

Abstract
Chromosomal positions of common fragile sites differ in lymphoblasts and fibroblasts, with positions dependent on the epigenetically determined density of replication origins at these loci. Because rearrangement of fragile loci and associated loss of fragile gene products are hallmarks of cancers, we aimed to map common fragile sites in epithelial cells, from which most cancers derive. Among the five most frequently activated sites in human epithelial cells were chromosome bands 2q33 and Xq22.1, which are not among top fragile sites identified in lymphoblasts or fibroblasts. FRA16D at 16q23 was among the top three fragile sites in the human epithelial cells examined, as it is in lymphoblasts and fibroblasts, while FRA3B at 3p14.2, the top fragile locus in lymphoblasts, was not fragile in most epithelial cell lines tested. Epithelial cells exhibited varying hierarchies of fragile sites; some frequent epithelial cell fragile sites are apparently not frequently altered in epithelial cancers and sites that are frequently deleted in epithelial cancers are not necessarily among the most fragile. Since we have reported that loss of expression of the FRA3B-encoded FHIT protein causes increased replication stress-induced DNA damage, we also examined the effect of FHIT-deficiency on markers of genome instability in epithelial cells. FHIT-deficient cells exhibited increases in fragile breaks and in γH2AX and 53BP1 foci in G1 phase cells, confirming in epithelial cells that the FHIT gene and encompassing FRA3B, is a “caretaker gene” necessary for maintenance of genome stability.
INTRODUCTION
Almost since their discovery common fragile sites (CFSs) have been the subject of debate concerning the cause of sensitivity to DNA damage at these loci and the biological consequences of damage to genes encompassing CFSs. Letessier et al. (2011) have recently reported that the fragility at FRA3B, encompassed by the FHIT gene, the most active CFS in human lymphoblasts, does not rely on fork slowing or stalling but rather on scarcity of replication initiation events within the locus. In lymphoblasts, but not in fibroblasts, initiation events are absent from the central fragile region of FRA3B, so that replication of this large region within the FHIT gene must be completed by convergence of flanking replication forks. Fibroblasts did not exhibit the fragility at FRA3B observed in the many lymphoblast cells tested over the years since CFS discovery. Nor was FRA16D, the second most active lymphoblast CFS, particularly fragile in fibroblasts, confirming earlier reports of tissue specificity of CFSs (Djalali et al., 1987; Murano et al., 1989a,b). An important conclusion of this study was that CFSs correspond to the initiation poor regions that finish replication latest in a given cell type (Huebner, 2011; Letessier et al., 2011; Debatisse et al., 2012).
The two genes FHIT and WWOX, at FRA3B and FRA16D, are among the most frequently altered by DNA deletion in precancerous and cancerous cells of epithelial origin (Sozzi et al., 1998; Mori et al., 2000; Bartkova et al., 2005; Gorgoulis et al., 2005) presumably due to exposure of tissues of these organs to replicative stress-causing agents. It has been argued that damage to genes at specific CFSs and loss of expression of the gene products contributes to selective growth of the precancers or cancers (Ohta et al., 1996; Huebner and Croce, 2001; Saldivar et al., 2012); counter arguments claim that the frequent deletions within these fragile loci means that loss of their expression occurs as unselected passenger events in cancers (Bignell et al., 2010; Negrini et al., 2010) and that loss of these genes does not contribute to the clonal expansion of precancerous cells. But what if these regions are not among the most fragile loci in epithelia? If not, this finding would support the argument that their very frequent deletion in precancers and cancers of epithelial tissues has contributed to progressive growth of the lesions. To initiate answers this question, we have mapped CFSs in human and murine cells of epithelial origin, in comparison to lymphoblasts.
In addition, we have previously shown, in normal, transformed, and cancer-derived cell lines, that FHIT depletion causes increased replication stress-induced DNA double-strand breaks. Depletion of FHIT protein did not activate the DNA damage response nor cause cell cycle arrest, allowing continued cell proliferation and ongoing chromosomal instability. Furthermore, cells established from Fhit knockout tissue showed rapid immortalization and selection of DNA deletions and amplifications, suggesting that FHIT loss-induced genome instability facilitates transformation (Saldivar et al., 2012; Miuma et al., unpublished data). Thus, we have also examined the effects of loss of expression of the FHIT/FRA3B locus on global genome stability in epithelial cells.
Thus, the goals of the current study were two-fold: (1) to determine the hierarchy of CFSs in epithelial tissue-derived cells; and (2) to confirm findings that FHIT protein deficiency increases expression of markers of global genome instability (Saldivar et al., 2012), increased CFS activation, and increased γH2AX and 53BP1 localization at nuclear foci, in epithelial cells. We have defined genomic locations of CFSs in established epithelial cell lines derived from mouse and human tissues to determine if they differ from those of lymphoblasts, are sites that are frequently broken or mutated in epithelial cancer cells, and if loss of FHIT protein expression causes increased genome instability in such cells. Knowledge of the most active CFSs of epithelial cells may contribute to understanding of the earliest genetic changes that occur in epithelial cells on the path to cancer development.
MATERIALS AND METHODS
Cell Lines and Reagents
Fhit+/+ and Fhit−/− mouse kidney epithelial cells from C57B1/B6 background mice were cultured in MEM with 10% FBS and 100 μg/ml gentamicin. The human mammary epithelial cell lines MCF10A (Soule et al., 1990) and 184A1 cells (Stampfer and Bartley, 1985) were, respectively, grown in F12/DMEM and HuMEC medium with 25 mg Bovine Pituitary Extract and supplements (Gibco/Invitrogen, Grand Island, NY cat#12753-018, 12754-016, 13028-014) (Keller et al., 2012); SV40 T antigen transformed BEAS2B lung epithelial cells and GM1500 lymphoblastoid cells from the Coriell Cell Repository (Camden, NJ) were grown in DMEM and RPMI, respectively, with addition of 10% FBS and gentamicin antibiotic. HCT116 colon cancer cells were grown in RPMI-1640 with addition of 10% FBS and gentamicin antibiotic.
shRNA Silencing
Short hairpin RNAs (shRNAs) can induce sequence-specific gene silencing in mammalian cells (Paddison et al., 2002). A lentiviral construct containing a puromycin resistance gene as well as shFHIT insert was used to target both alleles of FHIT in the MCF10A cells. MCF10A cells (60–80% confluent) were infected with lentiviral shRNAs targeting human FHIT or a nonspecific control shRNA (Santa Cruz Biotechnology, Dallas, TX) using the manufacturer’s recommended protocol. For each 60 mm dish, 1 mg of shRNAs and 6 μl of Lipofectamine 2000 (Invitrogen) were diluted in Opti-MEM (Gibco) and incubated for 45 min. Cells were washed in Opti-MEM, overlaid with the shRNA/Lipofectamine solution, and incubated overnight at 37°C. Verification of shRNA knockdown (KD) of FHIT expression by western blot was performed after selection in 2 μg/ml puromycin.
Immunofluorescence Assays
Cells were grown on eight-chamber slides, fixed with 4% paraformaldehyde, permeabilized with ice-cold 70% ethanol, and blocked in 1% BSA. Primary antisera, rabbit anti-γH2AX, or rabbit anti-53BP1 (Cell Signaling Technologies, Danvers, MA), diluted 1:200, were added and cells incubated with antisera overnight at 4°C. Slides were washed 3 × 10 min in PBS, and secondary antisera (AlexaFluor 488 or 594—conjugated donkey anti-rabbit IgG or anti-mouse IgG, 1:500, Molecular Probes, Grand Island, NY) were added and incubated for 1 hr at room temperature. Slides were washed and mounted using Fluoro-Gel II—with DAPI Images were acquired with an Olympus FV1000 confocal microscope and analyzed using Image J software. For all immunofluorescence assays 100 cells were analyzed in each of three independent experiments.
Western Blot Analysis
Cells were lysed with RIPA buffer supplemented with Protease Cocktail Inhibitors (Thermo Scientific, Pittsburgh, PA), and immunoblot analyses were performed as described previously (Saldivar et al., 2012). Proteins were separated by SDS gel electrophoresis, transferred to nylon membranes, and immunoblotted with antisera against human FHIT, GAPDH, and human TK1 (AbD Serotec, Oxford, UK).
Preparation of Metaphase Spreads and Fragile Site Analysis
Fragile sites were induced by exposure of cells to 0.4 μM aphidicolin (Aph) for 18 hr before harvest. Fhit+/+ or Fhit−/− mouse kidney cells at the 8th subculture, MCF10A1 shCtrl and shFHIT, 184A1, BEAS2B, HCT116, and GM1500 cells were harvested for chromosome preparation by standard conditions after a 2 hr colcemid treatment (0.01 μg/ml) to block cells in mitosis. Cells were trypsinized, pelleted, and resuspended in 0.075 M KCl hypotonic solution for 15 min at 37°C. Cells were fixed in methanol/acetic acid (3:1), dropped on glass slides, and allowed to air dry. G-banding was performed for identification of fragile sites (Seabright, 1971) at specific human and mouse chromosome bands. Chromosomes were analyzed using a Zeiss Axioskop Widefield LM at 100× magnification. Twenty to 44 metaphases were analyzed for assessment of numbers and positions of CFSs in these cells, as noted in the legend to Table 1.
TABLE 1.
CFSs in Order of High to Low Frequency in Epithelial Cells Versus Lymphoblasts and Fibroblasts
| MCF10A | MCF10A FHIT KD | 184A1 | HCT116 | BEAS-2B | GM1500 | Lympho blasts (Mrasek et al., 2010) |
MRC-5 fibroblasts (Letessier et al., 2011) |
Primary Fibroblasts (Murano et al., 1989b) |
|---|---|---|---|---|---|---|---|---|
| 16q23 | 16q23 | 16q23 | 16q23 | 3p14.2 | 16q23 | 3p14.2 | 3q13.3 | 3q26.2 |
| 2q33 | 7q22 | 3q26.2 | 2q33 | 2q33 | 3p14.2 | 16q23.2 | 1p31.1 | 7q11.2 |
| 5q15 | 2q33 | Xq22.1 | 4q21.3 | 16q23 | 4q35 | Xp22.3 | 16q23 | 16q23 |
| Xq22.1 | 9p22 | 12q22 | Xp22.3 | Xq22.1 | 8q22 | 2q32.1 | 2q22 | 1p31 |
| 1q23 | 5q15 | 14q24.3 | Xq22.1 | 13q32 | Xp22.1 | 1p21.3 | 3q28 | 10q11.2 |
| 2q21 | Xq22.1 | 7q11.2 | 7q31.2 | 1p31.2 | 6q25.1 | 6q26 | 3q12 | 12q23 |
| 12q22 | 2q21 | Xp21.2 | 3q25 | 7q31.2 | 1p22 | 7q31.1 | 7q31.1 | 7q31 |
| 1p22 | 1p22 | 13q14.3 | 4p15.2 | 7q11.2 | 9q12 | 7q32.2 | 7q11.2 | |
| 1p31 | 1q44 | 1p31.2 | 3p13 | 22q13 | 2q22 | 1q44 | 3p14.2 | |
| 2q13 | 2q23 | 13q22 | Xp21.2 | 1p22 | 3p25 | 4q31.1 | 13q31 | |
| 0.025 | 0.058 | 0.143 | 0.062 | 0.139 | 0.04 |
Numbers of metaphases analyzed for CFS enumeration in MCF10A, MCF10A FHIT KD, 184A1, HCT116, BEAS2B, and GM1500 is 36, 20, 20, 20, 44, and 20, respectively. Bold numbers in the last row indicate the average number of breaks per chromosome in the epithelial cells versus the lymphoblastoid GM1500 cells.
Copy Number Variation Analysis
DNA was isolated using a DNeasy Kit (Qiagen, cat# 69506, Germantown, MD). High-resolution copy number analysis (CNV) was performed in the Nationwide Childrens’ Hospital Research Institute core facility, using the Agilent Human Genome CGH Microarray 4x180K (Santa Clara, CA).
Statistical Analysis
For boxplots, the bottom and top of the box correspond to the 25th and 75th percentiles, respectively, and whiskers represent data points within 1.5 × IQR (interquartile range). Two-sided t-tests were used to determine significance for data with a normal distribution and equal variances. Non-parametric data were analyzed using the Mann-Whitney rank sum test for single comparisons or using the Kruskal-Wallis test for multiple comparisons. P values <0.05 were considered to indicate statistical significance.
RESULTS
CFSs in Cells of Human and Murine Epithelial Origin
We determined the most frequently activated fragile sites in epithelial cells, using cell lines that would grow for numerous subcultures, so that we could also examine the effect on fragility and genome instability in these cells when the FHIT gene was silenced. Two nonmalignant immortal breast cell lines were used: MCF10A breast epithelial cells immortalized after explanting fibrocystic tissue from a subcutaneous mastectomy surgery (Soule et al., 1990); the 184A1 line derived from normal reduction mammoplasty tissue and immortalized following exposure to a chemical carcinogen (Stampfer and Bartley, 1985); both cell lines are near diploid, estrogen receptor and p16/CDKN1A negative. As shown in the Figure 1 graph in the upper panel, for the MCF10A breast epithelial cell line, the CFSs identified were, in order of frequency: 16q23 (FRA16D/WWOX) > 2q33 (FRA2I), 5q15 (FRA5D) > 1q23, 2q21, 12q22, and Xq22 (representative karyotype in Supporting Information Fig. S1). For 184A1 cells the CFSs were 16q23 > 3q26.2, Xq22, 12q22, 14q24.3 > 7q11.2, and Xp21.2 (not shown graphically, see Table 1 for summary); for the HCT115 colon carcinoma-derived cells the top four CFSs were 16q23, 2q33, 4q21.3, Xp22.3, and Xq22.1, respectively (Table 1); and for comparison with lymphoblastoid cells, the CFSs observed for GM1500 were: 16q23 > 3p14.2 (FRA3B/FHIT) > 4q35 > 8q22, Xp22.1 > 6q25.1, 1p22, and 9q12 (data for all cells examined by us in comparison with lymphoblasts and fibroblasts examined by others are summarized in Table 1).
Figure 1.
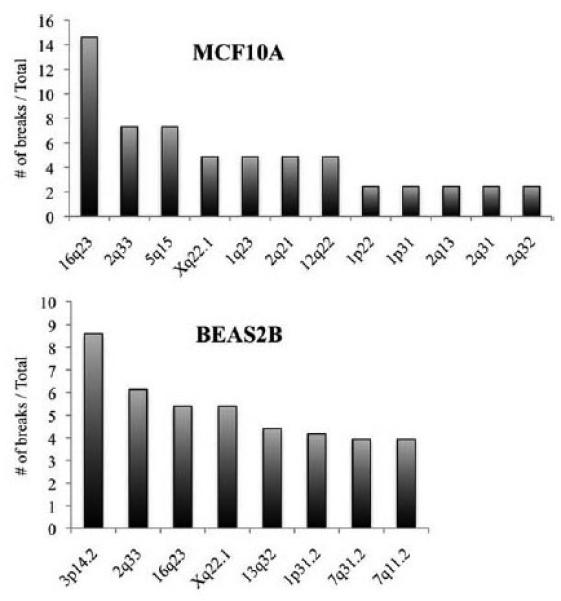
CFSs in human epithelia-derived cells. Upper panel, frequency of Aph-induced breaks at individual CFSs in MCF10A breast epithelial cells; the 16q23/WWOX locus is most frequently expressed in MCF10A cells. Lower panel, expression of CFSs in BEAS2B cells; note that the FRA3B/FHIT locus at 3p14.2 is the most fragile locus in these SV40 T immortalized lung epithelia-derived cells. Otherwise the range of fragile sites is similar to those in MCF10A.
BEAS2B cells, established by transformation of normal human lung bronchial epithelial cells by SV40 T antigen, exhibit three cytogenetically distinct subclones, two of which are near-tetraploid. In BEAS2B cells the most active CFSs were 3p14.2 > 2q33 > 16q23 > Xq22.1 > 13q32 > 1p31.2, 7q31.2, and 7q11.2 (Fig. 1 lower panel, Table 1 for comparative summary), which varies somewhat from active CFSs in lymphoblasts, fibroblasts and the other epithelial cells we examined.
Origins of Replication at CFSs of Epithelial Cells
A genome-scale approach—Repli Seq—has been developed (Hansen et al., 2010) to map temporally ordered replicating DNA using massively parallel sequencing and applied to study regional variation in human DNA replication time across multiple human cell types, providing high-resolution DNA replication patterns relative to cell-cycle time and genomic position; the investigators have shown that different cell types exhibit characteristic replication signatures with plasticity in regional replication time patterns over much of the human genome (Consortium et al., 2011, 2012). This Repli-seq database was used previously to show the association of paucity of replication origins and late timing of replication completion with fragility of FRA3B and FRA16D in lymphoblasts, while fibroblasts showed increased replication origins and earlier completion of replication in these regions (Letessier et al., 2011). We have used the Repli-seq database to compare density of replication origins and replication timing in three CFSs, FRA3B (3p14.2), FRA16D (16q23) and FRA2I (2q33) in human lymphoblast and epithelial cells (Fig. 2). In Figure 2, right panel, are the Repli-seq drawings for the replication origins and S-phase replication timing for a lymphoblast line in which both FRA3B and FRA16D show no replication origins within the fragile regions (bracketed by the vertical lines within the drawing). We know that FRA3B and FRA16D are highly fragile in lymphoblasts and would predict that FRA2I replication would be completed late in S phase in lymphoblasts with contribution of the S4 activated replication origin. The Repli-seq data reproduced in Figure 2, left panel, illustrates the replication programs of the same three regions in the epithelial NHEK cell (normal human epidermal keratinocytes). The replication program for FRA16D is compatible with fragility while FRA3B would be predicted not to be fragile in this epithelial cell, in accord with our data for the MCF10A and 184A1 cells. The replication program for FRA2I is also compatible with fragility in epithelial cells.
Figure 2.
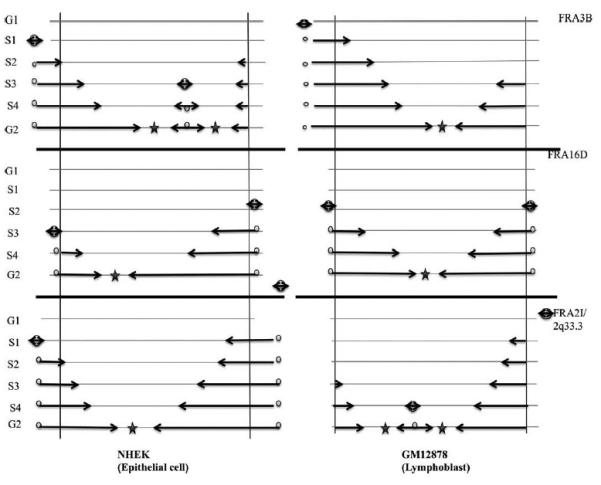
Repli-seq data for FRA3B, FRA16D, and FRA2I in human epithelial and lymphoblast cells. Drawings representative of replication dynamics in the core regions of FRA3B/3p14.2, FRA16D/16q23.3, and FRA2I/2q33.3 from ENCODE Repli-Seq database: Right panel. Human lymphoblast cell; core regions are delimitated by vertical lines and cell cycle phases are indicated on the left with S phase subdivided into four fractions; the cores are poor in initiation events in most lymphoblasts for FRA3B and FRA16D and during unperturbed S phase are replicated by long-traveling forks emanating from origins located in the flanking regions that fire around early or mid-S phase. Convergent forks merge in G2 phase, resulting in late completion of the core replication. At FRA2I/2q33.3, encompassing the PARD3B gene, lymphoblasts show one replication origin at the end of S phase, possibly accounting for a low frequency of CFS activity at 2q33.3 in lymphoblasts compared with FRA3B and FRA16D. Repli-Seq data have been used from Replication Timing by Repli-seq from ENCODE/University of Washington database according to the ENCODE data release policy (Consortium, 2011, 2012). Left panel depicts the core regions for the same loci, FRA3B, FRA16D, and FRA2I, in the epithelial cell line, NHEK.
Effect of Loss of FHIT Protein Expression on CFS Activity in Epithelial Cells
We had previously observed that fibroblasts derived from Fhit knockout mice showed a two-fold increase in average numbers of gaps per chromosome (Turner et al., 2002) and have now shown that loss of FHIT protein expression can lead to global genome instability (Saldivar et al., 2012). Thus, we have examined the effect of FHIT protein expression deficiency on frequency and hierarchy of CFS activation in epithelial cells.
Cytogenetics of noncancerous mouse kidney (MK) epithelia-derived cell lines established from explants of baby mouse kidney of a wild-type (WT) Fhit+/+ and Fhit−/− (KO) mouse (Saldivar et al., 2012; Miuma et al., unpublished data) were examined with and without Aph treatment. In comparisons of the locations and frequencies of activation of CFSs in MK cells with and without Aph treatment (Figs. 3 and 4), we observed approximately twofold increases in the frequencies of fragile breaks in the Fhit−/− cells. Figure 4 (upper panel) shows the distribution and frequencies of activation of the fragile sites in +/+ versus −/− MK cells. Note that the mouse band 8E1 corresponds to FRA16D/WWOX (Krummel et al., 2002) in human and band 14A2 is equivalent to human FRA3B/FHIT. The hierarchy of frequent CFSs differs slightly in the Fhit−/− versus +/+ MK cells, though we do not know if the difference is Fhit-specific. Moreover, MK Fhit silenced cells showed substantially increased levels of chromosomal aneuploidy, including chromosome losses and gains (not shown).
Figure 3.
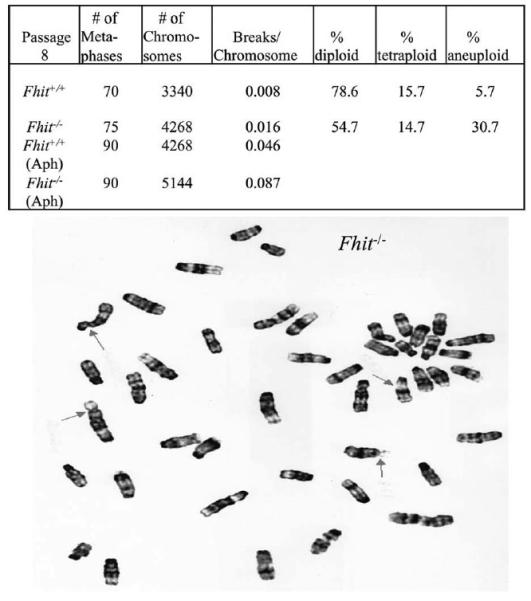
Spontaneous and Aph-induced breaks in Fhit+/+ and Fhit−/− MK cells. Upper panel. Mean overall spontaneous chromosome gaps and breaks per chromosome in Fhit+/+ (WT) and Fhit−/− MK cells. There is an approximately twofold increase in breaks per chromosome in Fhit−/− MK cells compared with the Fhit+/+ cells and an increase in % aneuploidy and tetraploidy in Fhit−/− MK cells relative to the Fhit+/+ cells. Cells were subcultured eight times and metaphase chromosomes prepared and counted (n 5 70 for Fhit+/+; n 5 75 for Fhit−/− metaphases). Lower panel. Representative breaks on Giemsa-stained metaphase of Fhit−/− cells treated with 0.4 lM Aph for 18 hr. Arrows indicate chromosome breaks.
Figure 4.
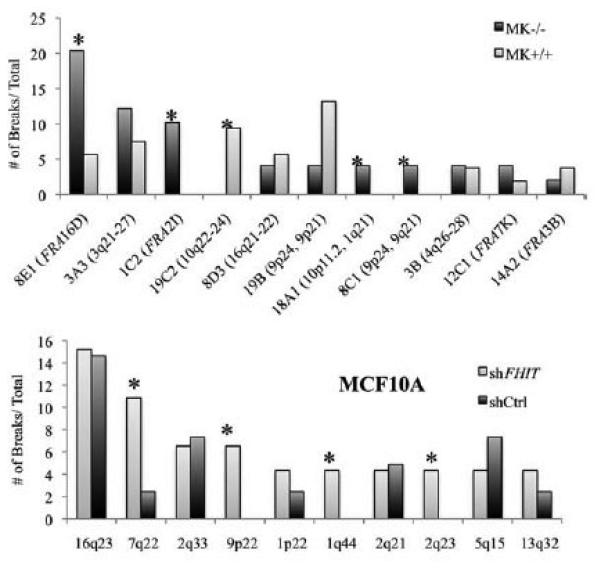
CFSs in epithelial cells with and without FHIT protein expression. Upper panel, comparison of frequencies of Aph-induced breaks at individual CFSs in the Fhit−/− and Fhit+/+ MK cells. Asterisks note fragile sites that vary extensively in frequency in the cells with absence of Fhit expression. The human homologous region for each mouse CFS is noted in parentheses. Lower panel, a similar comparison of numbers of breaks at CFSs in MCF10A cells infected with shCtrl or shFHIT lentivirus vectors; the order of frequency of the most fragile loci varies slightly in the two cell types, but the 16q23/WWOX locus remains the most frequently expressed in both cell lines.
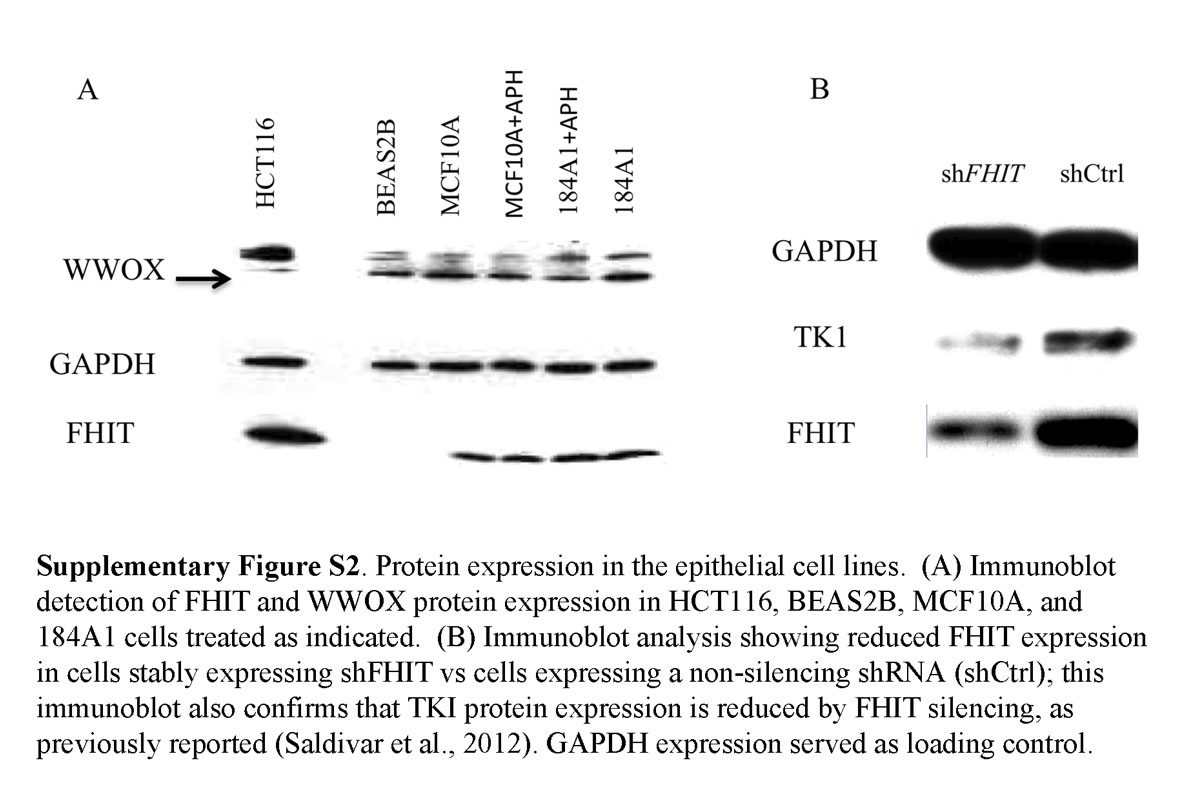
We also examined the frequency of CFS activation after treatment with Aph in the immortal non-cancerous MCF10A cells after FHIT protein expression KD by shRNA. MCF10A cells were transfected with FHIT-specific shRNA to study the effect of FHIT protein KD on CFSs in epithelial cells (see Supporting Information Fig. S2B for illustration of FHIT protein KD by shRNA); we found that there were variations in the frequency of activation of specific CFSs in the FHIT positive and deficient MCF10A cells after 18 hr treatment with 0.4 μm Aph (Fig. 4, lower panel). Karyotypic analysis revealed that the FHIT KD cells exhibited an approximately twofold increase in frequency of gaps and breaks compared with FHIT-positive cells (breaks per chromosome in MCF10A compared with MCF10A/FHIT KD, 0.027/0.058).
Additional Markers of FHIT Loss-Induced Genome Instability in Epithelial Cells
Sister chromatid exchanges (SCEs) can be induced by various genotoxic treatments (Hagmar et al., 1998; Sonoda et al., 1999), suggesting that SCEs reflect a DNA repair process and a link between SCE and DNA replication. CFSs are hot-spots for SCE formation, which are formed by the action of homologous recombination during replication (Sonoda et al., 1999). To measure the frequency of SCEs in FHIT-deficient cells, Fhit−/− and Fhit+/+ MK cells were labeled with BrdU for two full cell cycles (40–48 hr). The frequency of spontaneous SCEs in +/+ MK cells was 0.27 exchanges per chromosome (Figs. 5A and 5B), while MK Fhit−/− cells showed 0.48 exchanges per chromosome, illustrating that loss of FHIT protein in epithelial cells is sufficient to cause a dramatic increase in chromosomal instability, even in the absence of exogenous genotoxic agents.
Figure 5.
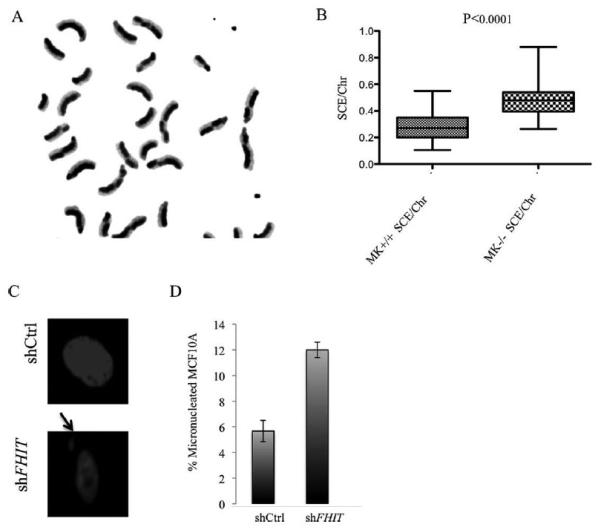
Sister chromatid exchange in mouse kidney cells. (A) Fhit−/− MK metaphase illustrating differential staining of sister chromatids and sites of sister-chromatid exchange (SCE). (B) SCE frequency is increased in Fhit−/− cells relative to Fhit+/+ cells. Statistical significance was assessed using two-sided student’s t-test. (C) Effect of FHIT knockdown on development of micronuclei in MCF10A cells. Representative images of DAPI-stained nuclei in shCtrl and shFHIT cells are shown. Arrow marks a micronucleus. (D) Quantification of micronucleated cells 7 days after shRNA transfections in MCF10A cells. Bar graphs represent the means, and error bars mark the standard deviations. P values determined using a two-sided t-test (P < 0.005).
To address the mechanism for increased chromosomal instability in these FHIT-deficient cells, we hypothesized that it is due to DSBs during DNA replication, as shown previously for several cell types (Saldivar et al., 2012). After FHIT KD in MCF10A cells by shFHIT lentiviral infection, western blot analysis revealed a decrease in the level of FHIT and thymidine kinase 1 (TK1, Supporting Information Fig. S2B) which would cause pyrimidine pool disequilibrium and contribute to genome instability of FHIT-deficient epithelial cells. There was also an increase in the basal levels of phospho-53BP1 and γH2AX, markers of DNA breaks, with an average of 26% of 53BP1 positive shFHIT cells versus 15% positive shCtrl cells (Figs. 6A and 6B) and for γH2AX foci 60% positive shFHIT versus 37% positive shCtrl cells (Figs. 6C and 6D), suggesting that MCF10A FHIT KD cells are not only more sensitive to genotoxic agents but are also undergoing spontaneous chromosomal alterations, supporting our proposal that FHIT-deficient cells accumulate genetic lesions.
Figure 6.
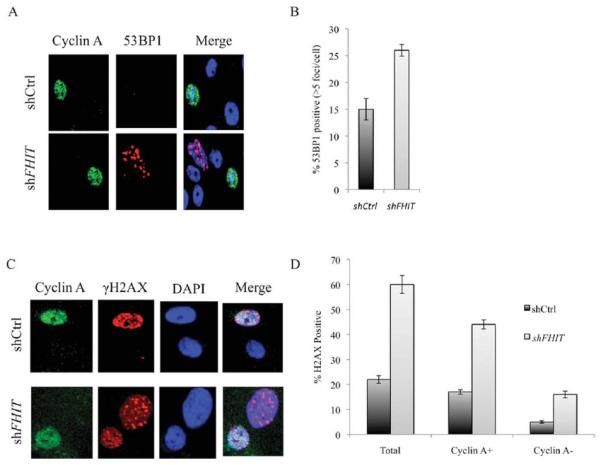
γH2AX foci and 53BP1 bodies in FHIT-deficient MCF10A cells. (A) Immunofluorescent detection of Cyclin A and 53BP1 in MCF10A cells after FHIT knockdown. Cyclin A-positive cells identify S phase cells and establish that the 53BP1 bodies are mostly in G1 cells. Representative images are shown. (B) Histograms of 53BP1 nuclear bodies/G1 phase cell 7 days following shRNA transfection. Data obtained were quantified from three independent experiments and statistical significance determined using a two-sided t-test. Bar graphs represent the means, and error bars mark the standard deviation. The data show that there is a significant association of level of 53BP1 bodies with FHIT-deficiency in MCF10A cells (P < 0.05). (C) Coimmunostaining of MCF10A cells for Cyclin A and γH2AX. (D) Histograms of γH2AX foci show a significant increase in numbers of γH2AX foci in FHIT KD cells compared with FHIT-expressing cells.
To complement the study of markers of genome instability in MCF10A cells after FHIT KD, we have maintained shFHIT silenced MCF10A cells for 30 days, prepared DNA of MCF10A control and MCF10A FHIT KD cells and have assessed chromosome copy number alterations in the MCF10A KD DNA versus MCF10A control DNA by Comparative Genome Hybridization Microarray analysis. In this comparison, we observed copy number gains and losses specific for FHIT KD as listed in Table 2, which also lists genes that are included in the lost and gained loci. It is interesting that even though the MCF10A cells are immortal cells with numerous previous genetic alterations (Soule et al., 1990; Worsham et al., 2006; Kadota et al., 2010), loss of FHIT expression even for only 30 days, induces additional genome instability.
TABLE 2.
Copy Number Alterations Induced by FHIT Knockdown in MCF10A Cells
| Chromosome | Position on chromosome | Fold change | Cytoband | Representative genes altered |
|---|---|---|---|---|
| Loss | ||||
| 3 | 61002345–61080836 | −0.62 | p14.2 | FHIT |
| 7 | 141750430–141785258 | −2.58 | q34 | MGAM |
| 9 | 21817082–21885202 | −4.93 | p21.3 | C9orf53 |
| 12 | 122634121–133779076 | −1.72 | q24.31–q24.33 | NCOR2 |
| 15 | 60687251–61144210 | −0.74 | q22.2 | RORA |
| 16 | 83721255–84451167 | −0.38 | q23.3–q24.1 | CDH13, ADAD2, WFDC1 |
| 22 | 32092559–32239249 | −0.85 | q12.2–q12.3 | DEPDC5 |
| X | 1770348–36331235 | −0.41 | p22.33–p21.1 | ZBED1, CD99 |
| Gain | ||||
| 3 | 51929623–51941665 | 1.29 | p21.2 | IQCF1 |
| 9 | 137332375–137332434 | 1.35 | q34.2 | RXRA |
| 10 | 38240258–38240317 | 1.31 | p11.1 | ZNF25 |
| 11 | 35269915–35276999 | 1.43 | p13 | SLC1A2 |
| 11 | 66006136–134927114 | 0.49 | q13.2–q25 |
PACS1, RAB1B, CD248, RIN1,
B3GNT1, CTSF, SLC29A2 |
| X | 116707254–155232214 | 0.297 | q24–q28 | VAMP7, KLHL13, WDR44 |
Fold change: ~−0.5 indicates hemizygous deletion ~−1 homozygous deletion; ~1.5, three copies gained, ~1.0, 2 copies gained, ~0.5, one copy gained. These FHIT loss-induced CNVs are in addition to those that had already occurred in the parental MCF10A and have been described previously (Soule et al., 1990; Worsham et al., 2006; Kadota et al., 2010).
Genetically unstable cells are known to exhibit abnormal nuclear structures, such as micronuclei (Iarmarcovai et al., 2008). To characterize further the unstable phenotype of the FHIT KD cells, we assessed numbers of micronuclei. Cells were stained with DAPI to visualize chromatin using fluorescence microscopy and subsequently scored for the presence of this type of nuclear alteration. Micronucleus formation in MCF10A cells carrying shFHIT versus shCtrl was increased >2-fold, with 12% of FHIT-silenced cells vs 5.6% of control cells showing micronuclei (Figs. 5C and 5D), further supporting the finding that reduction in FHIT protein expression in established epithelial cell lines leads to the development of chromosomal instability.
DISCUSSION
CFSs in Epithelial Cells
It is of interest that 16q23 (FRA16D/WWOX) shows the greatest fragility in all the epithelial cell lines except BEAS2B where it is the second most active site. This CFS is also in the top three in lymphoblast and fibroblast cell lines, and the encompassing gene, WWOX, is reduced in expression in many types of cancer, including breast cancer (Guler et al., 2004). It is possible that the in vitro growth of the mammary gland cells involved a selective process that led to expansion of clones with the CFS characteristics observed, though it is generally observed that CFSs in primary, cultured, and even EBV-transformed lymphoblasts are similar, so there has so far not been a suggestion that the hierarchy of CFSs in a particular cell type is influenced by selective growth conditions. Likewise it is interesting that 3p14.2 (FRA3B/FHIT) was most fragile in the lung epithelial cell since FHIT protein is very frequently lost or reduced in lung and other preneoplasias (Sozzi et al., 1998; Mori et al., 2000; Wistuba et al., 2000; Bartkova et al., 2005; Gorgoulis et al., 2005), likely influenced by sensitivity of the site to replication stress, and indeed FHIT expression is lost in the BEAS2B cells (Supporting Information Fig. S2A). This suggests that the transformation of the bronchial epithelial parental cells by SV40 T antigen involved selective growth of cell clones with FRA3B fragility and loss of FHIT expression. On the other hand, 2q33 (FRA2I), in the top three CFSs in four of the epithelial cell lines examined, is not a frequently altered CFS in lymphoblasts and thus has not been considered an important CFS previously, though this locus and others identified as CFSs in these epithelial cells may encompass genes that have been reported to be cancer-associated as summarized in Table 3. In all epithelial cells examined at least two CFSs in the top five are the same. Table 3 lists possible tumor suppressor genes that were reportedly deleted in cancer cell lines and are near or possibly within some of the epithelial cell CFSs observed in this study.
TABLE 3.
Candidate Cancer-Relevant Genes Near Epithelial CFS Loci
| CFSs | Putative tumor suppressor genes | Cancer cell lines | Reference |
|---|---|---|---|
| 1p22 | BCL10 | Breast, colon | Lee et al., 1999 |
| 2q21 | R3HDM, LCT, LRP1B | Oral | Stankov et al., 2004; Cengiz et al., 2007 |
| 2q33 | PARD3B, PLCL2 | Lung, breast, glioblastoma | Kohno et al., 1996; Rao et al., 2004; Rothenberg et al., 2010 |
| 5q15 |
POU5F2, KIAA0825,
ANKRD32, MCTP1 |
H209 (Lung), HEC-1 uterus, KYSE-30 esoph |
Finch et al., 2005; Brown et al., 2009; Rothenberg et al., 2010 |
| 7q22 |
COL1A2, PRKAR2B, PSMC2,
PIK3CG, DLX5, DLX6, PCOLCE, PMS2P1, SERPINE1, TRIP6, CUX1, ORC5, LHFPL3, RELN |
Breast, uterine, gastric | Dohi et al., 2010; Mrasek et al., 2010; Boberg et al., 2012 |
| 9p22 | Kidney, oral, bladder | Worsham et al., 1993; Simoneau et al., 2000; Grady et al., 2001 |
|
| 13q32 | FARP1, STK24 | NSCLC (VMRC-LCD, H1975), ESSC (CNV) |
Jongsma et al., 2002; Rothenberg et al., 2010; Kim et al., 2011 |
| Xq22.1 | TCEAL7 | Ovarian, breast, lung | Chien et al., 2008 |
Though these new epithelial cell-associated CFSs have not been precisely mapped within the given chromosome bands, the individual chromosome bands harbor genes, listed in column two, that might be near or within the fragile regions. Column three lists types of epithelial cancer-derived cells that exhibit alterations at the specific chromosome bands as reported in the references listed in column 4.
In comparing the murine MK−/− and +/+ cells, three of the five most active Aph-induced CFSs are the same though the hierarchy of frequency within the top five differs. These are cell lines at early tissue culture passage, p8, but may have been selected for expansion of specific clones. If so, it is interesting that the Fra14A2/Fhit locus is among the CFSs observed in the MK+/+ but not in the −/− cells where its loss could not provide a selective advantage due to knockout of its expression. The murine equivalent (8E1) of the human 16q23/FRA16D/WWOX locus is among the top four CFSs (as in human epithelial cells) in the MK cells and is the most active in the MK−/− cells. Also of interest, the murine equivalent (1C2) of the human 2q33/FRA2I locus is among the top three CFSs in the MK−/− cells; these results may suggest that growth in tissue culture has influenced the growth of variant clones but that there is overall similarity in selection in the murine and human epithelial cells, with variations perhaps dependent on combinations of tissue of origin and transforming influence (loss of FHIT, loss of CDKN2A, gain of SV40 T, with concomitant inactivation of RB1 and TP53 by T antigen).
Would some of the variation in Aph-induced fragile site frequency and hierarchy among the epithelial cells disappear if we studied normal proliferative cells from the various epithelial organs rather than immortalized lines? The answer is not readily apparent although there is near universal agreement that the top two CFSs in lymphoblasts, whether primary or EBV transformed, are FRA3B and FRA16D and FRAXB/Xp22 is always in the top five. We note that we also assayed for Aph-induced CFSs in HCT116 colon cancer cells, a cancer cell line that expresses abundant FHIT protein (Supporting Information Fig. S2A). For this cell line, too, 16q23/FRA16D/WWOX and FRA2I/2q33 are the top CFSs.
Because a top CFS of each epithelial cell line, regardless of the altered gene (shFHIT, CDKN2A mutant, and SV40 T), is FRA16D, there is no indication of a large influence of a specific transforming agent, just as there is not for the EBV-transformed lymphoblasts.
An interesting observation in MCF10A cells following FHIT KD was that the twofold increase in chromosome breaks/gaps included induction of several novel fragile loci including 7q22, 9p22, 1q44, and 2q23. What these loci have in common that make them particularly sensitive to FHIT absence is unknown. Perhaps their expression is a consequence of the reduced replication fork velocity and increased replication fork stalling and collapse in FHIT-deficient cells.
Repli-Seq Data for Epithelial Cells vs Lymphoblasts and Fibroblasts
It is also of interest that the Repli-seq data for three of our top epithelial fragile regions support the model of fragility dependent on placement of DNA replication origins in cells derived from specific tissues (Letessier et al., 2011). As noted in the results section, the FRA3B locus of NHEK epithelial cells shows Repli-seq data that would suggest it is less fragile in these cells than in lymphoblasts as we have observed in the cytogenetics experiments, while FRA16D shows very similar distributions of replication origins in the epithelial and lymphoblast cells from the Repli-seq database. FRA2I/2q33 should be more fragile in epithelial cells and less fragile in lymphoblast cells, showing concordance between our findings and the Repli-seq examples shown in Figure 2.
In summary, epithelial cells show differences and similarities in active CFSs when compared with lymphoblasts, with FRA16D among the most active in both and thus among the most sensitive to replication stress in cells of epithelial and lymphoblast origin. Expression of the WWOX protein is very frequently reduced or absent in human solid tumors, particularly breast cancers (Guler et al., 2004). Breast cancer cells also show reduced or absent expression of FHIT in >60% of cancers and yet the FRA3B/FHIT locus is apparently not fragile in breast epithelial cells. This finding may lend some weight to the idea that loss of FHIT expression is selected for during breast cancer initiation or progression.
Genome Instability in FHIT-Deficient Epithelial Cells
We have previously shown (Saldivar et al., 2012) that loss of expression of FHIT leads to genomic instability in mouse embryo fibroblasts, established mouse kidney cells, and human embryonic kidney cells transformed by Adenovirus 5 T antigen (293 cells after siFHIT), but had not carried out extensive studies of cells derived from non cancerous epithelia of various organs, particularly in combination with cytogenetic studies of CFS activation. Since most solid human cancers, carcinomas and adenocarcinomas, derive from the epithelia of major internal organs, our aim was to examine genome instability and its link to fragile site activation in non-malignant epithelia-derived cell lines. In each experiment in which FHIT expression was reduced, we observed increasing levels of instability whether measured by activity of CFSs or by markers such as SCE, γH2AX, and 53BP1 foci, or micronucleus frequency. Even in cells in which FHIT was absent or reduced, such as BEAS2B, the average number of CFS breaks per chromosome was more than fivefold greater than in MCF10A cells; and when FHIT was knocked down in MCF10A cells the average number of CFS breaks per chromosome was doubled, as in MK−/− cells versus MK+/+ cells.
The CGH array analysis results provide further evidence for increased genome instability in MCF10A FHIT KD cells as copy number alterations in these cells were increased compared with control cells after 30 days of FHIT KD. Parental MCF10A cells exhibit a t(3;9)(pl3;p22) that results in complete loss of the CDKN2A and CDKN2B loci on chromosome 9 (Soule et al., 1990; Kadota et al., 2010), leading to immortality of the cells, though xenografts of the cells are non-tumorigenic (Worsham et al., 2006). After silencing FHIT in MCF10A cells, we observed chromosomal losses or gains at 3p14.2, 7q34, 9q21.3, 15q22.2, 16q23.3, and 3p21.2, which carry tumor suppressor genes or oncogenes, respectively which are associated with cancer development. For example, the RORA gene at FRA15A (15q22.2), deleted after FHIT KD, is expressed in normal breast, prostate and ovarian epithelium and is frequently inactivated in cancers that arise from these organs (Zhu et al., 2006). Increased copy number variations and deletion of possible tumor suppressors after FHIT KD support our proposal that FHIT loss leads to genomic instability and contributes to cancer development.
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors thank Dr. Tsonwin Hai of the Department of Molecular and Cellular Biochemistry at OSU for the MCF10A cells, Dr. Bert Vogelstein of Johns Hopkins University for the HCT116 cells and members of the Huebner lab for helpful discussions. The authors also thank Teresa Druck for MS editing and expert help with preparation of illustrations.
Supported by: US National Institutes of Health, Grant numbers: CA120516, CA154200, and CA132453 (to K.H.); Pelotonia Postdoctoral Fellowship, Ohio State University Comprehensive Cancer Center (to S.A.H.); US National Institutes of Health, Grant numbers: F31CA157150 and T32GM068412 (to J.C.S.); Director, Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy, Grant numbers: DE-AC02-05CH11231 (to M.R.S.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, Teh BT, Deloukas P, Yang F, Campbell PJ, Futreal PA, Stratton MR. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boberg DR, Batistela MS, Pecharki M, Ribeiro EM, Cavalli IJ, Lima RS, Urban CA, Furtado-Alle L, Souza RL. Copy number variation in ACHE/EPHB4 (7q22) and in BCHE/MME (3q26) genes in sporadic breast cancer. Chem Biol Interact. 2013;203:344–347. doi: 10.1016/j.cbi.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Brown KK, Alkuraya FS, Matos M, Robertson RL, Kimonis VE, Morton CC. NR2F1 deletion in a patient with a de novo paracentric inversion, inv(5)(q15q33.2), and syndromic deafness. Am J Med Genet A. 2009;149:931–938. doi: 10.1002/ajmg.a.32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cengiz B, Gunduz M, Nagatsuka H, Beder L, Gunduz E, Tamamura R, Mahmut N, Fukushima K, Ali MA, Naomoto Y, Shimizu K, Nagai N. Fine deletion mapping of chromosome 2q21–37 shows three preferentially deleted regions in oral cancer. Oral Oncol. 2007;43:241–247. doi: 10.1016/j.oraloncology.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Chien J, Narita K, Rattan R, Giri S, Shridhar R, Staub J, Beleford D, Lai J, Roberts LR, Molina J, Kaufmann SH, Prendergast GC, Shridhar V. A role for candidate tumor-suppressor gene TCEAL7 in the regulation of c-Myc activity, cyclin D1 levels and cellular transformation. Oncogene. 2008;27:7223–7234. doi: 10.1038/onc.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012;28:22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Djalali M, Adolph S, Steinbach P, Winking H, Hameister H. A comparative mapping study of fragile sites in the human and murine genomes. Hum Genet. 1987;77:157–162. doi: 10.1007/BF00272384. [DOI] [PubMed] [Google Scholar]
- Dohi O, Takada H, Wakabayashi N, Yasui K, Sakakura C, Mitsufuji S, Naito Y, Taniwaki M, Yoshikawa T. Epigenetic silencing of RELN in gastric cancer. Int J Oncol. 2010;36:85–92. [PubMed] [Google Scholar]
- Finch R, Moore HG, Lindor N, Jalal SM, Markowitz A, Suresh J, Offit K, Guillem JG. Familial adenomatous polyposis and mental retardation caused by a de novo chromosomal deletion at 5q15-q22: Report of a case. Dis Colon Rectum. 2005;48:2148–2152. doi: 10.1007/s10350-005-0177-7. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Grady B, Goharderakhshan R, Chang J, Ribeiro-Filho LA, Perinchery G, Franks J, Presti J, Carroll P, Dahiya R. Frequently deleted loci on chromosome 9 may harbor several tumor suppressor genes in human renal cell carcinoma. J Urol. 2001;166:1088–1092. [PubMed] [Google Scholar]
- Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, McCue P, Huebner K. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100:1605–1614. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- Hagmar L, Bonassi S, Stromberg U, Brogger A, Knudsen LE, Norppa H, Reuterwall C. Chromosomal aberrations in lymphocytes predict human cancer: A report from the European Study Group on Cytogenetic Biomarkers and Health (ESCH) Cancer Res. 1998;58:4117–4121. [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner K. DNA fragility put into context. Nature. 2011;470:46–47. doi: 10.1038/470046a. [DOI] [PubMed] [Google Scholar]
- Huebner K, Croce CM. FRA3B and other common fragile sites: The weakest links. Nat Rev Cancer. 2001;1:214–221. doi: 10.1038/35106058. [DOI] [PubMed] [Google Scholar]
- Iarmarcovai G, Bonassi S, Botta A, Baan RA, Orsiere T. Genetic polymorphisms and micronucleus formation: A review of the literature. Mutat Res. 2008;658:215–233. doi: 10.1016/j.mrrev.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Jongsma AP, Piek JM, Zweemer RP, Verheijen RH, Klein Gebbinck JW, van Kamp GJ, Jacobs IJ, Shaw P, van Diest PJ, Kenemans P. Molecular evidence for putative tumour suppressor genes on chromosome 13q specific to BRCA1 related ovarian and fallopian tube cancer. Mol Pathol. 2002;55:305–309. doi: 10.1136/mp.55.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, Dunn BK, Wakefield LM, Lee MP. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One. 2010;5:e9201. doi: 10.1371/journal.pone.0009201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, Smith AE, Prat A, Perou CM, Gilmore H, Schnitt S, Naber SP, Garlick JA, Kuperwasser C. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci USA. 2012;109:2772–2777. doi: 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Lachuer J, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nobusawa S, Nakazato Y, Tanaka Y, Vital A, Mariani L, Ohgaki H. Alterations in the RB1 pathway in low-grade diffuse gliomas lacking common genetic alterations. Brain Pathol. 2011;21:645–651. doi: 10.1111/j.1750-3639.2011.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Otsuka T, Inazawa J, Abe T, Yokota J. Breakpoint junction of interstitial homozygous deletion at chromosome 2q33 in a small cell lung carcinoma. DNA Res. 1996;3:421–424. doi: 10.1093/dnares/3.6.421. [DOI] [PubMed] [Google Scholar]
- Krummel KA, Denison SR, Calhoun E, Phillips LA, Smith DI. The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1. Genes Chromosomes Cancer. 2002;34:154–167. doi: 10.1002/gcc.10047. [DOI] [PubMed] [Google Scholar]
- Lee SH, Shin MS, Kim HS, Park WS, Kim SY, Lee HK, Park JY, Oh RR, Jang JJ, Park KM, Han JY, Kang CS, Lee JY, Yoo NJ. Point mutations and deletions of the Bcl10 gene in solid tumors and malignant lymphomas. Cancer Res. 1999;59:5674–5677. [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- Mori M, Mimori K, Shiraishi T, Alder H, Inoue H, Tanaka Y, Sugimachi K, Huebner K, Croce CM. Altered expression of Fhit in carcinoma and precarcinomatous lesions of the esophagus. Cancer Res. 2000;60:1177–1182. [PubMed] [Google Scholar]
- Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol. 2010;36:929–940. doi: 10.3892/ijo_00000572. [DOI] [PubMed] [Google Scholar]
- Murano I, Kuwano A, Kajii T. Cell type-dependent difference in the distribution and frequency of aphidicolin-induced fragile sites: T and B lymphocytes and bone marrow cells. Hum Genet. 1989a;84:71–74. doi: 10.1007/BF00210675. [DOI] [PubMed] [Google Scholar]
- Murano I, Kuwano A, Kajii T. Fibroblast-specific common fragile sites induced by aphidicolin. Hum Genet. 1989b;83:45–48. doi: 10.1007/BF00274145. [DOI] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–An evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012;28:295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PH, Arias-Pulido H, Lu XY, Harris CP, Vargas H, Zhang FF, Narayan G, Schneider A, Terry MB, Murty VV. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer. 2004;4:5. doi: 10.1186/1471-2407-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SM, Mohapatra G, Rivera MN, Winokur D, Greninger P, Nitta M, Sadow PM, Sooriyakumar G, Brannigan BW, Ulman MJ, Perera RM, Wang R, Tam A, Ma XJ, Erlander M, Sgroi DC, Rocco JW, Lingen MW, Cohen EE, Louis DN, Settleman J, Haber DA. A genome-wide screen for microdeletions reveals disruption of polarity complex genes in diverse human cancers. Cancer Res. 2010;70:2158–2164. doi: 10.1158/0008-5472.CAN-09-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J, Wheeler LJ, Mathews CK, Huebner K. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012;8:e1003077. doi: 10.1371/journal.pgen.1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971;2:971–972. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- Simoneau M, LaRue H, Aboulkassim TO, Meyer F, Moore L, Fradet Y. Chromosome 9 deletions and recurrence of superficial bladder cancer: Identification of four regions of prognostic interest. Oncogene. 2000;19:6317–6323. doi: 10.1038/sj.onc.1204022. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol. 1999;19:5166–5169. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Ghirelli C, Tornielli S, Sard L, Huebner K, Pierotti MA, Croce CM, Pilotti S. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res. 1998;58:5032–5037. [PubMed] [Google Scholar]
- Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc Natl Acad Sci USA. 1985;82:2394–2398. doi: 10.1073/pnas.82.8.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankov K, Pastore A, Toschi L, McKay J, Lesueur F, Kraimps JL, Bonneau D, Gibelin H, Levillain P, Volante M, Papotti M, Romeo G. Allelic loss on chromosomes 2q21 and 19p 13.2 in oxyphilic thyroid tumors. Int J Cancer. 2004;111:463–467. doi: 10.1002/ijc.20259. [DOI] [PubMed] [Google Scholar]
- Turner BC, Ottey M, Zimonjic DB, Potoczek M, Hauck WW, Pequignot E, Keck-Waggoner CL, Sevignani C, Aldaz CM, McCue PA, Palazzo J, Huebner K, Popescu NC. The fragile histidine triad/common chromosome fragile site 3B locus and repair-deficient cancers. Cancer Res. 2002;62:4054–4060. [PubMed] [Google Scholar]
- Wistuba, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, Fondon JW, III, Garner HR, McKay B, Latif F, Lerman MI, Lam S, Gazdar AF, Minna JD. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60:1949–1960. [PubMed] [Google Scholar]
- Worsham MJ, Benninger MJ, Zarbo RJ, Carey TE, Van Dyke DL. Deletion 9p22-pter and loss of Y as primary chromosome abnormalities in a squamous cell carcinoma of the vocal cord. Genes Chromosomes Cancer. 1993;6:58–60. doi: 10.1002/gcc.2870060111. [DOI] [PubMed] [Google Scholar]
- Worsham MJ, Pals G, Schouten JP, Miller F, Tiwari N, van Spaendonk R, Wolman SR. High-resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res Treat. 2006;96:177–186. doi: 10.1007/s10549-005-9077-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25:2901–2908. doi: 10.1038/sj.onc.1209314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.