 10.1016/s0014-2999(96)00919-3. [DOI] [PubMed] [Google Scholar]
- Cahill CM, McClellan KA, Morinville A, Hoffert C, Hubatsch D, O'Donnell D, et al. Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J Comp Neurol. 2001;440:65–84. doi: 10.1002/cne.1370. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Zhang L, Mascarella SW, Navarro HA, Rothman RB, Cantrell BE, et al. Discovery of the first N-substituted 4beta-methyl-5-(3-hydroxyphenyl)morphan to possess highly potent and selective opioid delta receptor antagonist activity. J Med Chem. 2004;47:281–284. doi: 10.1021/jm030419a. [DOI] [PubMed] [Google Scholar]
- Clark MJ, Furman CA, Gilson TD, Traynor JR. Comparison of the relative efficacy and potency of mu-opioid agonists to activate Galpha(i/o) proteins containing a pertussis toxin-insensitive mutation. J Pharmacol Exp Ther. 2006;317:858–864. doi: 10.1124/jpet.105.096818. [DOI] [PubMed] [Google Scholar]
- Contet C, Gaveriaux-Ruff C, Matifas A, Caradec C, Champy MF, Kieffer BL. Dissociation of analgesic and hormonal responses to forced swim stress using opioid receptor knockout mice. Neuropsychopharmacology. 2006;31:1733–1744. doi: 10.1038/sj.npp.1300934. [DOI] [PubMed] [Google Scholar]
- Cox BM, Christie MJ, Lakshmi D, Toll L, Traynor JR. Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br J Pharmacol. 2015;172:317–323. doi: 10.1111/bph.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondio G, Ronzoni S, Eggleston DS, Artico M, Petrillo P, Petrone G, et al. Discovery of a novel class of substituted pyrrolooctahydroisoquinolines as potent and selective delta opioid agonists, based on an extension of the message-address concept. J Med Chem. 1997;40:3192–3198. doi: 10.1021/jm9608218. [DOI] [PubMed] [Google Scholar]
- Dortch-Carnes J, Potter DE. Bremazocine: a kappa-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005;11:195–212. doi: 10.1111/j.1527-3458.2005.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faget L, Erbs E, Le Merrer J, Scherrer G, Matifas A, Benturquia N, et al. In vivo visualization of delta opioid receptors upon physiological activation uncovers a distinct internalization profile. J Neurosci. 2012;32:7301–7310. doi: 10.1523/JNEUROSCI.0185-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filaretov AA, Bogdanov AI, Yarushkina NI. Stress-induced analgesia. The role of hormones produced by the hypophyseal-adrenocortical system. Neurosci Behav Physiol. 1996;26:572–578. doi: 10.1007/BF02359502. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Opioid receptor genes inactivated in mice: the highlights. Neuropeptides. 2002;36:62–71. doi: 10.1054/npep.2002.0900. [DOI] [PubMed] [Google Scholar]
- Hargreaves KM, Mueller GP, Dubner R, Goldstein D, Dionne RA. Corticotropin-releasing factor (CRF) produces analgesia in humans and rats. Brain Res. 1987;422:154–157. doi: 10.1016/0006-8993(87)90550-6. [DOI] [PubMed] [Google Scholar]
- Holdridge SV, Cahill CM. Spinal administration of a delta opioid receptor agonist attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Eur J Pain. 2007;11:685–693. doi: 10.1016/j.ejpain.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Iyengar S, Kim HS, Wood PL. Kappa opiate agonists modulate the hypothalamic-pituitary-adrenocortical axis in the rat. J Pharmacol Exp Ther. 1986;238:429–436. [PubMed] [Google Scholar]
- Jainudeen MR. The use of etorphine hydrochloride for restraint of a domesticated elephant (Elephas maximus) J Am Vet Med Assoc. 1970;157:624–626. [PubMed] [Google Scholar]
- Kastenberger I, Lutsch C, Herzog H, Schwarzer C. Influence of sex and genetic background on anxiety-related and stress-induced behaviour of prodynorphin-deficient mice. PLoS ONE. 2012;7:e34251. doi: 10.1371/journal.pone.0034251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavaliers M, Innes D. Stress-induced opioid analgesia and activity in deer mice: sex and population differences. Brain Res. 1987;425:49–56. doi: 10.1016/0006-8993(87)90482-3. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. Opioid receptors: from binding sites to visible molecules in vivo. Neuropharmacology. 2009;56(Suppl. 1):205–212. doi: 10.1016/j.neuropharm.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen I, Pinker SR. Antagonism of swim-stress-induced antinociception by the delta-opioid receptor antagonist naltrindole in adult and young rats. Br J Pharmacol. 1990;100:685–688. doi: 10.1111/j.1476-5381.1990.tb14076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp RJ, Santoro G, De Leon IA, Lee KB, Edsall SA, Waite S, et al. Structure-activity relationships for SNC80 and related compounds at cloned human delta and mu opioid receptors. J Pharmacol Exp Ther. 1996;277:1284–1291. [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. 2009;89:1379–1412. doi: 10.1152/physrev.00005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KO, Akil H, Woods JH, Traynor JR. Differential binding properties of oripavines at cloned mu- and delta-opioid receptors. Eur J Pharmacol. 1999;378:323–330. doi: 10.1016/s0014-2999(99)00460-4. [DOI] [PubMed] [Google Scholar]
- Leri F, Franklin KB. Diazepam in the ventral striatum dissociates dopamine-dependent and dopamine-independent place conditioning. Neuroreport. 2000;11:2553–2557. doi: 10.1097/00001756-200008030-00041. [DOI] [PubMed] [Google Scholar]
- Lewis JW, Husbands SM. The orvinols and related opioids–high affinity ligands with diverse efficacy profiles. Curr Pharm Des. 2004;10:717–732. doi: 10.2174/1381612043453027. [DOI] [PubMed] [Google Scholar]
- Li JG, Benovic JL, Liu-Chen LY. Mechanisms of agonist-induced down-regulation of the human kappa-opioid receptor: internalization is required for down-regulation. Mol Pharmacol. 2000;58:795–801. [PubMed] [Google Scholar]
- Li JG, Zhang F, Jin XL, Liu-Chen LY. Differential regulation of the human kappa opioid receptor by agonists: etorphine and levorphanol reduced dynorphin A- and U50,488H-induced internalization and phosphorylation. J Pharmacol Exp Ther. 2003;305:531–540. doi: 10.1124/jpet.102.045559. [DOI] [PubMed] [Google Scholar]
- Luger TJ, Hayashi T, Weiss CG, Hill HF. The spinal potentiating effect and the supraspinal inhibitory effect of midazolam on opioid-induced analgesia in rats. Eur J Pharmacol. 1995;275:153–162. doi: 10.1016/0014-2999(94)00759-z. [DOI] [PubMed] [Google Scholar]
- Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, et al. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci. 2003;23:10331–10337. doi: 10.1523/JNEUROSCI.23-32-10331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. [Review] [44 refs] Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez L, Andres-Trelles F, Hidalgo A, Baamonde A. Gender and test dependence of a type of kappa mediated stress induced analgesia in mice. Gen Pharmacol. 1994;25:903–908. doi: 10.1016/0306-3623(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL 1 receptor. Nature. 1995;377(6549):532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Miaskowski C, Taiwo YO, Levine JD. Kappa- and delta-opioid agonists synergize to produce potent analgesia. Brain Res. 1990;509:165–168. doi: 10.1016/0006-8993(90)90327-8. [DOI] [PubMed] [Google Scholar]
- Minami M, Satoh M. Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci Res. 1995b;23:121–145. doi: 10.1016/0168-0102(95)00933-k. [DOI] [PubMed] [Google Scholar]
- Minami M, Hosoi Y, Toya T, Katao Y, Maekawa K, Katsumata S, et al. In situ hybridization study of kappa-opioid receptor mRNA in the rat brain. Neurosci Lett. 1993;162:161–164. doi: 10.1016/0304-3940(93)90585-9. [DOI] [PubMed] [Google Scholar]
- Minami M, Maekawa K, Yabuuchi K, Satoh M. Double in situ hybridization study on coexistence of æ-, ë- and kappa-opioid receptor mRNAs with preprotachykinin A mRNA in the rat dorsal root ganglia. Brain Res Mol Brain Res. 1995a;30:203–210. doi: 10.1016/0169-328x(94)00290-u. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Grisel JE, Reinscheid KK, Civelli O, Belknap JK, Grandy DK. Orphanin FQ is a functional anti-opioid peptide. Neuroscience. 1996;75:333–337. doi: 10.1016/0306-4522(96)00338-7. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KV, Lariviere WR, et al. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci U S A. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa SA, Bopaiah CP, Stein C, Schafer M. Involvement of corticotropin-releasing hormone receptor subtypes 1 and 2 in peripheral opioid-mediated inhibition of inflammatory pain. Pain. 2003;106:297–307. doi: 10.1016/S0304-3959(03)00302-6. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lee Y, Maidment NT. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999;832:168–170. doi: 10.1016/s0006-8993(99)01425-0. [DOI] [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K. Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J Pharmacol Exp Ther. 1998;286:362–375. [PubMed] [Google Scholar]
- Nemmani KV, Ramarao P. Role of benzodiazepine-GABAA receptor complex in attenuation of U-50,488H-induced analgesia and inhibition of tolerance to its analgesia by ginseng total saponin in mice. Life Sci. 2002;70:1727–1740. doi: 10.1016/s0024-3205(02)01496-0. [DOI] [PubMed] [Google Scholar]
- Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, et al. Unrestrained nociceptive response and dysregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J. 1997;16:1858–1864. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payza K. Binding and activity of opioid ligands at the cloned human delta, mu and kappa receptors. In: Chang K-J, Porecca F, Woods JH, editors. The Delta Receptor. New York: Marcel Dekker Incorporated; 2003. pp. 261–275. [Google Scholar]
- Perrine SA, Hoshaw BA, Untewald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Clarke PB. Comparison between delta-opioid receptor functional response and autoradiographic labeling in rat brain and spinal cord. J Comp Neurol. 2005;481:416–426. doi: 10.1002/cne.20378. [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. doi: 510.1016/j.tips.2011.1006.1008; [Epub 2011 Sep]; 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270(5237):792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry. 2012;71:232–238. doi: 10.1016/j.biopsych.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosland JH, Hunskaar S, Hole K. The effect of diazepam on nociception in mice. Pharmacol Toxicol. 1987;61:111–115. doi: 10.1111/j.1600-0773.1987.tb01786.x. [DOI] [PubMed] [Google Scholar]
- Russell SE, Rachlin AB, Smith KL, Muschamp J, Berry L, Zhao Z, et al. Sex differences in sensitivity to the depressive-like effects of the kappa opioid receptor agonist U-50488 in rats. Biol Psychiatry. 2014;76:213–222. doi: 10.1016/j.biopsych.2013.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. J Pharmacol Sci. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Schafer M, Mousa SA, Zhang Q, Carter L, Stein C. Expression of corticotropin-releasing factor in inflamed tissue is required for intrinsic peripheral opioid analgesia. Proc Natl Acad Sci U S A. 1996;93:6096–6100. doi: 10.1073/pnas.93.12.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A, et al. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 1998;17:886–897. doi: 10.1093/emboj/17.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, French AM. Age-related differences in sensitivity to the antinociceptive effects of kappa opioids in adult male rats. Psychopharmacology (Berl) 2002;162:255–264. doi: 10.1007/s00213-002-1102-6. [DOI] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–1275. [PubMed] [Google Scholar]
- Sternberg WF, Chesler EJ, Wilson SG, Mogil JS. Acute progesterone can recruit sex-specific neurochemical mechanisms mediating swim stress-induced and kappa-opioid analgesia in mice. Horm Behav. 2004;46:467–473. doi: 10.1016/j.yhbeh.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenz yl]-N,N-diethylbenzamide] and heroin. J Pharmacol Exp Ther. 2005;314:221–231. doi: 10.1124/jpet.104.082685. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Cerebral hemisphere regulation of motivated behavior. Brain Res. 2000;886:113–164. doi: 10.1016/s0006-8993(00)02905-x. [DOI] [PubMed] [Google Scholar]
- Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, et al. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr. 1998;178:440–466. [PubMed] [Google Scholar]
- Vazquez-DeRose J, Stauber G, Khroyan TV, Xie XS, Zaveri NT, Toll L. Retrodialysis of N/OFQ into the nucleus accumbens shell blocks cocaine-induced increases in extracellular dopamine and locomotor activity. Eur J Pharmacol. 2013;699:200–206. doi: 10.1016/j.ejphar.2012.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Voigtlander PF, Lewis RA. U-50,488, a selective kappa opioid agonist: comparison to other reputed kappa agonists. Prog Neuropsychopharmacol Biol Psychiatry. 1982;6:467–470. doi: 10.1016/s0278-5846(82)80130-9. [DOI] [PubMed] [Google Scholar]
- Wadenberg ML. A review of the properties of spiradoline: a potent and selective kappa-opioid receptor agonist. CNS Drug Rev. 2003;9:187–198. doi: 10.1111/j.1527-3458.2003.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang X, Partilla JS, Bishop-Mathis K, Benaderet TS, Dersch CM, et al. Differential effects of opioid agonists on G protein expression in CHO cells expressing cloned human opioid receptors. Brain Res Bull. 2008;77:49–54. doi: 10.1016/j.brainresbull.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki PA, Evans CJ. ORL-1: an awkward child of the opioid receptor family. Neuroscientist. 1998;4:172–184. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 U50,488H antinociceptive response across sex and genotype. Baseline withdrawal thresholds were taken before drug injection. Male and female WT, MOP KO and MOP/NOP KO mice were treated with U50,488H (10 mg·kg−1, s.c.). Tail withdrawal latencies were measured 40 min after drug injection. While males are tended to have higher U50,488H antinociceptive responses (particularly in MOP/NOP KO), no significant difference between genotype or sex was found when compared with a two-way anova (n = 7–26). Error bars = SEM.
Figure S2 Etorphine antinociception is revealed in the presence of naltrindole in males, but not females. Tail withdrawal latencies were tested 40 min after etorphine (0.3–10 mg·kg−1, s.c.). Etorphine alone did not produce antinociception in either males or females (results presented as raw tail flick response, in seconds). Pretreatment with naltrindole (10 mg·kg−1, s.c.) 20 min prior to etorphine injection revealed significant antinociception in males, but not females when compared with a two-way repeated measures anova (n = 7–10 per group). Error bars = SEM. **P < 0.01.
Table S1 Baseline tail withdrawal latencies in 49°C water.