

Abstract
Flavobacterium psychrophilum is an important fish pathogen, responsible for Cold Water Disease, with a significant economic impact on salmonid farms worldwide. In spite of this, little is known about the bacterial physiology and pathogenesis mechanisms, maybe because it is difficult to manipulate, being considered a fastidious microorganism. Mutants obtained using a Tn4351 transposon were screened in order to identify those with alteration in colony morphology, colony spreading and extracellular proteolytic activity, amongst other phenotypes. A F. psychrophilum mutant lacking gliding motility showed interruption of the FP1638 locus that encodes a putative type-2 glycosyltransferase (from here on referred to as fpgA gene, Flavobacterium psychrophilum glycosyltransferase). Additionally, the mutant also showed a decrease in the extracellular proteolytic activity as a consequence of down regulation in the fpgA mutant background of the fpp2-fpp1 operon promoter, responsible for the major extracellular proteolytic activity of the bacterium. The protein glycosylation profile of the parental strain showed the presence of a 22 kDa glycosylated protein which is lost in the mutant. Complementation with the fpgA gene led to the recovery of the wild-type phenotype. LD50 experiments in the rainbow trout infection model show that the mutant was highly attenuated. The pleiotropic phenotype of the mutant demonstrated the importance of this glycosyltranferase in the physiology and virulence of the bacterium. Moreover, the fpgA mutant strain could be considered a good candidate for the design of an attenuated vaccine.
Introduction
Flavobacterium psychrophilum is a Gram-negative bacterium and it is a member of the Cytophaga-Flavabacterium-Bacteroides (CFB) group. This microorganism is the etiological agent of Cold Water Disease (CWD) (as well as rainbow trout fry syndrome), one of the most important infectious pathologies, causing significant economic losses in salmonids on fish farms worldwide. Outbreaks occur at temperatures below 14 °C and mainly affect fingerlings. Currently there are no efficient commercial vaccines to protect against CWD. Besides, other approaches such as the use of immunostimulants, probiotics or lytic bacteriophages do not provide sufficient infection control. Therefore, disease management is based on fish monitoring strategies and the use of antimicrobial therapy [1,2].
In spite of being considered a fastidious bacterium because it is difficult to isolate and manipulate [3], several advances have been reported during the last few years in culture [4], experimental infection techniques [5], typing [6-9], molecular diagnosis [10-15] and genomics [16]. Furthermore, genetic manipulation systems based on transposition mutagenesis [17] and disruption of the specific genes by homologous recombination have been developed [18]. However, the mechanisms of pathogenicity of this bacterium are still unknown and little data exist about the genetic basis of virulence [19,20].
Even though protein glycosylation was once considered to be a specifically eukaryotic phenomenon, nowadays it is clear that it is widespread in prokaryotes [21,22]. In fact, several glycoproteins described in Gram-negative pathogenic bacteria have been associated with virulence [23,24]. In the case of F. psychrophilum, a glycosylated protein, OmpA, has been purified and characterized [25,26]. In addition, 35 genes encoding putative glycosyltransferases have been identified in the genome of strain JIP02/86 [16]. Moreover, some members of the CFB group showed glycosylated proteins [27,28]. In particular, Bacteroides fragilis, forming a relevant part of the human intestinal microbiota, presented, at least eight glycoproteins which are related to the competitive colonization of mammalian intestines [28,29]. All of this suggests a relevant role of glycosyltransferases in the biology of bacteria belonging to this group.
The ability of pathogenic bacteria to cause disease depends to a large extent on the expression of specific gene-encoding factors which help them to invade the host tissues, develop the infection and evade host defenses. Amongst other characteristics, bacterial motility [30-32] and extracellular proteolytic activity [33] are considered to be involved in the infection caused by different Gram-negative bacteria. With the aim of achieving a deeper insight into the pathogenesis mechanisms of F. psychrophilum, a set of mutants deficient in gliding motility and extracellular proteolytic activity were isolated using the Tn4351-mutagenesis system [19]. In this study, a mutant with a change in the FP1638 locus was further analyzed. The interrupted gene, which encodes a type-2 glycosyltransferase, was found to be involved in the glycosylation of a 22 kDa protein. The modification of protein glycosylation in the mutant strain caused, amongst other effects, high attenuation of the virulence of the bacterium.
Material and methods
Bacterial strains and growth conditions
The strains, plasmid and primers used in this study are listed in Table 1. Escherichia coli strains S17-1 λpir [34] and BW19851 [35] were used to transfer DNA into F. psychrophilum THC02/90. E. coli strains were grown at 37 °C in 2 × TY medium (10 g of tryptone per liter, 10 g of yeast extract per liter, 5 g of NaCl per liter) with 20 g of agar per liter added for solid medium. F. psychrophilum THC02/90 was grown at 12 °C or 18 °C in nutrient broth (NB; Pronadisa, Madrid, Spain) or NBF [36]. Nutrient agar (NA: NB containing 1.5 g of agar/L), or nutrient agar charcoal (NAC; NA supplemented with 0.05% activated charcoal), were used for solid cultures as previously described [36]. Growth in liquid culture was carried out at 250 rpm and 12 °C and determined by measuring OD525nm at different times. Stock cultures were kept in NB containing 25% glycerol at −80 °C. To observe colony spreading, F. psychrophilum strains were grown as previously described in 1/6NA [37]. Extracellular proteolytic activity on solid medium was visualized using NBF containing 1.5 g/mL of agar and supplemented with 0.75% gelatin [19]. Azocasein assays were performed according to Secades et al., [38] where one unit of enzyme activity [32] was defined as the amount that yielded an increase in the OD420nm of 0.01 in two hours at 30 °C. For selective growth of E. coli S17-1 λpir, 50 μg/mL streptomycin was used and transformants were selected with 100 μg/mL ampicillin. Selection of F. psychrophilum transconjugants was carried out with 10 μg/mL erythromycin and 10 μg/mL tetracycline.
Table 1.
Bacterial strains, plasmids and primers used in this study
| Plasmid, bacterial strain or primer | Description or sequence | Source or reference |
|---|---|---|
| Plasmidsa | ||
| pEP4351 | Ori R6K dependent protein pir,; RP4 oriT; Cmr Tcr (Emr); Tn4351 vector transfer. | [39] |
| pCP23 | ColE1 ori, (pCP1 ori), Apr (Tcr), E. coli–F. psychrophilum shuttle plasmid | [40] |
| pCP23-fpgA | FP1638 gene, derived from pCP23. | This study |
| pCP23-Gfpp2 | pCP23-G carrying fpp2-fpp1 promoter | [41] |
| Bacterial strains | ||
| F. psychrophilum | ||
| THC02/90 | Wild-type. | [42] |
| fpgA− | FP1638 mutant created by Tn4351 transposition. | This study |
| fpgA+ | Mutant ΔFP1638 carrying pCP23-FP1638 plasmid, complemented strain. | This study |
| THC02/90-G | THC02/90 strain carrying pCP23-G plasmid | [41] |
| THC02/90 -fpp2 | THC02/90 strain carrying pCP23-Gfpp2 plasmid | [41] |
| fpgA− -fpp2 | ΔFP1638 strain carrying pCP23-Gfpp2 plasmid | [41] |
| E. coli | ||
| S17-1 λpir | λpir hsdR pro thi; RP4-2 Tc::Mu Km::Tn7 | [34] |
| BW19851 | RP4-2tet::Mu-1kan::Tn7 integrant; _uidA::pir_ recA1 hsdR17 creB510 endA1 zbf-5 thi | [35] |
| Primersb | ||
| 26-F | 5′ACTGGGATCCAGTTTTAAGCCCGCAAA 3′ | This study |
| 26-R | 5′ ACTGCTGCAGCAATGAACTTCGTCTTG 3′ | This study |
| TN-1 | 5′ GGACCTACCTCATAGACAA 3′ | [19] |
| IS4351-F | 5′ TCAGAGTGAGAGAAAGGG 3′ | [19] |
| promfpp2-F | 5′ ATCAGGATCCGAGCACTACACTTTCTAGA 3′ | [41] |
| promfpp2-R | 5′ GATTGGATCCTGTTCGGTAGTGTAGCA 3′ | [41] |
aAntibiotic resistance phenotypes: ampicillin, Apr; tetracycline, Tcr; erythromycin, Emrb. Antibiotic resistance phenotypes and other features listed in parentheses are those expressed by F. psychrophilum but not by E. coli.
bRestriction sites for cloning are in bold.
DNA technology
Genomic DNA extractions were performed with the Gen Elute Bacterial DNA (Sigma-Aldrich Co., St-Louis, MO, USA) extraction kit. Plasmid DNA was purified with the Gen Elute Plasmid miniprep (Sigma-Aldrich Co.) kit.
PCR amplification products were separated on 1.5% agarose gels and bands were purified with the Illustra™ GFX, PCR DNA and the Gel Band Purification Kit gel extraction system.
Identification of Tn4351 interrupted locus and sequencing of the surrounding DNA region
The insertion of Tn4351 into the genome of the fpgA mutant strain was confirmed by Southern blot analysis. Total DNA from the mutant and parental strains was isolated and digested with HindIII and XbaI restriction enzymes. After agarose gel electrophoresis, DNA fragments were transferred to a nylon membrane (Amershan Bioscience, Uppsala, Sweden) and fixed with UV irradiation. A DIG DNA labeling and detection kit (Roche, Basel, Switzerland) was used to prepare the probe and to perform hybridization. As a probe, a 6.2 kb SalI fragment from pEP4351 containing Tn4351 transposon was used [19].
To isolate and sequence the genomic DNA flanking the Tn4351 chromosomal insertion, DNA of the mutant strain was digested with HindIII followed by a re-ligation process. The resulting circular molecules were used as a template to amplify by inverse PCR the sequences adjacent to the Tn4351 insertion site using a specific pair of primers TN-1/IS4351-F (Table 1) and the Certamp long amplification kit (Biotools B&M Laboratories, Madrid, Spain).
Automated fluorescence sequencing of the PCR amplified products was performed at the Oviedo University DNA analysis facility using BigDye 3.1 Terminator chemistry on an ABI PRISM 3100 Genetic Analyzer platform (Applied Biosystems, CA, USA). Sequences were compared to databases using the Basic Local Alignment Search Tool (BLAST) from the National Center for Biotechnology Information (NCBI).
Complementation of the fpgA gene mutation
To complement the fpgA mutant, the DNA sequence corresponding to the encoding gene was amplified from the parental strain by PCR, using 26-F and 26-R primers and the Expand Long Template PCR System (Roche), obtaining a fragment of 1105 pb (Table 1). BamHI and PstI restriction sites were introduced into the sequences of 26-F and 26-R respectively, in order to clone the PCR product into the plasmid pCP23 that has promoter activity [41] (Table 1). The resulting plasmid was designated pCP23-fpgA (Table 1). Transfer of pCP23-fpgA to F. psychrophilum was carried out by conjugation as previously described [17] and pCP23-fpgA was recovered from the transconjugants, digested with BamHI and PstI and analyzed by agarose gel electrophoresis to confirm the presence of the insert.
Transcriptional fusion analysis based on gfp reporter gene
The previously constructed pCP23-Gfpp2 (Table 1), derived from the GFP-based reporter vector pCP23-G [41], was used for the analysis of the transcriptional activity of the fpp2-fpp1 operon [18] in the context of wild-type and fpgA− strains. The pCP23-Gfpp2 plasmid was introduced by transformation in E. coli S17 λpir and further conjugated into F. psychrophilum fpgA− and wild-type strains as described by Álvarez et al. [17], originating the fpgA−-fpp2 and THC02/90-fpp2 strains (Table 1). For flow cytometry analysis, 5 mL of NB supplemented with tetracycline were inoculated with 50 μL of mid-exponential-phase cultures from fpgA−-fpp2 and THC02/90-fpp2 strains. The THC02/90-G strain (Table 1) was used as the fluorescence emission negative control. They were incubated at 12 °C in NB supplemented with 10 mM CaCl2 in an orbital shaker at 250 rpm. Then, early-stationary phase cultures were washed once with PBS and resuspended in 500 μL of PBS prior to fluorescence emission analysis. Promoter expression quantification was assessed in a flow cytometer (Beckman Coulter Cytomics FC 500) with 488 Ar and 633 HeNe lasers. Green fluorescence was detected on FL-14 channel (505–545 nm) and 10 000 events were acquired from each sample. All cultures were analyzed in triplicate. Fluorescence emission, with and without calcium in culture media, was assessed in a Student’s t-test. Differences were considered significant when p-value < 0.05.
LD50 determination
For LD50 experiments rainbow trout (Oncorhynchus mykiss) weighing between 5 and 7 g were kept in 60-liter tanks at 14 ± 1 °C in continually flowing dechlorinated water.
On arrival at the aquarium, the fish were acclimatized to experimental conditions and 5% were selected randomly and screened for any bacteria. Samples from liver, muscle, blood and spleen were taken and plated onto permissive media (TSA and NAC) and incubated at 18 °C for 72 h. If bacterial growth was observed in these samples, the whole batch of fish was discarded for further experiments. Cultures of F. psychrophilum parental and fpgA− strains were grown to exponential phase, harvested by centrifugation and washed twice with PBS. Cells were resuspended in PBS and serial dilutions were prepared. Groups of 10 fish were challenged by intramuscular injection of 50 μL of dilutions containing 103-109 colony forming units (CFU) for the parental strain and 106-109 CFU for the fpgA− mutant strain, and LD50 was calculated ten days post-infection according to the method of Reed and Muench [43]. In the control group 10 fish were injected with 50 μL of PBS. To verify the presence of F. psychrophilum in dead fish, samples obtained from muscle (the injection zone), brain, liver and eyes were plated onto NAC plates (supplemented with the corresponding antibiotic for mutant strain) and incubated for 72 h at 18 °C. The assay was qualitative and was performed in order to confirm that the dead fish had a massive presence of the bacterium in different organs due to the development of septicemia. After culture, PCR was routinely used for the identification of the bacterium, using specific primers [13,19]. The results are the average of two independent experiments. Animal experiments were performed in accordance with the European legislation governing animal welfare, and they were authorized and supervised by the Animal Experimentation Ethics Committee of Universidad de Oviedo.
Analysis of protein glycosylation
The wild-type, fpgA− and fpgA− carrying pCP23-fpgA plasmid (from here, fpgA+) strains were grown in NB for 120 h at 12 °C, harvested by centrifugation (10 000 rpm, 10 min) and washed twice with Tris–HCl (50 mM pH 6.8). Pelleted cells were re-suspended in 1 mL of the same buffer and cells were broken by ten 12-s sonication with 1 min intervals in an ice bath. Then, the suspension was centrifuged (12 000 rpm, 20 min, 4 °C) and aliquots of the supernatant were used for protein analysis by 14% SDS-PAGE according to the Laemmli methods. Proteins were visualized by silver and Coomassie brilliant blue staining. Glycosylated proteins were detected using the Pro-Q Emerald 300 glycoprotein staining kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. The effect of proteinase K (50 μg/mL) and trypsin (12 μg/mL) on the glycosylated proteins was assessed by incubation of the total protein extract from the wild-type strain at 60 °C for 1 h before separation by SDS-PAGE. When a mixture of both enzymes was used, the reaction with proteinase K was carried out first. Then, an additional incubation in the presence of trypsin was performed under the same conditions. Controls without the presence of proteases were carried out simultaneously in all cases.
Genetic analysis
The BLAST program was used to compare protein sequences and Simple Modular Architecture Research Tools (SMART) [44] for detecting conserved domains. MotifScan software from MyHits was used to identify the motifs present in each sequence. The carbohydrate-active enzymes (CAZy) [45] bank was used to classify the glycosyltransferase. The ProtParam program [46] was used for molecular mass computation and SignalP3.0 [47] to predict the possible location of a signal peptide cleavage site.
Results
Analysis of the Tn4351-disrupted sequence of fpgA
A set of F. psychrophilum mutants were obtained by Tn4351 insertional mutagenesis as described by Álvarez et al. [17] and then in order to study the gliding-virulence relationship, a mutant strain, defective in colony spreading, was further analyzed. Southern blot analysis of this mutant revealed a single transposon insertion in its genome (data not shown). The DNA sequence surrounding the transposon insertion was obtained by inverse PCR as described by Álvarez et al. [19]. The disrupted sequence for the isogenic mutant corresponds to the FP1638 locus (Gen Bank accession no. NC_009613) of the F. psychrophilum genome [16], a gene of 965 bp that encodes for a 319-amino-acid protein with a predicted mass of 38 081 Da. The transposon was inserted after nucleotide 774. The protein function corresponds to a type-2 glycosyltransferase since it presents a catalytic domain between amino acids 4 and 178, characteristic of the subfamily 2 glycosyltransferases [16]. It also shows a high level of similarity with this kind of enzyme from a variety of species such as Dyadobacter fermentans DSM 18053 (Gen Bank accession no. YP_003088235.1), Methanosarcina barkeri Fusaro strain WcaA (Gen Bank accession no. YP_303801.1) and Pelosinus fermentas (Gen Bank accession no. WP_0079027).
The gene appears in the same orientation as FP1637 (Gen Bank accession no. NC_009613.3) and is separated from it by 4 bp in the genome of strain JIP02/86 of F. psychrophilum [16]. An identical situation was defined by PCR and further sequencing for the F. psychrophilum THCO2/90 strain. FP1637 codifies a protein of unknown function which exhibits high sequence identity to predicted WbhW protein of E. coli, similar to a glycosyltransferase enzyme [16]. A sigma 70-like promoter sequence was located twenty base pairs upstream of the FP1637 start codon (Figure 1). These results indicate that FP1637 and FP1638 loci could form an operon regulated by the promoter region mentioned.
Figure 1.
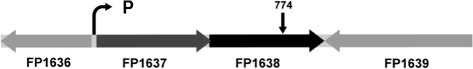
Genomic organization of the region surrounding the FP1638 ( fpgA ) gene in F. psychrophilum. The direction of transcription is indicated by arrows. The position of a putative promoter (P) involved in the regulation of both the FP1637 and FP1638 gene (separated by 4 bp) is indicated. Transposon insertion is located at position 774 of the fpgA locus.
Phenotypic characterization of fpgA− strain and complementation assays
With the aim of making a deeper analysis of the fpgA− strain, different phenotypic characteristics were studied. At 12 °C, the mutant presented a slightly lower growth rate and a decreased final cell density in comparison with the parental strain (Figure 2A). When extracellular proteolytic activity was analyzed, a significant decrease was observed in relation to the parental strain (Figure 2A).
Figure 2.
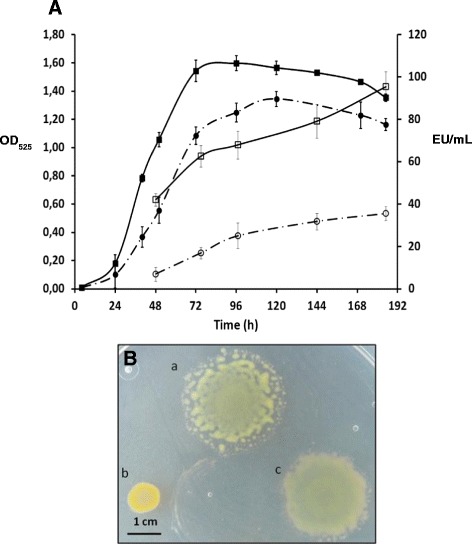
Growth, extracellular proteolytic activity and colony spreading of F. psychrophilum wild-type and fpgA − strains. (A) Growth curve at 12 °C in NBF medium, monitored by determining OD525. (■) wild-type and (●) fpgA− strains. Extracellular caseinolytic activity in cell-free supernatants was determined with azocasein as described by Secades et al. [38]. (□) wild-type and (○) fpgA− strains. EU, enzyme units. (B) Colony spreading of F. psychrophilum strains grown on 1/6NA for 96 h at 20 °C according to Pérez-Pascual et al. [37]. (a) wild-type; (b) fpgA−; and (c) fpgA+ (complemented strain).
In order to go further into the cause of the decrease in the extracellular proteolytic activity of the fpgA− strain, the pCP23-Gfpp2 plasmid, containing a transcriptional fusion between the fpp2 promoter, regulating the main gene driving caseinolytic activity in the bacterium under in vitro conditions [18], and the gfpmut3 gene, was introduced into fpgA−. Strain THC02-90/G with the plasmid pCP23-G harboring a promoterless gfpmut3 gene was utilized as a negative control and no fluorescence emission could be detected under the growth conditions tested. THC02/90-fpp2 and fpgA−-fpp2 showed 117.02 ± 49.42 fluorescence relative units (fru) and 17.46 ± 3.00 fru, respectively (p-value 0.013). Therefore, promoter expression quantification by flow cytometry shows that, in the fpgA− background, fpp2 promoter expression was around 85% lower than that found in the parental strain in the final-exponential phase of the growth curve.
Additionally, fpgA− showed colony spreading defects, forming non-spreading colonies with rounded edges, when grown on 1/6NA medium, whereas the parental strain presented the characteristic high spreading phenotype (Figure 2B). Complementation experiments using pCP23-derived plasmid show that the presence of the plasmid pCP23-fpgA in fpgA− restored both colony spreading (Figure 2B) and extracellular proteolytic activity (89.2 EU/mL at 168 h of incubation).
Protein glycosylation profile of wild-type, mutant and complemented strains
Taking account of the predicted FpgA protein function, an analysis of protein glycosylation profiles of both parental and mutant strains was carried out in order to further characterize its role. The parental strain presented a wide and diffuse glycosylated band of around 22 kDa, whereas fpgA− lacked this band, showing a new one of around 18 kDa (Figure 3A). This band could also be observed after silver staining (Figure 3B). The treatment of the extracts with different proteases show that in all proteolytic processes a new glycosylated band of about 18 kDa appeared (Figure 3C and D), suggesting a proteinaceous nature for the 22 kDa product. Different attempts were carried out in order to purify and further identify the peptide amino acid sequence of the 22 kDa glycosylated protein by mass spectrometry analysis but, unfortunately, they were unsuccessful. The complementation of the fpgA− strain with the pCP23 plasmid harboring the fpgA gene resulted in the recovery of the glycosylation profile of the wild-type strain, with the presence of a band of 22 kDa (Figure 3A).
Figure 3.
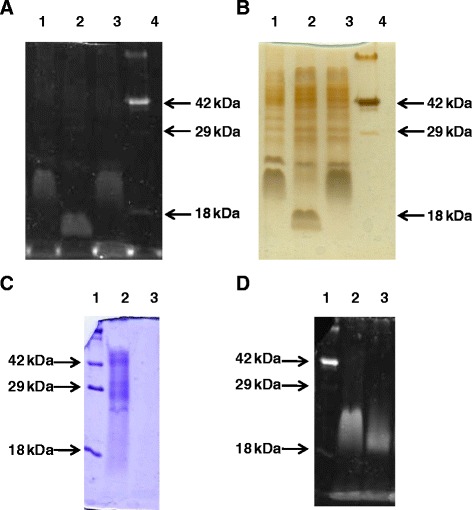
SDS-PAGE and gel staining of glycosylated proteins extracted from F. psychrophilum wild-type, fpgA − , and fpgA + (complemented fpgA − ) strains. Total extracted proteins were separated by 14% SDS-PAGE gels that were (A, D) Pro-Q Emerald specific protein glycosylation; (B) silver; and (C) Coomassie blue, stained. (A) and (B): lane 1, parental; lane 2, fpgA-; and lane 3, fpgA+. In the right of each image, the position of Candy Cane (Life Technologies, Carlasbad, Cal.) with the 42 kDa and 18 kDa glycosylated proteins is indicated (lane 4). (C) and (D): lane 2, wild-type strain; lane 3, wild-type strain extract treated with proteinase K (similar results were obtained when trypsin and a mixture of both enzymes were used). Lane 1, Candy Cane (Life Technologies, Carlasbad, Cal.) molecular mass markers.
LD50 experiments
In order to evaluate the effect of the fpgA mutation in the virulence of F. psychrophilum, LD50 experiments were carried out on rainbow trout. Ten days post-infection, the LD50 value was 2.63 × 106 CFU for the wild-type strain. Under the same conditions, fpgA− showed LD50 values higher than 109 CFU. Thus, the mutant strain for the fpgA locus could be considered as being highly attenuated compared to the wild-type strain. Additionally, visual examination for characteristic erosive skin lesions around the injection site showed that fpgA− infected fishes did not present any kind of tissue damage comparable to that seen in parental strain infections (data not shown). Cultures on NAC medium of samples from the different organs of dead fish show the presence exclusively of the characteristic yellow-pigmented colonies of F. psychrophylum, as was expected in the case of septicemia. Routine PCR analyses show that these colonies corresponded to F. psychrophilum (data not shown).
Discussion
F. psychrophilum mutants obtained by Tn4351 transposition were selected on the basis of the presence of alteration in colony spreading [19]. The fpgA− strain presented the disruption in a gene encoding a protein with a glycosyltransferase function. Its genetic context strongly suggests that the fpgA gene forms an operon with the FP1637 gene, due to the short distance between them (4 bp) and the presence of a promoter sequence upstream of FP1637.
Extracellular proteolytic activity and colony spreading were affected in fpgA−. Complementation experiments show that the fpgA gene is responsible for both effects. Involvement of glycosyltransferases in bacterial motility is not limited to F. psychrophilum; these kind of proteins usually positioned in the outer membrane, generate polysaccharides implicated, for example, in bacterial attachment [48], an essential step for cell movement by gliding. In addition, these enzymes participate in the slime biosynthesis processes in Myxococcus xanthus and, consequently, are involved in the adventurous motility (A motility), where some authors proposed that the bacterium glides over a trail of polysaccharide [49,50]. In the same way, the disruption of three different glycosyltransferases in the marine bacterium Synechococcus sp. resulted in a non-swimming phenotype in mutant strains, indicating that these enzymes are required for motility [51].
Under the assayed conditions, most of the extracellular proteolytic activity detected from the wild-type strain is caused by the metalloproteases Fpp1 and Fpp2 whose encoding genes fpp2-fpp1 form an operon [18]. As shown by GFP-based regulation experiments, the expression of this operon was found to be lower in the fpgA− context than that of the wild-type strain. This result suggests that the decrease of extracellular proteolytic activity detected in the fpgA mutant could be a consequence of down-regulation of, at least, the fpp2-fpp1 operon. As occurring in F. johnsoniae, mutation in genes involved in gliding (gld genes) determined a change in the secretion of extracellular enzymes [52-54]. Until now, it has not been possible to establish a relationship between these two phenomena. However, it seems that a change in regulation affecting extracellular proteolytic activity occurs in parallel to the loss of the ability to move, rather than a physical interaction between the gliding machinery and the secretion systems.
The disruption of the fpgA gene, encoding a glycosyltransferase, caused an altered glycosylation profile in the mutant strain. While the wild-type strain presented a glycosylated element of 22 kDa, fpgA− shows an 18 kDa glycoconjugate. In any case, care must be taken when assigning a molecular mass size by SDS-PAGE in glycosylated proteins. The effect of proteolytic enzymes on the 22 kDa glycosylated band strongly suggests that it has a proteinaceous nature. The fact that a glycosylated core of 18 kDa is still present after the proteolytic treatments could be a consequence of steric impediments that might make it difficult for the proteases to gain access to the whole protein; currently, it is suggested that the addition of carbohydrates to a protein could be related to major resistance to the proteolysis process [23,55]. Taking into account that the function ascribed to FpgA corresponds to a glycosyltransferase, the results suggest that the 22 kDa protein has, at least, double glycosylation modification. One of them is probably carried out by FpgA, since the interruption of the coding locus does not involve lack of glycosylation, but a reduction of the molecular mass of the 22 kDa glycosylated protein to 18 kDa. In this sense, B. fragilis a member of the CFB group, presents a protein with a triple glycosylation modification [29]. Complementation experiments confirm this hypothesis since, the introduction of the fpgA gene into fpgA− led to the recovery of the 22 kDa glycosylated protein. It is interesting to point out that several members of the CFB group show differential glycoprotein production during their growth [28]. Thus, in B. fragilis 8 glycoproteins have been identified [29]; in Elizabethkingia meningoseptica (former Flavobacterium meningosepticum) [56,57] and F. columnare, several secreted proteins showed glycosylations [58]. These data, together with the glycosylation of the F. psychrophilum outer membrane protein OmpA [25,26] and the proportion of putative glycosyltransferases present in the genome of F. psychrophilum, strongly suggest a greater relevance of this type of enzyme in the physiology of the CFB group in relation to other bacteria, as already proposed by Fernandez-Gomez et al. [48]. The importance of the glycosylation process in the virulence of the bacterium is clear since loss of virulence in the mutant strain was definitive. The pleiotropic phenotype of the mutant strain makes the determination of the phenotypic characteristic responsible for the loss of pathogenecity, difficult. Intriguingly, in many Gram-negative pathogenic bacteria most glycoproteins were associated with pathogenesis [59]. This is the case for the glycosylation of the HWM1 adhesin of Haemophilus influenzae [60], the flagella in Campylobacter jejuni [59,61,62] and the protein-forming type IV pili in Neisseria meningitidis [63], amongst others. The lack of virulence seen in the fpgA− strain indicates that this strain could be a suitable candidate for the development of attenuated vaccines for the prevention of CWD. According to the pleiotropic effect generated by the mutation of the fpgA gene, it could not be ruled out that FpgA could be involved in a chaperon system as suggested for some glycosylated proteins in B. fragilis [29].
This work, together with recent data, indicates that protein glycosylation in prokaryotes and, in particular, in the CFB group could play an important role on the biology of these microorganisms, as previously suggested [28,48]. All of this could suggest that this glycosyltransferase has a regulatory role. Additionally, the avirulent mutant strain could be considered a good candidate to be used as a live vaccine in order to control the CWD.
Acknowledgements
The author’s thanks to Dr. Beatriz Álvarez for the generously providing fpgA− mutant and to Paula Yagüe for her technical support in protein analysis. We would also like to thank Jessica Méndez, Pilar Reimundo, Roberto Navais and Desirée Cascales for their permanent help during the development of this work. Esther Gómez and David Pérez-Pascual were the recipients of respective grants from the Spanish Ministerio de Ciencia e Innovación. This project was founded by MICNN (grant AGL2009-07003).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DP-P and EG carried out experiments. JAG conceived the experiments and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
David Pérez-Pascual, Email: david.perez@jouy.inra.fr.
Esther Gómez, Email: uo167556@uniovi.es.
José A Guijarro, Email: jaga@uniovi.es.
References
- 1.Nematollahi A, Decostere A, Pasmans F, Haesebrouck F. Flavobacterium psychrophilum infections in salmonid fish. J Fish Dis. 2003;26(10):563–574. doi: 10.1046/j.1365-2761.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 2.Starliper CE. Bacterial coldwater disease of fishes caused by Flavobacterium psychrophilum. J Adv Res. 2011;2:97–108. doi: 10.1016/j.jare.2010.04.001. [DOI] [Google Scholar]
- 3.Michel C, Antonio D, Hedrick RP. Production of viable cultures of Flavobacterium psychrophilum: approach and control. Res Microbiol. 1999;150:351–358. doi: 10.1016/S0923-2508(99)80061-8. [DOI] [PubMed] [Google Scholar]
- 4.Cepeda C, Garcia-Marquez S, Santos Y. Improved growth of Flavobacterium psychrophilum using a new culture medium. Aquaculture. 2004;238:75–82. doi: 10.1016/j.aquaculture.2004.05.013. [DOI] [Google Scholar]
- 5.Garcia C, Pozet F, Michel C. Standardization of experimental infection with Flavobacterium psychrophilum, the agent of rainbow trout Oncorhynchus mykiss fry syndrome. Dis Aquat Organ. 2000;42:191–197. doi: 10.3354/dao042191. [DOI] [PubMed] [Google Scholar]
- 6.Izumi S, Aranishi F, Wakabayashi H. Genotyping of Flavobacterium psychrophilum using PCR-RFLP analysis. Dis Aquat Organ. 2003;56:207–214. doi: 10.3354/dao056207. [DOI] [PubMed] [Google Scholar]
- 7.Soule M, LaFrentz S, Cain K, LaPatra S, Call DR. Polymorphisms in 16S rRNA genes of Flavobacterium psychrophilum correlate with elastin hydrolysis and tetracycline resistance. Dis Aquat Organ. 2005;65:209–216. doi: 10.3354/dao065209. [DOI] [PubMed] [Google Scholar]
- 8.Arai H, Morita Y, Izumi S, Katagiri T, Kimura H. Molecular typing by pulsed-field gel electrophoresis of Flavobacterium psychrophilum isolates derived from Japanese fish. J Fish Dis. 2007;30:345–355. doi: 10.1111/j.1365-2761.2007.00809.x. [DOI] [PubMed] [Google Scholar]
- 9.Nicolas P, Mondot S, Achaz G, Bouchenot C, Bernardet JF, Duchaud E. Population structure of the fish-pathogenic bacterium Flavobacterium psychrophilum. Appl Environ Microbiol. 2008;74:3702–3709. doi: 10.1128/AEM.00244-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urdaci M, Chakroun C, Faure D, Bernardet J. Development of a polymerase chain reaction assay for identification and detection of the fish pathogen Flavobacterium psychrophilum. Res Microbiol. 1998;149:519–530. doi: 10.1016/S0923-2508(98)80006-5. [DOI] [PubMed] [Google Scholar]
- 11.Cepeda C, Santos Y. Rapid and low-level toxic PCR-based method for routine identification of Flavobacterium psychrophilum. Int Microbiol. 2000;3:235–238. [PubMed] [Google Scholar]
- 12.Wiklund T, Madsen L, Bruun MS, Dalsgaard I. Detection of Flavobacterium psychrophilum from fish tissue and water samples by PCR amplification. J Appl Microbiol. 2000;88:299–307. doi: 10.1046/j.1365-2672.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- 13.del Cerro A, Mendoza MC, Guijarro JA. Usefulness of a TaqMan-based polymerase chain reaction assay for the detection of the fish pathogen Flavobacterium psychrophilum. J Appl Microbiol. 2002;93:149–156. doi: 10.1046/j.1365-2672.2002.01661.x. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara-Nagata E, Eguchi M. Development and evaluation of a loop-mediated isothermal amplification assay for rapid and simple detection of Flavobacterium psychrophilum. J Fish Dis. 2009;32:873–881. doi: 10.1111/j.1365-2761.2009.01066.x. [DOI] [PubMed] [Google Scholar]
- 15.Strepparava N, Wahli T, Segner H, Petrini O: Detection and quantification ofFlavobacterium psychrophilumin water and fish tissue samples by quantitative real time PCR.BMC Microbiol 2014, 14:105. [DOI] [PMC free article] [PubMed]
- 16.Duchaud E, Boussaha M, Loux V, Bernardet JF, Michel C, Kerouault B, Mondot S, Nicolas P, Bossy R, Caron C, Bessières P, Gibrat JF, Claverol S, Dumetz F, Le Hénaff M, Benmansour A. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat Biotechnol. 2007;25:763–769. doi: 10.1038/nbt1313. [DOI] [PubMed] [Google Scholar]
- 17.Alvarez B, Secades P, McBride MJ, Guijarro JA. Development of genetic techniques for the psychrotrophic fish pathogen Flavobacterium psychrophilum. Appl Environ Microbiol. 2004;70:581–587. doi: 10.1128/AEM.70.1.581-587.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Pascual D, Gomez E, Alvarez B, Mendez J, Reimundo P, Navais R, Duchaud E, Guijarro JA. Comparative analysis and mutation effects of fpp2-fpp1 tandem genes encoding proteolytic extracellular enzymes of Flavobacterium psychrophilum. Microbiology. 2011;157:1196–1204. doi: 10.1099/mic.0.046938-0. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez B, Secades P, Prieto M, McBride MJ, Guijarro JA. A mutation in Flavobacterium psychrophilum tlpB inhibits gliding motility and induces biofilm formation. Appl Environ Microbiol. 2006;72:4044–4053. doi: 10.1128/AEM.00128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez B, Alvarez J, Menendez A, Guijarro JA. A mutant in one of two exbD loci of a TonB system in Flavobacterium psychrophilum shows attenuated virulence and confers protection against cold water disease. Microbiology. 2008;154:1144–1151. doi: 10.1099/mic.0.2007/010900-0. [DOI] [PubMed] [Google Scholar]
- 21.Schaffer C, Graninger M, Messner P. Prokaryotic glycosylation. Proteomics. 2001;1:248–261. doi: 10.1002/1615-9861(200102)1:2<248::AID-PROT248>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 22.Messner P. Prokaryotic protein glycosylation is rapidly expanding from “curiosity” to “ubiquity”. Chembiochem. 2009;10:2151–2154. doi: 10.1002/cbic.200900388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benz I, Schmidt MA. Never say never again: protein glycosylation in pathogenic bacteria. Mol Microbiol. 2002;45:267–276. doi: 10.1046/j.1365-2958.2002.03030.x. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt MA, Riley LW, Benz I. Sweet new world: glycoproteins in bacterial pathogens. Trends Microbiol. 2003;11:554–561. doi: 10.1016/j.tim.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Dumetz F, Lapatra SE, Duchaud E, Claverol S, Le Henaff M. The Flavobacterium psychrophilum OmpA, an outer membrane glycoprotein, induces a humoral response in rainbow trout. J Appl Microbiol. 2007;103:1461–1470. doi: 10.1111/j.1365-2672.2007.03359.x. [DOI] [PubMed] [Google Scholar]
- 26.Merle C, Faure D, Urdaci MC, Le Henaff M. Purification and characterization of a membrane glycoprotein from the fish pathogen Flavobacterium psychrophilum. J Appl Microbiol. 2003;94:1120–1127. doi: 10.1046/j.1365-2672.2003.01946.x. [DOI] [PubMed] [Google Scholar]
- 27.Nothaft H, Szymanski CM. Protein glycosylation in bacteria: sweeter than ever. Nat Rev Microbiol. 2010;8:765–778. doi: 10.1038/nrmicro2383. [DOI] [PubMed] [Google Scholar]
- 28.Coyne MJ, Fletcher CM, Chatzidaki-Livanis M, Posch G, Schaffer C, Comstock LE. Phylum-wide general protein O-glycosylation system of the Bacteroidetes. Mol Microbiol. 2013;88:772–783. doi: 10.1111/mmi.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fletcher CM, Coyne MJ, Villa OF, Chatzidaki-Livanis M, Comstock LE. A general O-glycosylation system important to the physiology of a major human intestinal symbiont. Cell. 2009;137:321–331. doi: 10.1016/j.cell.2009.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardel CL, Mekalanos JJ. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect Immun. 1996;64:2246–2255. doi: 10.1128/iai.64.6.2246-2255.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens JM, Galyov EE, Stevens MP. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol. 2006;4:91–101. doi: 10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
- 32.Wassenaar TM, Bleumink-Pluym NM, van der Zeijst BA. Inactivation of Campylobacter jejuni flagellin genes by homologous recombination demonstrates that flaA but not flaB is required for invasion. Embo J. 1991;10:2055–2061. doi: 10.1002/j.1460-2075.1991.tb07736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ingmer H, Brondsted L. Proteases in bacterial pathogenesis. Res Microbiol. 2009;160:704–710. doi: 10.1016/j.resmic.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 34.Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol. 1983;1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 35.Metcalf WW, Jiang W, Wanner BL. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6K gamma origin plasmids at different copy numbers. Gene. 1994;138:1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez B, Guijarro JA. Recovery of Flavobacterium psychrophilum viable cells using a charcoal-based solid medium. Lett Appl Microbiol. 2007;44:569–572. doi: 10.1111/j.1472-765X.2007.02126.x. [DOI] [PubMed] [Google Scholar]
- 37.Perez-Pascual D, Menendez A, Fernandez L, Mendez J, Reimundo P, Navais R, Guijarro JA. Spreading versus biomass production by colonies of the fish pathogen Flavobacterium psychrophilum: role of the nutrient concentration. Int Microbiol. 2009;12:207–214. [PubMed] [Google Scholar]
- 38.Secades P, Alvarez B, Guijarro JA. Purification and characterization of a psychrophilic, calcium-induced, growth-phase-dependent metalloprotease from the fish pathogen Flavobacterium psychrophilum. Appl Environ Microbiol. 2001;67:2436–2444. doi: 10.1128/AEM.67.6.2436-2444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper AJ, Kalinowski AP, Shoemaker NB, Salyers AA. Construction and characterization of a Bacteroides thetaiotaomicron recA mutant: transfer of Bacteroides integrated conjugative elements is RecA independent. J Bacteriol. 1997;179:6221–6227. doi: 10.1128/jb.179.20.6221-6227.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agarwal S, Hunnicutt DW, McBride MJ. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc Natl Acad Sci U S A. 1997;94:12139–12144. doi: 10.1073/pnas.94.22.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez E, Perez-Pascual D, Fernandez L, Mendez J, Reimundo P, Navais R, Guijarro JA. Construction and validation of a GFP-based vector for promoter expression analysis in the fish pathogen Flavobacterium psychrophilum. Gene. 2012;497:263–268. doi: 10.1016/j.gene.2012.01.069. [DOI] [PubMed] [Google Scholar]
- 42.Chakroun C, Grimont F, Urdaci MC, Bernardet JF. Fingerprinting of Flavobacterium psychrophilum isolates by ribotyping and plasmid profiling. Dis Aquat Organ. 1998;33:167–177. doi: 10.3354/dao033167. [DOI] [PubMed] [Google Scholar]
- 43.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–497. [Google Scholar]
- 44.SMART: Simple Modular Architecture Research Tools.http://smart.embl-heidelberg.de/.
- 45.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Expasy, ProtParam tool [http://www.expasy.ch/tools/protparam.html]
- 47.SignalP 3.0 Server [http://www.cbs.dtu.dk/services/SignalP/]
- 48.Fernandez-Gomez B, Richter M, Schuler M, Pinhassi J, Acinas SG, Gonzalez JM, Pedros-Alio C. Ecology of marine Bacteroidetes: a comparative genomics approach. Isme J. 2013;7:1026–1037. doi: 10.1038/ismej.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolgemuth C, Hoiczyk E, Kaiser D, Oster G. How myxobacteria glide. Curr Biol. 2002;12:369–377. doi: 10.1016/S0960-9822(02)00716-9. [DOI] [PubMed] [Google Scholar]
- 50.Yu R, Kaiser D. Gliding motility and polarized slime secretion. Mol Microbiol. 2007;63:454–467. doi: 10.1111/j.1365-2958.2006.05536.x. [DOI] [PubMed] [Google Scholar]
- 51.McCarren J, Brahamsha B. Swimming motility mutants of marine Synechococcus affected in production and localization of the S-layer protein SwmA. J Bacteriol. 2009;191:1111–1114. doi: 10.1128/JB.01401-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braun TF, Khubbar MK, Saffarini DA, McBride MJ. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J Bacteriol. 2005;187:6943–6952. doi: 10.1128/JB.187.20.6943-6952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braun TF, McBride MJ. Flavobacterium johnsoniae GldJ is a lipoprotein that is required for gliding motility. J Bacteriol. 2005;187:2628–2637. doi: 10.1128/JB.187.8.2628-2637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McBride MJ, Braun TF, Brust JL. Flavobacterium johnsoniae GldH is a lipoprotein that is required for gliding motility and chitin utilization. J Bacteriol. 2003;185:6648–6657. doi: 10.1128/JB.185.22.6648-6657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Upreti RK, Kumar M, Shankar V. Bacterial glycoproteins: functions, biosynthesis and applications. Proteomics. 2003;3:363–379. doi: 10.1002/pmic.200390052. [DOI] [PubMed] [Google Scholar]
- 56.Plummer TH, Jr, Tarentino AL, Hauer CR. Novel, specific O-glycosylation of secreted Flavobacterium meningosepticum proteins. Asp-Ser and Asp-Thr-Thr consensus sites. J Biol Chem. 1995;270:13192–13196. doi: 10.1074/jbc.270.22.13192. [DOI] [PubMed] [Google Scholar]
- 57.Reinhold BB, Hauer CR, Plummer TH, Reinhold VN. Detailed structural analysis of a novel, specific O-linked glycan from the prokaryote Flavobacterium meningosepticum. J Biol Chem. 1995;270:13197–13203. doi: 10.1074/jbc.270.22.13197. [DOI] [PubMed] [Google Scholar]
- 58.Vinogradov E, Perry MB, Kay WW. The structure of the glycopeptides from the fish pathogen Flavobacterium columnare. Carbohydr Res. 2003;338:2653–2658. doi: 10.1016/j.carres.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 59.Guerry P. Campylobacter flagella: not just for motility. Trends Microbiol. 2007;15:456–461. doi: 10.1016/j.tim.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Grass S, Buscher AZ, Swords WE, Apicella MA, Barenkamp SJ, Ozchlewski N, St Geme JW 3rd. The Haemophilus influenzae HMW1 adhesin is glycosylated in a process that requires HMW1C and phosphoglucomutase, an enzyme involved in lipooligosaccharide biosynthesis. Mol Microbiol. 2003;48:737–751. doi: 10.1046/j.1365-2958.2003.03450.x. [DOI] [PubMed] [Google Scholar]
- 61.Szymanski CM, Michael FS, Jarrell HC, Li J, Gilbert M, Larocque S, Vinogradov E, Brisson JR. Detection of conserved N-linked glycans and phase-variable lipooligosaccharides and capsules from campylobacter cells by mass spectrometry and high resolution magic angle spinning NMR spectroscopy. J Biol Chem. 2003;278:24509–24520. doi: 10.1074/jbc.M301273200. [DOI] [PubMed] [Google Scholar]
- 62.Young NM, Brisson JR, Kelly J, Watson DC, Tessier L, Lanthier PH, Jarrell HC, Cadotte N, St Michael F, Aberg E, Szymanski CM. Structure of the N-linked glycan present on multiple glycoproteins in the Gram-negative bacterium, Campylobacter jejuni. J Biol Chem. 2002;277:42530–42539. doi: 10.1074/jbc.M206114200. [DOI] [PubMed] [Google Scholar]
- 63.Virji M. Post-translational modifications of meningococcal pili. Identification of common substituents: glycans and alpha-glycerophosphate–a review. Gene. 1997;192:141–147. doi: 10.1016/S0378-1119(97)00082-6. [DOI] [PubMed] [Google Scholar]