

Abstract
A major obstacle to successful treatment of hepatocellular carcinoma (HCC) is its high resistance to cytotoxic chemotherapy due to overexpression of multidrug resistance genes. Activation of the AKT pathway is known to be involved in chemoresistance in HCC; however, the underlying mechanisms modulating the AKT pathway by chemopreventive agents remain unclear. In the present study, we found that indole-3-carbinol (I3C) treatment for tumor cells repressed the AKT pathway by increasing the expression of phosphatase and tensin homolog (PTEN) in HCC xenograft tumor and HCC cell lines. qRT-PCR data showed that the expression of miR-21 and miR-221&222 was significantly reduced by I3C in HCC cells in vitro and in vivo. Reactivation of the AKT pathway via restoration of miR-21 was reversed by I3C. Ectopic expression of miR-21 mediated-accelerated wound healing was abrogated by I3C. Moreover, reducing the expression of miR-21 by anti-miR decreased the resistance of HCC cells to I3C. These results provide experimental evidences that I3C could function as a miR-21 regulator, leading to repression of the PTEN/AKT pathway and opening a new avenue for eradication of drug-resistant cells, thus potentially helping to improve the therapeutic outcome in patients diagnosed with HCC.
Keywords: Indole-3-carbinol, Hepatocellular carcinoma, PTEN, miR-21, miR-221&222
1. Introduction
Liver cancer, primarily hepatocellular carcinoma (HCC), is the third leading cause of cancer death worldwide [1,2]. HCC has strikingly increased in the U.S. over the past several years, reaching an incidence rate of 30,000 new cases each year and is about twice as common in men than in women. HCC is often diagnosed at late-stage when it is ineligible for potentially curative surgical therapies [3]. Moreover, intrinsic resistance to chemotherapeutic agents contributes to the low efficacy of current therapeutic regimens for this cancer type [4]. Many molecular alterations, known to be involved in the malignant transformation and progression, may also be responsible for tumor drug resistance [5]. Among various molecular defects that help HCC cells evade drug-induced apoptosis signaling, constitutive activation of AKT has drawn much attention [6–8]. Activation of the AKT signaling pathway has been reported in more than 40% of human hepatocellular carcinoma [7,8]. Cell proliferation and survival regulated by the AKT pathway plays a critical role in metastasis, thus, making it a potential therapeutic target to combat metastasis.
The chemopreventive potential of indole-3-carbinol (I3C) (Fig. 1A), a naturally occurring phytochemical in cruciferous vegetables [9], is promising due to its unique ability to perturb many oncogenic signaling pathways [10–12]. Research on the mechanisms by which consumption of indole-3-carbinol might influence cancer incidence has mainly focused on its ability to alter estrogen metabolism. Meanwhile, increasing evidence shows that I3C arrests cell growth and induces apoptosis by targeting a broad range of signaling pathways pertinent to cell proliferation, survival, apoptosis [13] and [14] cell cycle regulation [15]. Although clinical trials of I3C have been mostly conducted with estrogen-associated cancers such as prostate [10] cervical dysplasia [16], vulvar intraepithelial neoplasia [17] and breast cancer [18], the precise molecular mechanism(s) by which I3C exerts its inhibitory effects on other cancers such as in lung and liver has not been fully elucidated.
Fig. 1.
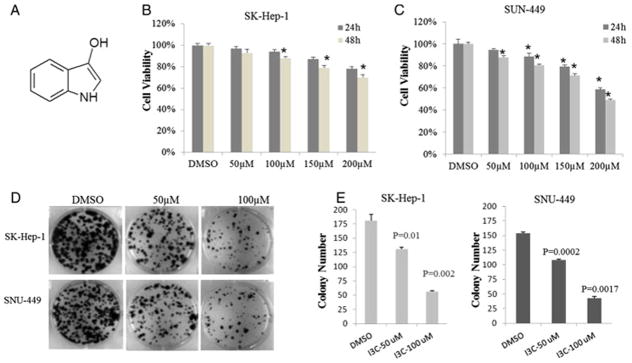
I3C inhibits tumorigenic properties of HCC cells in vitro. (A) Chemical structure of I3C. (B, C) I3C reduces HCC cell proliferation in culture. 1 × 104 SK-Hep-1 cells and SNU-449 cells were seeded in 96-well plates, respectively, and incubated overnight to allow attachment before starting the treatments with DMSO alone or different doses of indole-3-carbinol (I3C) dissolved in DMSO. Cell viability was assessed at indicated time points by MTT. (D) Clonogenic survival of HCC cells decreased when exposed to I3C. 5 × 102 SK-Hep-1 and SNU-449 cells were separately seeded in 6-well plates and allowed to grow for 24 h. The cells were then treated with medium containing 50 and 100 μM of I3C or DMSO. After 2 weeks, HCC cells were washed with PBS twice, fixed with methanol, and then stained with crystal violet and counted. The data are mean ± SD values of triplicates from a three representative experiments (D, E) or mean ± SD values from three independent experiments (A, B), respectively (*P < 0.05; **P < 0.01).
MicroRNAs (miRNAs), a class of highly conserved small non-coding RNAs of ~22–25 nucleotides, have recently emerged as important regulators of gene expression at the post-transcriptional level by either impeding translation or destabilizing target mRNAs [19]. MiRNAs are involved in crucial biological processes including development, differentiation, apoptosis, and proliferation [20,21] through combinatorial regulation either a miRNA may have multiple different mRNA targets or a mRNA target might similarly be targeted by multiple different miRNAs. It is well documented that miRNA expressions are deregulated in almost all human tumors and specific miRNAs play a causal role in cancer pathogenesis [19,20]. OncomiRs such as miR-21, miR-221&222 and miR-181b [22–25] are found to be up-regulated in liver cancer patients. MiRNA overexpression in tumors may contribute to tumorigenesis by down-regulating tumor suppressor genes such as PTEN. Recent studies have shown that natural agents including indole-3-carbinol [26], isoflavone [27] and [28], curcumin [29], 3,3′-diindolylmethane [27] and resveratrol [30] could alter miRNA expression profiles, leading to the inhibition of cancer cell proliferation, promotion of apoptosis, or enhancement of efficacy of conventional cancer therapeutics. Therefore, miRNAs have become attractive targets for liver cancer chemoprevention and chemotherapy.
In this study, we sought to elucidate the roles of I3C in modulating the PTEN expression and activation of AKT pathway by targeting miRNAs in human HCC cell lines.
2. Materials and methods
2.1. Reagents
I3C with >98% purity was purchased from Sigma. A 200 mmol/L stock solution of I3C was prepared in DMSO and stored at −20 °C. The stock solution was diluted in cell culture medium for desirable concentrations. MTT, Tris, glycine, NaCl, SDS and bovine serum albumin were purchased from Sigma-Aldrich. MEM, RPMI-1640, FBS, 0.4% trypan blue vital stain, and antibiotic–antimycotic mixture were obtained from Invitrogen. Rabbit polyclonal antibodies to STAT3 and mouse monoclonal antibodies against PTEN were obtained from Santa Cruz Biotechnology. Phospho-AKT (Ser 473), (Thr 308) and phospho-GSK-3β (Ser9) AKT antibodies were purchased from Cell Signaling Technology. Ki67 antibody was purchased from BD Biosciences. Goat anti-rabbit–horseradish peroxidase (HRP) conjugate and goat anti-mouse HRP were purchased from Sigma-Aldrich. Anti-miR, miR mimic, PTEN siRNA and scrambled control RNAs were obtained from Applied Biosystems Inc. Human HCC cell lines SK-Hep-1 and SNU-449 were obtained from the American Type Culture Collection.
2.2. Cell lines and transfection
The human HCC cell lines, SK-Hep-1 and SNU-449 were cultured in MEM and RPMI 1640 containing 10% FBS and penicillin–streptomycin, respectively. The cells were seeded at a density of 3 × 105 cells in a six-well culture dish. After 24 h, cells were treated with various concentrations of I3C dissolved in DMSO. Control cells were treated with 0.1% DMSO alone. Transfection with 100 nM anti-miR-21 or negative control RNA (Ambion), 100 nM PTEN specific siRNA and negative control (Invitrogen) was conducted with Lipofectamine 2000 according to manufacturer’s instructions. Cells plated at 50–60% confluence in 6-well plates were transfected at 0 and 48 h and then split into two 6-well plates. On the next day, the cells were either treated with DMSO or I3C as described above. The cells were harvested by trypsinization, stained with 0.4% trypan blue, and live and dead cells were counted using a hemocytometer.
2.3. Western blotting
For detection of phosphoproteins, I3C-treated whole-cell extracts were lysed in lysis buffer [31]. Lysates were then centrifuged at 14,000 rpm for 10 min to remove insoluble material and resolved on a 12% SDS-PAGE gel. After electrophoresis, the proteins were blotted onto a PVDF membrane, blocked with 5% nonfat milk, and probed with primary antibodies overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally visualized by a chemiluminescent detection system (Pierce).
2.4. RNA isolation and real-time PCR (qRT-PCR)
Total RNA was extracted using TriZol reagent as recommended by the manufacturer (Invitrogen). To verify the alterations in the expression of specific miRNAs, TaqMan MicroRNA Assay Kit was used. 10 ng of total RNA from each sample was subjected to reverse transcription with a specific miRNA primer. Real-time RT-PCR (qRT-PCR) was then carried out in a total of 25 μl reaction mixture in an ABI Step One Plus real time PCR system. The PCR program was initiated by 10 min at 95 °C before 40 thermal cycles, each at 15 s at 95 °C and 1 min at 60 °C. Data were analyzed according to the comparative Ct method and normalized by U6 expression in each sample [28]. Real-time RT-PCR was performed using SYBR Green as the detection fluorophore. Forward (F) and reverse (R) primers used were as follows: PTEN-F 5′-CGGC AGCATCAAATGTTTCAG-3′ and PTEN-R 5′-AACTGGCAGGTAGAAGGCAA CTC-3′; β-actin-F 5′-CCAAGGCCAACCGCG AGAAGATGAC-3′, and β-actin-R: 5′-AGGGTACATGGTGGTGCCGCCAGAC-3′.
2.5. Plasmid construct
The 3′-UTR of the human PTEN gene with miR-21 binding site (PTEN-wt) and miR-21 binding site deleted (PTEN-del) was PCR amplified from human genomic DNA (wt: forward, 5′-CCG CTC GAG TTC ACA TCC TAC CCC TTT GC-3′ and reverse, 5′-ATA AGA ATG CGG CCG CGG TCC AGA GTC CAG CAT AAA-3′; del: forward, 5′-CCG CTC GAG GCA GTT GGC TAA GAG AGG TT-3′ and reverse, 5′-ATA AGA ATG CGG CCG CGG TCC AGA GTC CAG CAT AAA-3′), and cloned into the Xho1 and Not1 of psiCHECK-2-control vector (Promega, Madison, WI, USA), which was designated psiCHECK-2-PTEN-wt and psiCHECK-2-PTEN-del after sequencing.
2.6. Luciferase assay
Cells were seeded in 24-well plates 24 h before transfection. SK-Hep-1 cells were transiently transfected with wild-type (psiCHECK-2-PTEN-wt) or deletion (psiCHECK-2-PTEN-del) in the presence or absence of I3C using Lipofectamine 2000. Luciferase assays were performed 48 h post-transfection using the dual-luciferase assay system (Promega, Madison, WI, USA) and they were normalized for transfection efficiency with cotransfected Renilla luciferase. All experiments were performed in triplicate.
2.7. Apoptosis assay
For apoptosis assays, floating and adherent cells were harvested at 24 and 48 h after transfection, and then combined and washed with PBS. Annexin V in combination with PI (Santa Cruz Biotechnology) was added to the cells and samples were analyzed within 20 min after staining by a Beckman Coulter EPICS XL (Beckman Coulter Inc., CA, USA) t. Fluorescence was detected by flow cytometry. Experiment was repeated in triplicate.
2.8. Cell colony formation assay
5 × 102 SK-Hep-1 and SNU-449 cells were separately seeded in 6-well plates and allowed to grow for 24 h. Cells were then treated with various concentrations of I3C or 0.1% DMSO. After 2 weeks, cells were washed with phosphate-buffered saline (PBS) twice, fixed with methanol, stained with crystal violet and counted.
2.9. MTT assay
The antiproliferative effect of I3C on HCC cells was determined by the MTT dye uptake method [30]. Briefly, the cells (5 × 103/ml) were incubated in triplicate in a 96-well plate in the presence or absence of the indicated concentration of I3C in a final volume of 0.2 ml for different time intervals at 37 °C. Thereafter, 10 μl of MTT solution (5 mg/ml in PBS) was added to each well. After a 2 hr incubation period at 37 °C, 0.1 ml lysis buffer (20% SDS, 50% dimethylformamide) was added followed by an additional 1 hr incubation at 37 °C, and then the optical density (OD) at 570 nm was measured by Tecan plate reader within 5 min.
2.10. Wound healing assay
For wound healing assay, SK-Hep-1 cells were seeded at a density of 3 × 105 cells in a six-well culture dish. After 24 h, cells were transfected with anti-miR-21, PTEN specific siRNA or respective negative control RNA along with the indicated concentration of I3C or DMSO were plated after 24 h post-transfection. A scratch wound was inflicted diagonally in the monolayer with a pipette tip. Image of the wound was captured at the beginning and every 24 h under the microscope.
2.11. PIP3 ELISA assay
PIP3 ELISA mass Elisa (Echelon Biosciences, Inc.) was used to determine the relative amount of PIP3 present in untreated and 200 μm I3C treated cells at indicated time. Briefly, lipids were extracted from cells and the PIP3 analysis was carried out as described in the manufacturer’s instructions, with the colorimetric signal measured at 450 nm using Tecan plate reader.
2.12. HCC xenograft study
Female athymic nude mice (5–7 weeks of age, from Harlan Laboratory) were used for investigating the antitumor efficacy in vivo. All experimental procedures using mice were done in accordance with protocols approved by The Ohio State University Institutional Animal Care and Use Committee. Briefly, approximately 5 × 106 SK-Hep-1 cells were injected subcutaneously into the right flanks of the nude mice. When tumors reached ~50 mm3, mice were randomly divided into two treatment groups (n = 7) that received the following treatment twice by I. P. per week: 1) I3C at 25 mg/kg body weight or 2) 1% DMSO in PBS. Tumor volumes were calculated as ab2/2 (where a is the longest diameter and b is the shortest diameter) [10]. The mice were sacrificed after 4 weeks of treatment. On sacrifice, tumor tissue from each mouse was harvested and cut into two pieces: one part was snap-frozen in liquid nitrogen for molecular analysis and the other part was fixed in formalin and embedded in paraffin for immunohistochemical staining.
2.13. Histological evaluations
Formalin-fixed tumor sections (4 μm) were immunostained with antibodies against Ki67 [32]. The percentage of Ki67-positive tumor cells was counted in 10 randomly chosen fields (~400 cells) from representative tumor samples from each experimental group. Tumor tissues were also stained with hematoxylin and eosin (H&E) by standard procedures and examined microscopically.
2.14. Statistics
Experiments were repeated three times, each performed in triplicate, and the data were presented as mean ± S.D. Statistical analysis of the data was performed using Student’s t-test. P < 0.05 was considered significant.
3. Results
3.1. I3C inhibits proliferation of SK-Hep-1 and SNU-449 cells
I3C is produced by the breakdown of the glucosinolate glucobrassicin, which can be found at relatively high levels in cruciferous vegetables [9]. The antiproliferation effects of I3C have been assessed in various human cancers [10,12–18]. To demonstrate the broad antiproliferation effects on different liver cancer cells, we used the hepatocellular carcinoma cell lines SK-Hep-1 and SUN-449 to examine the effects of I3C on cell proliferation. For a short term study, SK-Hep-1 and SNU-449 cells were treated with DMSO or 50, 100, 150 and 200 μM I3C dissolved in DMSO (final concentration of 0.1%) for 24 and 48 h, respectively, and then subjected to the MTT assay. As shown in Fig. 1B and C, both cell lines were to the antiproliferative effect of I3C in a dose- and time-dependent manner. SNU-449 cell proliferation was inhibited up to 50% by I3C after a 48 hr treatment at 200 μM I3C (Fig. 1C). Similarly, clonogenic survival of HCC cells was significantly reduced in a dose dependent manner when treated with I3C at 50 and 100 μM for 2 weeks (Fig. 1D and E). Moreover, the size of the colonies was markedly smaller in drug-treated cell cultures. Thus, I3C inhibits both growth and clonogenic survival of HCC cells. I3C has been reported to induce apoptosis and cell cycle arrest in many human cancers such as prostate cancer (10,12,15), breast cancer (11) and myeloid and leukemia cells (14). However, to date, a limited number of study focused on hepatocellular carcinoma. To evaluate the role of apoptosis in the growth-inhibitory effects of I3C, we then characterized the apoptotic cell death induced by I3C. We stained the cells with Annexin V-FITC and the PI dye, and then analyzed the cells by flow cytometry after I3C treatment. Representative results are shown in Fig. S1. After 200 μM I3C treatment, the frequency of apoptotic cells markedly increased to 10.92% (24 h), 27.42% (48 h) and 21.42% (48 h) for SNU-449 and SK-Hep-1 cells, respectively. Quantitative assessment of the percentage of Annexin V-positive cells from 3 independent experiments showed that apoptotic cell death was significantly increased by I3C treatment in HCC cell lines (Fig. S1B). These findings agree with previous studies on I3C-induced apoptosis in other cancers.
3.2. I3C targets the PTEN/AKT signaling pathway and downregulates miR-21, miR-221&222
It has been reported previously that I3C inhibits growth of many types of cancer cells in culture by targeting the AKT signaling pathway, which is activated in response to growth factor stimuli [33]. The proto-oncogene product AKT plays a major role in the cell survival pathway by regulating cell growth and apoptosis. Activation of the AKT signaling pathway is known to initiate from membrane-bound receptors via PI3K and subsequent phosphorylation of downstream signaling targets (e.g. GSK3β), inducing its inactivation and, in turn, inhibiting apoptosis in response to growth factor stimuli.
In this study, we investigated whether activation of the AKT protein was altered in I3C-treated SK-Hep-1 and SNU-449 cells. HCC cell lines treated with I3C did not show any changes at the steady-state levels of total AKT protein, whereas phosphorylations at serine 473 and threonine 308 were significantly inhibited in a time-dependent manner by I3C (Fig. 2A). The dose–response effect on phosphorylation for the key downstream target, GSK-3β, was observed in both cell lines. No significant change was found in total GSK-3 level. The basal level of PTEN, which acts as an inhibitor of the AKT pathway, was slightly detectable in both cell lines. Interestingly, PTEN expression increased significantly with prolonged time of I3C treatment both in cultured SK-Hep-1 and SNU-449 cells. To confirm these results, qRT-PCR analysis of PTEN from DMSO-treated cells or DMSO plus I3C-treated cells revealed PTEN mRNA level was significantly up-regulated in I3C-treated cells versus DMSO-treated mice (Fig. S2). The change of PTEN mRNA is consistent with the alterations in the protein level. PTEN counterbalances PI3K mediated generation of phosphatidylinositol (3–5)-trisphosphate (PIP3) and hence the level of AKT phosphorylation at ser 473 and thr 308. To demonstrate that I3C abrogated the production of PIP3, the cellular content of PIP3 was quantified using a PIP3-competitive ELISA. We confirmed that the amount of PIP3 in the SK-Hep-1 and SNU-449 cell lines was gradually downregulated post treatment by I3C at 24 h and 48 h, respectively (Fig. 2B).
Fig. 2.
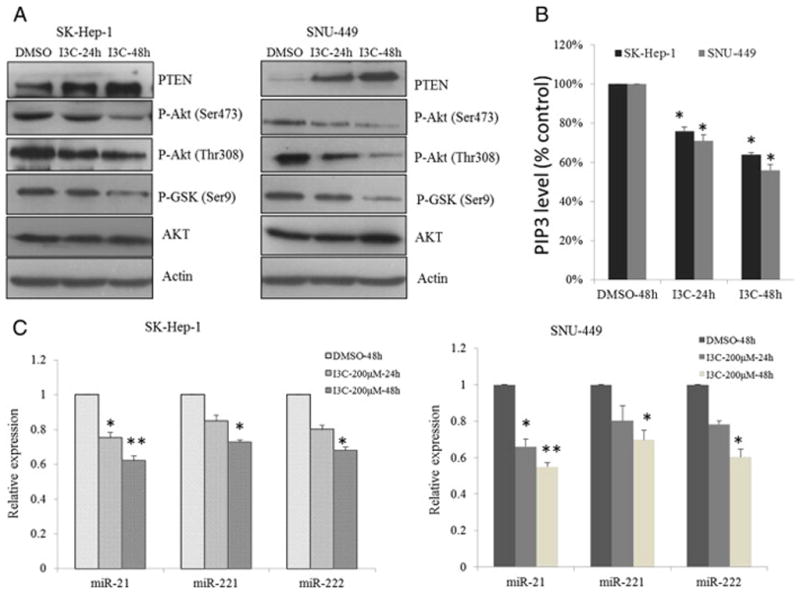
I3C inhibits PTEN/AKT signaling. (A) Inhibition of the PTEN/AKT signaling axis by indole-3-carbinol in HCC cells. Western blot analysis of the time-dependent effects of I3C on phosphorylation of AKT and its downstream targets GSK3β in SK-Hep-1 and SNU-449 cells exposure to 200 μM I3C in 10% FBS-containing DMEM. (B) Using a competitive PIP3 ELISA kit, level of PIP3 in cells untreated or treated with 200 μM of I3C at indicated time was examined. (C) Real-time analysis of the time-dependent effects of I3C on the expression levels of miR-21, miR-221&222 in SK-Hep-1 and SNU-449 cells exposed to 200 μM I3C in 10% FBS-containing DMEM (*P < 0.05; **P < 0.01).
PTEN was shown to be a direct target of miR-21 and contributes to miR-21-induced cell invasion [34]. Previous studies also demonstrated that miR-221&222 enhanced cellular migration through the activation of the AKT pathway and metallopeptidases by targeting PTEN and TIMP3 tumor suppressors [23]. Moreover, miR-21 and miR-221&222 were reported to highly overexpress in primary HCC tissues and cell lines [23,34]. Inhibition of miR-21 in cultured HCC cells increased the expression of PTEN tumor suppressor and decreased tumor cell proliferation, migration, and invasion. To examine whether miRNAs are involved in I3C-induced PTEN up-regulation, we compared the expression profiles of miR-21, miR-221&222 in cells treated with I3C and DMSO. The expression levels of these miRNAs were significantly altered by I3C in both SK-Hep-1 and SUN-449 cells. Compared with cells treated with DMSO, down-regulation of miR-21, miR-221&222 reached 38%, 27%, and 32% in SK-Hep-1 cells (Fig. 2C, left), respectively. This reduction was also obvious in SNU-449 cells with a similar trend (Fig. 2C, right), which was consistent with the aforementioned results of cell proliferation inhibition. Altogether, these findings suggest that PTEN up-regulation could be potentially modulated by I3C activity.
3.3. Inhibition effect of I3C on miR-21-activated PTEN/AKT pathway
To further investigate whether miRNA-mediated regulation of the PTEN/AKT pathway was affected by I3C, SK-Hep-1 cells were transfected with miR-21 and anti-miR21 respectively, and further exposed to I3C for 48 h. Transfection of miR-21 elevated the miR-21 level and decreased PTEN protein accompanied by increased phospho-AKT at Ser473 and Thr308 and phospo-GSK-3β at Ser9 (Fig. 3A and B). Exposure to I3C declined the miR-21 expression by 20% and partially restored PTEN. Consequently, the miR-21-activated AKT pathway was inhibited by I3C (Fig. 3B). In contrast, depletion of miR-21 by anti-miR-21 in SK-Hep-1 cells significantly reduced the phosphorylation of AKT, downstream targets of PTEN, that are key mediators of tumor cell survival (Fig. 3C and D). Cells transfected with anti-miR-21 exposed to I3C show much lower miR-21 expression and higher PTEN expression followed by lower phosphorylation of AKT (Fig. 3C and D). Clearly, miR-21 is involved in the I3C-induced inhibition of the PTEN/AKT pathway. To study whether I3C induced downregulation of miR-21 directly regulates PTEN, we cloned 3′UTR of PTEN with or without miR-21 binding site (PTEN-wt and PTEN-del) to psiCHECK-2 vector and performed a luciferase assay. As shown in Fig. 3E, luciferase activity of psiCHECK-2-PTEN-wt was significantly decreased compared to the vector control in the absence of I3C, thus suggesting PTEN is a direct target of miR-21 (***P < 0.001) which is consistent with previous findings (34). In the presence of I3C, decreased luciferase activity of psiCHECK-2-PTEN-wt was abrogated, and this could be due to I3C-induced miR-21 inhibition (Fig. 2B). As expected, luciferase activity of psiCHECK-2-PTEN-del was not affected by I3C. These results confirmed that I3C induced downregulation of miR-21 directly regulates PTEN. Cells treated with miR-221&222 or anti-miR-221&222, along with I3C did not show obvious change of PTEN/AKT pathway compared to cells treated with miR-221&222 or anti-miR-221&222 alone, which suggested that miR-221&222 may not be involved in inhibition of PTEN/AKT pathway by I3C (data not shown).
Fig. 3.

Inhibitory effect of I3C on miR21- activated PTEN/AKT pathway. (A) I3C abrogated upregulation of miR-21 in HCC cells. HCC cells transfected with 100 nM miR-21 or negative control were exposed to 200 μM I3C for 48 h. (B) miR-21 mediated activation of PTEN/AKT pathway was reversed by I3C. HCC cells transfected with miR-21 or negative control were treated with DMSO or 200 μM I3C for 48 h and subjected to Western blot analysis. (C) I3C enhanced downregulation of miR-21 mediated by anti-miR-21 in HCC cells. HCC cells transfected with 100 nM anti-miR-21 or negative control were exposed to 200 μM I3C for 48 h. (D) anti-miR-21 mediated inhibition of PTEN/AKT pathway was enhanced by I3C. HCC cells transfected with anti-miR-21 or negative control were treated with DMSO or 200 μM I3C for 48 h and subjected to Western blot analysis. (E) The wt or del reporter plasmid was transfected into SK-Hep-1 cells with I3C (I3C+) or without I3C (I3C−). The normalized luciferase activity in the control group was set as relative luciferase activity. Luciferase activity of psiCHECK-2-PTEN-wt was significantly decreased compared with vector control in the absence of I3C (***P < 0.001). In the presence of I3C, decreased luciferase activity of psiCHECK-2-PTEN-wt was abrogated. Luciferase activity of psiCHECK-2-PTEN-del was not affected by I3C. The data are representative of three independent experiments. (F) Accelerated wound healing caused by ectopic expression of miR-21 was reversed by I3C. HCC cells transfected with miR-21 were treated with DMSO or 200 μM for 48 h, and subjected to migration analysis. Results are representative of at least three independent experiments.
To further confirm the above results, wound healing assay was conducted with cells transfected with miR-21, anti-miR-21 or negative control RNA and cultured in medium containing DMSO or 200 μM I3C, and monitored to assess wound healing after 48 h (Fig. 3F). The rate of wound healing of cells transfected with miR-21 was accelerated compared with cells treated with the respective control. Accelerated wound healing was inhibited markedly by I3C indicating that I3C can abolish wound healing in miR-21-overexpressing cells. Conversely, anti-miR-21 decreased wound healing rate in SK-Hep-1 cells with I3C exposure further enhancing this effect. This result was further confirmed by the slow wound healing rate inhibited by both anti-miR-21 and I3C.
3.4. Down-regulation of miR-21 reduces resistance of HCC cells to I3C
The refractoriness of cancer cells to chemotherapy continues to be a major clinical obstacle for successful treatment of a number of cancers including liver cancer. Recently, there has been considerable interest in the potential use of anti-miR oligos as anticancer agents for HCCs because of their preferential accumulation in the liver [35]. As shown in Fig. 4A, the survival rate of SK-Hep-1 cells treated with anti-miR-21 significantly decreased when exposed to I3C at concentrations ranging from 50 to 200 μM. miR-21 expression was reduced by 81% in HCC cells transfected with anti-miR-21, which was further reduced by an additional 10% upon exposure to I3C (Fig. 4A). In contrast, ectopic expression of miR-21 to SK-Hep-1 cells decreased sensitivity to I3C (Fig. 4B). These data indicate that anti-miR-21 can sensitize HCC cells to I3C. We also examined whether miR-221&222 have similar effect on SK-Hep-1 cell to I3C. As expected, miR-221&222 did not enhance the sensitivity of SK-Hep-1 cells to I3C (Fig. 4 A and B), which further confirm the results that no obvious changes of PTEN/AKT pathway were observed between cells treated with miR-221&222 or anti-miR-221&222 along with I3C and cells treated with miR-221&222 or anti-miR-221&222 alone.
Fig. 4.

Inhibition of miR-21, miR-221&222 reduces resistance of HCC cells to I3C. (A, B) MTT assay of HCC cells in the presence of I3C. SK-Hep-1 cells seeded in 96-well plates with a density of 1 × 104 cells/well were treated with anti-miR-21, anti-miR-221&222 or miR-21, miR-221&222 mimic. After 48 h, I3C was added at various concentrations (0–200 μM) and cells were allowed to grow for another 48 h followed by MTT assay. The absorbance at 595 nm of treated cells was divided by that of the untreated cells (which was taken as 100%) to assess the percentage of growth, which was then plotted as a function of the I3C concentration (*P < 0.05; **P < 0.01).
3.5. I3C suppresses the growth of HCC xenograft tumors in vivo
To assess the inhibition of I3C on HCC growth in vivo, SK-Hep-1 cells were used to generate xenograft tumors in athymic nude mice. Athymic nude mice bearing established subcutaneous SK-Hep-1 tumors around 50 mm3 were treated with I3C twice a week at 25 mg/kg by I.P. injection for one month (n = 6). As shown in Fig. 5, the tumor size measured by a caliper demonstrated significant inhibition of SK-Hep-1 tumor growth as early as 9 days after the first injection of I3C. Notably, I3C treatment inhibited tumor size by 50% and weight by 60%, respectively, relative to vehicle-treated controls (P < 0.01) (Fig. 5B and C).
Fig. 5.
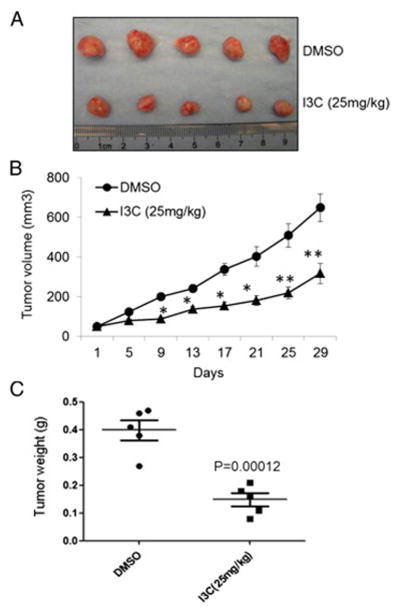
I3C suppresses tumor growth in nude mice. Athymic nude mice bearing SK-hep-1 xenograft tumors were treated with I3C at 25 mg/kg twice a week. (A) Photos of representative xenograft tumors harvested at the end of the treatment. (B) Suppressive effect of I3C on the growth of established SK-Hep-1 xenograft tumors in nude mice relative to that in vehicle-treated controls (n = 7). (C) Statistical analysis of tumor weight (*P < 0.05; **P < 0.01).
3.6. I3C inhibited PTEN/AKT pathway in vivo
We next sought to investigate whether the tumor-suppressive mechanism of I3C identified in vitro also occurs in vivo by evaluating representative intratumoral biomarkers (e.g. PTEN/AKT pathways) and miR-21, miR-221&222. Real time PCR analysis indicated miR-21, miR-221&222 were down-regulated by I3C up to 40% in SK-Hep-1 xenograft tumors (Fig. 6A). Importantly, relative to the DMSO-treated controls, PTEN was markedly increased in the tumors treated with I3C, and accompanied reduction of AKT phosphorylation at Ser 473 and Thr 308 and the levels of p-GSK (Ser 9) (Fig. 6B) which represent hallmark bio-markers of I3C-induced inhibition of PTEN/AKT. Moreover, the suppression of SK-Hep-1 tumor growth by I3C was reflected in a significant reduction in the number of proliferating cells in the tumor, as determined by Ki67 immunostaining (Fig. 6C and D). In summary, these findings suggest that I3C inhibited PTEN/AKT pathway, simultaneously down-regulated miR-21, miR-221&222 in vivo.
Fig. 6.

The inhibitory effect of I3C on PTEN/AKT pathway in vivo is mediated partly through miR-21, miR-221&222. (A) Real-time PCR analysis of miR-21, miR-221&222 expressions in tumors. (B) Western blot analysis of PTEN/AKT pathway in tumors developed in mice treated with I3C. (C) Photographs of H&E-stained sections of tumors formed in nude mice treated as indicated (top) and representative Ki67-immunostained tumor sections. (D) Proliferation indices of SK-Hep-1 tumors from each treatment group, as determined by the percentage of Ki67-positive cells in representative tumor samples (n = 3).
4. Discussion
Despite rapid progress in detection and therapy, advanced HCC still remains a major clinical problem due to the drug resistance. Seeking agents or molecules to enhance cancer cell sensitivity to therapy is the long-term goal to improve the therapeutic efficacy. HCC exhibits alterations in the abundance of specific miRNAs with oncogenic and tumor suppressor activities [23,36–38]. The relationship between aberrant miRNA expression and HCC development indicates that miRNAs could be the potential targets for chemopreventive and chemotherapeutic agents. Accordingly, miR-21 and miR-221&222 are the most frequently up-regulated microRNAs associated with cancer development, including liver [23,34], lung [23], glial [24], and colorectal cancer [22]. It has been shown that inhibition of miR-21 by anti-oligos decreased tumor cell proliferation, migration, and invasion in cultured HCC cells via an increase in PTEN expression and downstream events involving AKT phosphorylation [34]. Moreover, miR-221&222 was reported to induce TRAIL resistance and enhance cellular migration through the activation of the AKT pathway by targeting PTEN. The tumor suppressor PTEN regulates the PI3K/AKT, a major cell survival pathway that plays a key role in the development of drug resistance, including TRAIL [23]. Thus, we studied the role of miR-21, miR-221&222 in I3C induced growth inhibition in HCC cells.
In this study, we found that expression of miR-21, miR-221&222 was significantly down-regulated by the chemopreventive drug I3C in vitro and in HCC xenograft tumors (Figs. 1 and 6). Moreover, ectopic expression of miR-21 mediated down-regulation of PTEN and activation of AKT/GSK and abrogated I3C-induced wound healing reduction. As expected, depletion of miR-21 by anti-miR-21 along with exposure to I3C significantly increased PTEN, inhibited AKT/GSK pathway and potentiated I3C-induced wound healing reduction. In the presence of I3C, decreased luciferase activity of psiCHECK-2-PTEN-wt was abrogated and this may be due to I3C-induced miR-21 inhibiton (Fig. 3E) which further confirmed that I3C induced down-regulation of miR-21 directly regulates PTEN. These results suggested the combination of I3C and anti-miR-21 treatment has a synergetic effect on inhibition of PTEN/AKT pathway respectively relative to either depletion of miR-21 or exposure to I3C alone. These findings suggested that HCC growth inhibition caused by I3C could be mediated, at least in part, by modulating miR-21 and its targets. Our data are in accordance with the results reported by other researchers [13,16]. Omar et al. has shown the down-regulation of AKT and up-regulation of downstream factors such as phosphorylated GSK, CCND1 and C-MYC during HCC development and reversion of the effects by chemopreventive agents, [1-(4-Chloro-3-nitrobenzenesulfonyl)-1H-indol-3-yl]-methanol (OSU-A9), an I3C derivative in a mouse model of hepatocellular carcinoma [13]. Similarly, I3C administration up-regulated the PTEN expression by reducing the expression of miR-21 in vinyl carbamate-induced lung cancer model [26]. In contrast, it has been reported that DIM, a primary I3C derivative, significantly reduces cell proliferation of human breast cancer cells via up-regulation miR-21, which subsequently down-regulated CDC25A [39]. These findings indicate that miR-21 functions as a pleiotropic oncomiR in cancer development either by reducing the expression of tumor suppressors such as PTEN [26] or oncogenes such as CDC25A [39]. Mounting evidence indicates that the role of miRNAs in carcinogenesis appears to be complicated, in the sense that both oncogenic and tumor suppressive effects were reported in different cancers. The expression level of miR-221&222 altered in I3C-treated cell lines and tumors suggest that these two microRNAs might also contribute to the I3C-induced reduction of PTEN. However, combination of either ecotopic expression of miR-221&222 or depletion of miR-221&222 with I3C treatment did not show significant change on PTEN and AKT/GSK pathway compared with single treatment. Importantly, exogenous delivery of miR-21, miR-221&222 enhanced resistance of HCC to I3C and inhibition of miR-21, miR-221&222 inversely decreased HCC sensitivity to I3C. These results indicated that miR-221&222 are involved in the I3C induced HCC tumorigenic property inhibition but not by regulating PTEN and AKT/GSK pathway. In contrast, combined treatment with miR-21 and I3C contributes to reduced HCC tumorigenicity by activating PTEN and inhibiting AKT/GSK pathway. Taken together, by definition of a sensitivity enhancer for cancer therapy, the given miRNAs themselves (at certain expression levels) may not have any, or have only minor effects on cancer cell proliferation, apoptosis, or cell cycle; however, in combination with another treatment, additive or synergistic effects can be observed. This effect of miRNAs provides a novel platform for targeting resistant cancer cells.
5. Conclusions
In summary, to the best of our knowledge this is the first report on the down-regulation of miR-21 by the chemopreventive drug I3C in a HCC xenograft model and HCC cell culture, and this can explain inhibition of the PTEN/AKT pathway induced by I3C. Our findings could be used to examine the efficacy of chemopreventive agents and the mechanisms through which chemopreventive agents inhibit carcinogenesis. Another potential use of miRNAs in cancer chemoprevention is to detect and monitor the toxic effects of chemopreventive agents. Our results clearly demonstrate that specific targeting of miRNAs by I3C could open a new avenue for eradication of drug-resistant cells, thus potentially improving survival outcome in patients diagnosed with malignancies.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This work was supported by NSF Nanoscale Science and Engineering Center (NSEC) grant EEC-0425626 and NIH grants R01DK088076 and R21CA086978.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamcr.2014.10.017.
References
- 1.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Blum HE. Hepatocellular carcinoma: therapy and prevention. World J Gastroenterol. 2005;11:7391–7400. doi: 10.3748/wjg.v11.i47.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas M. Molecular targeted therapy for hepatocellular carcinoma. J Gastroenterol. 2009;44:136–141. doi: 10.1007/s00535-008-2252-z. [DOI] [PubMed] [Google Scholar]
- 5.D’Alessandro N, Poma P, Montalto G. Multifactorial nature of hepatocellular carcinoma drug resistance: could plant polyphenols be helpful? World J Gastroenterol. 2007;13:2037–2043. doi: 10.3748/wjg.v13.i14.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH, Chen PJ, Cheng AL. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2011;337:155–161. doi: 10.1124/jpet.110.175786. [DOI] [PubMed] [Google Scholar]
- 7.Hu TH, Huang CC, Lin PR, Chang HW, Ger LP, Lin YW, Changchien CS, Lee CM, Tai MH. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer. 2003;97:1929–1940. doi: 10.1002/cncr.11266. [DOI] [PubMed] [Google Scholar]
- 8.Nakanishi K, Sakamoto M, Yamasaki S, Todo S, Hirohashi S. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer. 2005;103:307–312. doi: 10.1002/cncr.20774. [DOI] [PubMed] [Google Scholar]
- 9.Beier RC. Natural pesticides and bioactive components in foods. Rev Environ Contam Toxicol. 1990;113:47–137. doi: 10.1007/978-1-4612-3366-4_2. [DOI] [PubMed] [Google Scholar]
- 10.Chinni SR, Sarkar FH. Akt inactivation is a key event in indole-3-carbinol induced apoptosis in PC-3 cells. Clin Cancer Res. 2002;8:1228–1236. [PubMed] [Google Scholar]
- 11.Rahman KM, Li Y, Sarkar FH. Inactivation of Akt and NF-kappaB play important roles during indole-3-carbinol-induced apoptosis in breast cancer cells. Nutr Cancer. 2004;48:84–94. doi: 10.1207/s15327914nc4801_12. [DOI] [PubMed] [Google Scholar]
- 12.Weng JR, Tsai CH, Kulp SK, Wang D, Lin CH, Yang HC, Ma Y, Sargeant A, Chiu CF, Tsai MH, Chen CS. A potent indole-3-carbinol derived antitumor agent with pleiotropic effects on multiple signaling pathways in prostate cancer cells. Cancer Res. 2007;67:7815–7824. doi: 10.1158/0008-5472.CAN-07-0794. [DOI] [PubMed] [Google Scholar]
- 13.Omar HA, Sargeant AM, Weng JR, Wang D, Kulp SK, Patel T, Chen CS. Targeting of the Akt-nuclear factor-kB signaling network by [1-(4-chloro-3-nitrobenzenesulfonyl)-1H-indol-3-yl]-methanol(OSU-A9), a novel indole-3-carbinol derivative, in a mouse model of hepatocellular carcinoma. Mol Pharmacol. 2009;76:957–968. doi: 10.1124/mol.109.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takada Y, Andreeff M, Aggarwal BB. Indole-3-carbinol suppresses NF-κB and IκBα kinase activation, causing inhibition of expression of NF-κB-regulated antiapoptotic and metastatic gene products and enhancement of apoptosis in myeloid and leukemia cells. Blood. 2005;106:2641–2649. doi: 10.1182/blood-2004-12-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 16.Bell MC, Crowley-Nowick P, Bradlow HL, Sepkovic DW, Schmidt-Grimminger D, Howell P, Mayeaux EJ, Tucker A, Turbat-Herrera EA, Mathis JM. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynecol Oncol. 2000;78:123–129. doi: 10.1006/gyno.2000.5847. [DOI] [PubMed] [Google Scholar]
- 17.Naik R, Nixon S, Lopes A, Godfrey K, Hatem MH, Monaghan JM. A randomized phase II trial of indole-3-carbinol in the treatment of vulvar intraepithelial neopla-sia. Int J Gynecol Cancer. 2006;16:786–790. doi: 10.1111/j.1525-1438.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- 18.Reed GA, Peterson KS, Smith HJ, Gray JC, Sullivan DK, Mayo MS, Crowell JA, Hurwitz A. A phase I study of indole-3-carbinol in women: tolerability and effects. Cancer Epidemiol Biomarkers Prev. 2005;14:1953–1960. doi: 10.1158/1055-9965.EPI-05-0121. [DOI] [PubMed] [Google Scholar]
- 19.Bushati N, Cohen SM. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 20.Calin GA, Croce CM. MicroRNA signatures in human cancer. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 21.Friedman JM, Jones PA. MicroRNAs: critical mediators of differentiation, development and disease. Swiss Med Wkly. 2009;139:466–472. doi: 10.4414/smw.2009.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 23.Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16:498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Kano M, Seki N, Kikkawa N, Fujimura L, Hoshino I, Akutsu Y, Chiyomaru T, Enokida H, Nakagawa M, Matsubara H. miR-145, miR-133a and miR-133b: tumor-suppressive miRNAs target FSCN1 in esophageal squamous cell carcinoma. Int J Cancer. 2010;127:2804–2814. doi: 10.1002/ijc.25284. [DOI] [PubMed] [Google Scholar]
- 25.Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, Ghoshal K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2010;29:1787–1797. doi: 10.1038/onc.2009.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 2010;31:252–258. doi: 10.1093/carcin/bgp208. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Kong D, Ahmad A, Bao B, Dyson G, Sarkar FH. Epigenetic deregulation of miR-29a and miR-1256 by isoflavone contributes to the inhibition of prostate cancer cell growth and invasion. Epigenetics. 2012;7:940–949. doi: 10.4161/epi.21236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7:464–473. doi: 10.1158/1535-7163.MCT-07-2272. [DOI] [PubMed] [Google Scholar]
- 30.Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31:1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Yu B, Wu Y, Lee RJ, Lee LJ. Efficient down-regulation of CDK4 by novel lipid nanoparticle-mediated siRNA delivery. Anticancer Res. 2011;31:1619–1626. [PubMed] [Google Scholar]
- 32.Sargeant AM, Rengel RC, Kulp SK, Klein RD, Clinton SK, Wang YC, Chen CS. OSU-HDAC42, a histone deacetylase inhibitor, blocks prostate tumor progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008;68:3999–4009. doi: 10.1158/0008-5472.CAN-08-0203. [DOI] [PubMed] [Google Scholar]
- 33.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JK, Kogure T, Nuovo GJ, Jiang J, He L, Kim JH, Phelps MA, Papenfuss TL, Croce CM, Patel T, Schmittgen TD. miR-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer Res. 2011;71:7608–7616. doi: 10.1158/0008-5472.CAN-11-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai S, Nasser MW, Wang B, Hsu SH, Datta J, Kutay H, Yadav A, Nuovo G, Kumar P, Ghoshal K. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284:32015–32027. doi: 10.1074/jbc.M109.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, Schmittgen TD, Croce C, Ghoshal K, Jacob ST. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–1161. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin Y. 3,3′-Diindolylmethane inhibits breast cancer cell growth via miR-21-mediated Cdc25A degradation. Mol Cell Biochem. 2011;358:345–354. doi: 10.1007/s11010-011-0985-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.