

Abstract
The past fifteen years has seen a revolution in our understanding of ionotropic glutamate receptor (iGluR) structure, starting with the first view of the ligand binding domain (LBD) published in 1998, and in many ways culminating in the publication of the full-length structure of GluA2 in 2009. These reports have revealed not only the central role played by subunit interfaces in iGluR function, but also myriad binding sites within interfaces for endogenous and exogenous factors. Changes in the conformation of inter-subunit interfaces are central to transmission of ligand gating into pore opening (itself a rearrangement of interfaces), and subsequent closure through desensitization. With the exception of the agonist binding site, which is located entirely within individual subunits, almost all modulatory factors affecting iGluRs appear to bind to sites in subunit interfaces. This review seeks to summarize what we currently understand about the diverse roles interfaces play in iGluR function, and to highlight questions for future research.
Introduction
The three main subfamilies of iGluRs (AMPA, kainate and NMDA) all respond to the binding of glutamate by opening an integral cation-selective pore. This leads to both neuronal excitation and a range of subtype-specific physiological effects (Traynelis et al. 2010). While structural studies have focused on iGluR subunits, these do not function in isolation. As obligate tetramers, each subunit is in direct contact with at least two neighbours (Fig. 1A; Sobolevsky et al. 2009). In addition, they interact with the lipid bilayer, and associate with accessory proteins such as TARPs and cornicons for AMPA receptors (Milstein & Nicoll, 2008; Schwenk et al. 2009; Straub & Tomita, 2012) and NETOs for kainate receptors (Copits & Swanson, 2012). In terms of contacts within the tetramer, both the N-terminal domain (NTD) and the LBD in the extracellular part of the receptor form dimers with adjacent subunits, while in the transmembrane domain (TMD), subunits interact with their two neighbours. These interfaces are central to receptor assembly and function, helping to control subunit composition and mediate the transduction of glutamate binding into the transient opening of the pore. Advances in our knowledge of iGluR structure have helped us understand some of the conformational changes defining both assembly and function (see recent reviews by Sukumaran et al. 2012; Kumar & Mayer, 2013; Sobolevsky, 2013). This review will focus specifically on the functional role of iGluR interfaces and the molecules that bind to them.
Figure 1. Domain organization in GluA2cryst.
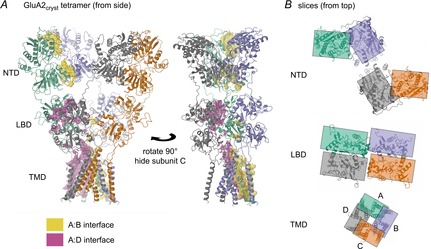
A, the GluA2cryst tetramer (PDB (protein data bank) code 3kg2; Sobolevsky et al. 2009), showing two side-views, rotated 90 deg. Subunits are coloured green (A), blue (B), orange (C) and grey (D). Interfaces between subunit A and neighbouring subunits are shown as magenta (the A:D interface) and yellow (the A:B interface) surfaces. The A:D interface is most visible in the left panel and consists of contacts in the LBD and TMD. The A:B interface is most visible in the right panel and mainly consists of contacts in the NTD and TMD. B, sectional views from above the tetramer, centred on the three main domains. Rectangles indicate the approximate extent of the subunits to show the switch from A:B and C:D contacts in the NTD to A:D and B:C contacts in the LBD. These views also highlight the change from (pseudo) 4-fold to 2-fold symmetry between the TMD and extracellular domains, as well as the right-handed subunit ‘twist’ (moving up from TMD to NTD), which is greatest for the B and D subunits. Structure cartoons in this and other figures were generated using CCP4mg (McNicholas et al. 2011). Interface surfaces were calculated by the PISA service (http://www.ebi.ac.uk; Krissinel & Henrick, 2007) called from within CCP4mg.
Structure overview of iGluR interfaces
Since the cloning of the first iGluR subunit nearly 25 years ago, our knowledge of their structure has progressed in zigzags, with unexpected twists coupled with the discovery of unique features. Initial topology models assumed homology with the pentameric nACh superfamily, but iGluRs were revealed to be tetramers with a distinct fold; the ligand binding domain is formed from two non-contiguous segments flanking the first three membrane domains (Hollmann et al. 1994; Stern-Bach et al. 1994). Further surprises have come from the first structure of a full-length iGluR (Sobolevsky et al. 2009). This not only confirmed that these receptors have a unique switch in symmetry (from 4-fold to 2-fold), but revealed an entirely unpredicted cross-over in the subunit pairings between the NTD and LBD (Fig. 1B).
Of the interfaces evident in the GluA2cryst structure, we have the most information on the contacts between neighbouring pairs of LBD and NTD domains. This knowledge has mostly come from X-ray crystallographic studies, first for the LBD (Armstrong & Gouaux, 2000) and more recently for the NTD (Jin et al. 2009; Kumar et al. 2009; Furukawa, 2012). Including the delta (GluD) subfamily, which is structurally but not functionally homologous, there are four (iGluR) subfamilies (Traynelis et al. 2010). Structural information is available for all four. To date a total of 253 LBD structures have been determined for subunits from all the subfamilies, along with 25 NTD structures representing all iGluR subfamilies except delta (Table 1). Of these, the majority have contained the two distinct 2-fold symmetric dimers (hereafter simply ‘dimers’) present in the GluA2cryst structure. The LBD has a bi-lobate fold, with the ligand bound in the cleft between lobes. The dimer is formed ‘back-to-back’, through interactions between the upper (‘D1’) lobes only. This results in an interface area averaging 860, 960 and 1100 Å2 for wild-type AMPA, kainate and NMDA receptor LBDs respectively (average interface-areas determined from WT structures using PISA web interface (http://www.ebi.ac.uk).
Table 1.
iGluR domain structures in protein data bank (PDB)
| AMPA (GluAn) |
Kainate (GluKn) |
NMDA (GluNn) |
Delta (GluDn) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subunita | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 5 | 1 | 2A | 2B | 2D | 3A | 3B | 2 |
| #NTD | 1 | 7 | 2 | 1 | 0 | 5* | 1 | 4* | 4* | 0 | 4* | 0 | 0 | 0 | 0 |
| (w. dimers) | (1) | (7) | (2) | (1) | — | (5) | (1) | (4) | (4) | — | (4) | — | — | — | — |
| #LBD | 0 | 130 | 17 | 6 | 34 | 29 | 8 | 0 | 14* | 7* | 0 | 5 | 4 | 2 | 2 |
| (w. dimers) | — | (127) | (17) | (6) | (27) | (23) | (1) | — | (7) | (7) | — | (0) | (0) | (0) | (1) |
| [w. allosteric] | — | [45] | [3] | [0] | [0] | [0] | [0] | — | [0] | [0] | — | [0] | [0] | [0] | [0] |
Total number of structures in the PDB (at end of April 2014), with the number containing dimeric complexes in parentheses. A dash means not applicable.
Includes heteromeric complexes between NTDs from GluK2/GluK5 (2 structures) and GluN1/GluN2B (2) and LBDs from GluN1/GluN2A (5).
No structures had been deposited for GluK4, GluN2C or GluD1 at time of writing.
The NTD also has a cleft between upper and lower lobes, although it is less pronounced. While zinc ions have been shown to bind at the cleft entrance in the GluN2B subunit (Karakas et al. 2009), no ligand has yet been identified that binds within the cleft. In AMPA and kainate receptor NTD structures the domains are arranged as dimers in a ‘side-to-side’ orientation, with close contacts between the adjacent upper and lower lobes (Fig. 2A). Structures for NMDA receptor NTDs exhibit a range of dimer conformations with contacts mainly between the upper lobes (Fig. 2B; Furukawa, 2012). The NTD dimer interfaces average 1290 and 1410 Å2 for AMPA and kainate receptor NTDs, but only 1130 Å2 for NMDA receptor NTDs (calculated as for LBD interface), reflecting the less extensive lower-lobe contacts. The functional roles of these interfaces are considered in more detail later.
Figure 2. NTD dimer interfaces.
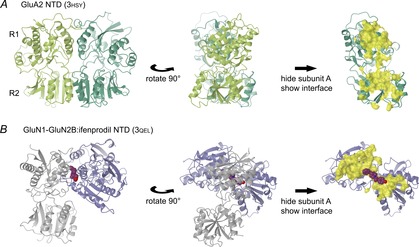
A, views of the GluA2 NTD dimer (PDB code 3hsy; Rossmann et al. 2011). Domains are shown in cartoon representation, with N-terminal residues at the top. The left and central panels show both protomers (light and dark green), with the view rotated 90 deg. The top and bottom lobes of the NTD are commonly designated as R1 and R2, so the interfaces form as R1:R1 and R2:R2. In the right panel only protomer B is shown, along with the interface between A and B (yellow surface) as calculated by PISA from within CCP4mg. B, views of the GluN1–GluN2B NTD dimer in complex with ifenprodil (3qel; Karakas et al. 2011). The Xenopus GluN1 and rat GluN2B protomers (light grey and blue, respectively) and the interface surface are shown as in A. The interface largely consists of R1:R1 interactions. Ifenprodil is shown in purple space-fill. The initial orientation was fixed by aligning protomer A (GluN1) with the equivalent GluA2 protomer, leaving protomer B oriented with R2 pointing towards the viewer in the left panel.
The switch in dimer pairings between the NTD and LBD was one of the key findings from the GluA2cryst structure (Fig. 1B). This arrangement has been confirmed for kainate receptors (Das et al. 2010), as well as an AMPA subunit without the linker deletions present in GluA2cryst (Midgett et al. 2012). It is reasonable to assume NMDA receptors assemble in the same way, although this has yet to be demonstrated. One question that the GluA2cryst structure does not answer, however, is how subunits are arranged in a heteromeric receptor. Subunits could be arranged A/B/A/B or A/A/B/B around the pore. This affects which dimer contacts are formed: in the former arrangement both LBD and NTD dimers must be heteromeric, while in the latter arrangement only one or the other would be. An alternating subunit arrangement has been determined for AMPA receptors (Mansour et al. 2001), but for NMDA receptors there is evidence in support of both alternating (Rambhadran et al. 2010; Salussolia et al. 2011; Riou et al. 2012) and paired arrangements (Balasuriya et al. 2013).
From a purely structural perspective, evidence on this aspect of receptor stoichiometry is incomplete; NTD dimers almost certainly preferentially assemble as heteromers, but the evidence is less clear-cut for LBD dimers. Hetero-dimerization of NTDs has been directly demonstrated both for kainate (Kumar et al. 2011) and NMDA receptors (Karakas et al. 2011; Lee & Gouaux, 2011), while heteromerization in AMPA receptors has been shown to be driven by interactions in the NTD (Shanks et al. 2010; Rossmann et al. 2011). Although homodimeric NTD structures have been observed for subunits which form obligate heteromers (i.e. GluK5, GluN1 and GluN2B), it is reasonable to ascribe these to the combined effects of crystal packing and high protein concentration. There is less information for LBD dimers, however. Heteromeric LBD structures have only been determined for GluN1 in complex with GluN2A, and there is biochemical evidence that this is the native form (Furukawa et al. 2005). However, both of these subunits have also been observed in crystal structures as homodimers, so although the alternating arrangement seems most likely for all receptor subtypes, definitive structural proof is still required.
Interface movements during receptor gating
The first AMPA LBD structures led to the development of a gating model for non-NMDA (i.e. AMPA and kainate) receptors in which the LBD dimer plays a literally pivotal role in channel activation and desensitization (Sun et al. 2002). In this model the dimer interface enables ligand binding to be transmitted into channel opening, while desensitization results from dissociation of the dimer interface, relieving strain on the pore and allowing closure. Consistent with this, dimer stability is inversely correlated with the rate of desensitization in both AMPA (Sun et al. 2002) and kainate receptors (Zhang et al. 2006; Chaudhry et al. 2009b). At the extreme, macroscopic desensitization can be blocked either with stabilizing point mutations (Stern-Bach et al. 1998; Nayeem et al. 2009) or introduced disulphides (Priel et al. 2006; Weston et al. 2006). Conversely, a disulphide link at the base of the dimer interface blocks GluA2 function, resulting in a conformation with a dissociated D1:D1 interface that has been ascribed to the desensitized state (Armstrong et al. 2006).
One consequence of this model is that the ‘normal’ dimer conformation is generally considered both the resting and active conformation. This fits the observations of several electron microscopy studies (Midgett et al. 2012; Schauder et al. 2013). These show symmetry-related density thought to correspond to NTD and LBD dimers. In other studies, however, LBD dimers appear not to be present in the resting (i.e. apo) state (Shanks et al. 2010). Experiments using luminescence resonance energy transfer to measure LBD separation in AMPA subunits lacking the NTD came to a similar conclusion (Gonzalez et al. 2010): dimers were only observed when antagonist was added, or with a non-desensitizing construct (GluA4 L505Y). If confirmed, these results would represent a significant challenge to the accepted gating model, which depends on an intact LBD dimer to enable channel opening. While it is tempting to look for possible technical factors, such as residual glutamate causing desensitization in the ‘apo’ forms (and with antagonist allowing recovery to a true resting state), another twist in the iGluR story cannot be ruled out.
When considering what the LBD dimer interface looks like in the active state there are variations in the LBD structures, both from mutations and ligand-specific conformational shifts (Nayeem et al. 2011). Recent single-channel recordings of non-desensitizing kainate receptor mutants show a close link between the dimer conformation and function. Although the cross-linked GluK2 Y521C–L783C mutant blocks macroscopic desensitization, it does not open either continuously or to the main conductance level (Daniels et al. 2013). This is actually consistent with the associated LBD structure (Weston et al. 2006), which was described as ‘relaxed’ compared to wild type (WT; Fig. 3A). In contrast, a second non-desensitizing mutant, GluK2 D776K, does open continuously to the main conductance level (Dawe et al. 2013). This mutant exhibits a tighter association at the top of the dimer interface than GluK2 WT (Fig. 3A; Nayeem et al. 2011), probably representing the active state of the receptor. Equivalent single-channel experiments have not been reported for non-desensitizing AMPA mutations, although for GluA2 L483Y the LBD dimer conformation is also similar to WT (Fig. 3B).
Figure 3. Dimer conformations of LBDs containing non-desensitizing iGluR mutants.
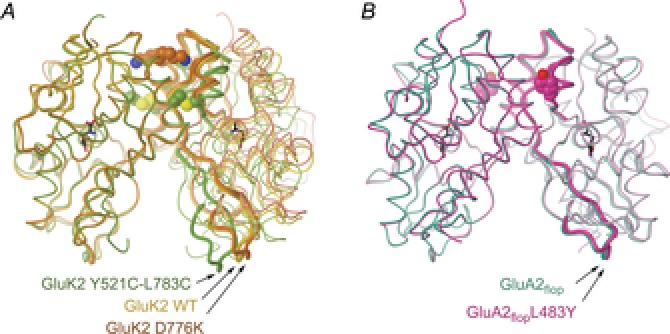
A, comparison of LBD dimer conformations for GluK2 WT, D776K (PDB codes 2xxr and 2xxx; Nayeem et al. 2011) and Y521C–L783C (2i0c; Weston et al. 2006). Polypeptide is shown as a tube, with mutations shown as spheres and ligand (glutamate) as black sticks. Dimers are aligned on the left protomer. Key residue stretches at the top (P769–W798) and bottom (A518–S670) of the right protomer are displayed as thicker tubes to highlight relative movements. B, LBD dimers for GluA2flop WT (1ftm; Armstrong & Gouaux, 2000) and L483Y (1lb8; Sun et al. 2002) in complex with AMPA, displayed as in panel A (highlighted residue stretches in the right protomer are T480–S635 and P737–G757). Here and elsewhere residue numbering follows the general convention; full-length except for GluA2, where it is for the mature polypeptide.
Of interfaces in the other domains, the NTD is not thought to play a significant role in non-NMDA receptor function, but does have an effect in NMDA receptors. Receptor properties such as single channel open probabilities and ligand potency are defined by the GluN2 subunit, and it has been shown that the NTD dominates this effect (reviewed in Hansen et al. 2010; Paoletti, 2011). This appears to be related to the greater conformational flexibility of the NTD in NMDA subunits (Karakas et al. 2011), although how conformational changes in the NTD are transmitted to the TMD is not known. Unsurprisingly given the functional role of the NMDA NTD, a number of endogenous and exogenous factors have been found to bind there. These are discussed in the following sections. Structural information on interfaces between subunit TMDs is currently limited to the closed conformation observed for GluA2cryst, but for obvious reasons they are central to iGluR function. In addition, accessory proteins interacting with non-NMDA subunits appear to do so via the TMD (Shanks et al. 2010) but also modulate the function of the LBD (Straub & Tomita, 2012). Further structural information on this region is therefore an essential prerequisite for a full understanding of iGluR function.
Physiological binding sites within domain interfaces
A number of endogenous factors interact with iGluRs in addition to glutamate (and glycine for NMDA receptors). These include polyamines and a wide range of ions. Polyamines affect the responses of all three receptor subtypes by binding to the TMD, while Mg2+ ions selectively block the NMDA receptor (Traynelis et al. 2010). In terms of the domains described here, both the NMDA receptor NTDs and kainate receptor LBDs have binding sites for endogenous modulators. Polyamines, H+ and Zn2+ modulate NMDA receptor responses by binding to the NTD (Paoletti & Neyton, 2007), and monovalent cations and anions affect GluK1 and GluK2 responses through sites in the LBD (Paternain et al. 2003; Wong et al. 2007). In the other receptor families, Ca2+ binds GluD2 at a site equivalent to the cation site in kainate subunits (Naur et al. 2007; Hansen et al. 2009) while Zn2+ binds to GluK3 at a site lower down the dimer interface (Veran et al. 2012). Binding sites for many of these factors have been identified in X-ray structures; these are described below.
Polyamines and zinc both bind to the NTD of NMDA receptors. The binding site for polyamines has been identified as the NTD dimer interface, where they act by increasing conformational freedom (Mony et al. 2011). Zinc binds with high affinity to GluN2A-containing NMDA receptors, and with lower affinity to receptors containing the GluN2B subunit (Paoletti & Neyton, 2007). Karakas et al. (2009) crystallized the GluN2B NTD in the presence of Zn2+, revealing five binding sites per protomer. A Zn2+ ion bound at the mouth of the cleft between lobes was identified as the physiologically relevant site. However, this ion is chelated by only two sidechains and is 50% occupied. A structure of the GluN1–GluN2A heteromeric NTD complex with zinc bound will be more convincing, and may even reveal an alternative binding mode. Indeed, a modulatory binding site outside of the various subunit interfaces would be the exception for iGluRs.
The other main binding sites for endogenous factors are those for monovalent ions in kainate receptors (Plested & Mayer, 2007; Plested et al. 2008; Bowie, 2010). In both GluK1 and GluK2 subunits, monovalent cations bind to two sites on either side of the LBD dimer 2-fold symmetry axis, while a single anion binds to a site on the axis (Fig. 4A). The cation sites are contained within individual protomers, while the anion site spans the two protomers. Binding of Na+ and Cl− ions have been shown to stabilize the dimer, and thereby slow desensitization (Chaudhry et al. 2009a). However, mutations to residues forming these sites give a more mixed picture. While elimination of Na+ binding invariably disrupts the dimer, as expected (Plested et al. 2008; Nayeem et al. 2011), mutations to residues forming the anion binding site have varied functional effects. Most mutations do accelerate the rate of desensitization (Plested & Mayer, 2007; Wong et al. 2007), but three mutants have been identified where the rate of desensitization is slowed (Nayeem et al. 2013). For two of these, sidechain rearrangements can explain the phenotype, but for one mutant (GluK2 R775A), the loss of chloride binding was the only apparent structural change. One possible explanation is that the charge balance in the WT receptor tends to destabilize the interface, and the loss of two basic sidechains counteracts this (Nayeem et al. 2013).
Figure 4. Ion binding to the LBD dimer interface in iGluR subunits.
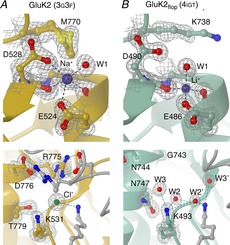
A, cation and anion binding sites in GluK2 (PDB code 3g3f; Chaudhry et al. 2009b) shown in the upper and lower panels respectively. In both cases residues and solvent in contact with the ion are indicated, along with the associated electron density (grey mesh; contoured at 1.5σ). For clarity in the lower panel density is only shown for ions, solvent and residues in the further protomer. B, equivalent views for GluA2flop WT (4igt; Assaf et al. 2013), in which lithium binding is observed at the ‘cation’ site, but no anion is present. Symmetry-related waters are indicated with a prime (′). Structure factors for electron density maps were downloaded from the Electron Density Server (eds.bmc.uu.se/eds/; Kleywegt et al. 2004) from within CCP4mg.
AMPA receptors do not respond to external monovalent ions in the same way as kainate receptors, although many of the residues in the two binding sites are conserved. Two recent structures show Li+ bound at the equivalent of the cation site (Fig. 4B; Assaf et al. 2013; Harms et al. 2013). The significance of this, if any, is unclear. Identification of lithium requires a high resolution structure, so it might have been missed before (33 AMPA LBD structures have been crystallized in the presence of lithium). On the other hand, there has been no indication from other structures that Na+ or another physiological cation binds at this site. This highlights one of the difficulties of using X-ray structures to identify binding sites. Not counting those sites described earlier, over 100 of the reported iGluR domain structures have a potentially physiological ion bound at one or more sites (e.g. 77 structures with Zn2+ bound; 8 with Na+; 35 with Cl−). Crystallization conditions are often far from physiological; in the reported iGluR structures pH varies between 4.0 and 9.5, for example. Ions such as sulphate or lithium can be present in the crystallization buffer at levels of several molar, while ions found in the brain can be present at concentrations very different from their normal physiological levels. In addition to those ions present as presumed artefacts of crystallization, real sites may be missed if they are not being explicitly looked for, particularly at lower resolutions. The discovery of further modulatory sites cannot therefore be ruled out.
Drug binding to domain interfaces
While the number of therapeutically useful drugs targeting iGluRs is currently limited (e.g. memantine for Alzheimer's disease and perampanel for epilepsy), there is no shortage of potential therapeutic targets (Bowie, 2008). In addition to compounds acting competitively at the agonist binding sites, there are non-competitive antagonists acting at all three receptor subtypes, and allosteric potentiators of AMPA receptors. Of these, several classes are thought to bind to the region between the LBD and the pore. This includes non-competitive antagonists of non-NMDA receptors such as GYKI53655 and LY 300164 (Balannik et al. 2005), as well as more recently identified NMDA antagonists (Acker et al. 2011; Hansen & Traynelis, 2011). Given the close apposition of subunit polypeptides in this region (Fig. 1A), it is a reasonable assumption that these compounds will bind to more than one subunit. However, the extent to which subunit interfaces are directly involved in their functional effects will have to await more detailed structural information for the TMD.
Two other classes of iGluR modulators have been definitively shown to bind to domain interfaces. Phenylethanolamines such as ifenprodil, a GluN2B-selective non-competitive antagonist, bind at the GluN1–GluN2 NTD dimer interface (Karakas et al. 2011), while AMPA potentiatiors typified by cyclothiazide bind in the LBD dimer interface (Sun et al. 2002). In the former case, NMDA receptor function is apparently inhibited through restrictions in NTD conformational freedom (Karakas et al. 2011). It seems unlikely that similar inhibition can occur in non-NMDA receptors, where the NTD has no reported effect on function. AMPA potentiators, in contrast, act by stabilizing the LBD dimer interface (Sun et al. 2002), binding to the lower half of the D1:D1 interface. Increased dimer stability, perhaps coupled with steric block, slows the rate of macroscopic desensitization. While equivalent compounds acting at kainate receptors have not been described, there appears no intrinsic reason a similar effect cannot be achieved.
Three binding modes have so far been identified for AMPA potentiators using X-ray crystallography (Fig. 5). Some compounds, including cyclothiazide itself, bind to two sites on either side of the dimer 2-fold symmetry axis (Fig. 5A). Others bind in two overlapping conformations to a more central site (Fig. 5B), while chemically symmetric compounds have now been identified that bind to a central site with a single conformation (Fig. 5C; Timm et al. 2011). In all cases the modulators interact with equivalent residues on both subunits, and will therefore bind to homomeric receptors preferentially. This is necessarily the case for any compound which binds to multiple sites related by 2-fold symmetry. However, by breaking the symmetry of compounds in the third class, it should be possible to develop AMPA receptor modulators which are selective for heteromeric receptors. Ultimately this level of selectivity may prove essential in developing safe and effective therapies targeting these ubiquitous receptor classes.
Figure 5. Allosteric potentiator binding to AMPA subunits.
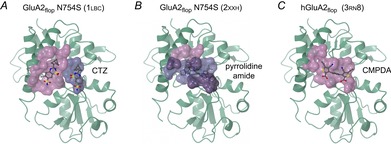
A, two molecules of cyclothiazide (CTZ) bind within the GluA2flop N754S LBD dimer interface (PDB code 1lbc; Sun et al. 2002). The main interface is shown as a pink surface and the minor interface as a blue surface. As the two binding sites are related by 2-fold symmetry, equivalent binding surfaces are present on the other protomer (not shown). The N745S mutation results in a ‘flip-like’ binding profile for allosteric modulators. B, several AMPA modulators bind to a single site on the dimer 2-fold axis, but in two orientations. In the example shown (2xxh; Ward et al. 2011), an amide designated 10a is bound in overlapping conformations, with an occupancy of 0.5 in each. Surfaces are shown as in A for the two binding modes, with areas of overlap in purple. C, allosteric potentiators with 2-fold symmetry have been identified that bind in a single orientation (Timm et al. 2011). The structure shown (3rn8) is of human GluA2flop in complex with phenyl-1,4-bis-alkylsulfonamide (CMPDA). Breaking such ligands’ symmetry could potentially lead to compounds with selectivity for specific heteromeric receptor complexes.
Conclusion
Structural studies on isolated subdomains have brought us a surprising way towards understanding how glutamate binding to iGluRs is translated into channel opening. They have led directly to models of receptor activation explaining properties ranging from ligand affinity and efficacy to desensitization and the effects of endogenous ions. In all of these, domain interfaces, and an understanding of how their conformation varies, have proved to be of central importance. The full-length GluA2cryst structure has served to confirm the utility of the isolated LBD and NTD structures, showing that the same dimeric associations exist in the intact receptor. It has also highlighted their limitations, revealing an unpredicted cross-over between the two extracellular domains. Despite this progress, significant gaps remain, not least how the conformation of the TMD changes when the receptor is activated, and the mechanisms by which accessory proteins modify this process. Future protein structures will undoubtedly play a role in answering these questions, although it remains to be seen whether structures of isolated domains still have more to reveal, or additional full-length iGluR structures will instead be required.
Glossary
- iGluR
ionotropic glutamate receptor
- LBD
ligand binding domain
- NTD
N-terminal domain
- TMD
transmembrane domain
- WT
wild type
Biographies
Tim Green studied for his Ph.D. at the University of Sheffield, before undertaking postdoctoral research in the laboratories of Nigel Unwin at the LMB in Cambridge and Steve Heinemann at the Salk Institute, La Jolla, USA. His research encompassed the quaternary structure of GABAA and 5-HT3 receptors in Cambridge, moving on to study ionotropic glutamate receptors at the Salk. In 2001 he returned to the UK, since when he has continued his work on structure-function relationships in kainate-selective ionotropic glutamate receptors at the University of Liverpool.
Naushaba Nayeem obtained her Ph.D. working with Eric Barnard at the MRC Molecular Neurobiology Unit, Cambridge. She then undertook postdoctoral research at the BBSRC Babraham research laboratory, Cambridge, and the Scripps Research Institute in La Jolla, USA. Returning to the UK, she has worked at the MRC Prion Unit, London, and now collaborates with Dr Green at the University of Liverpool.
Additional information
Competing interests
None declared.
Funding
None declared.
References
- Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC. Traynelis SF. Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-d-aspartate receptor subunit-selective modulators. Mol Pharmacol. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N. Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M. Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Assaf Z, Larsen AP, Venskutonyte R, Han L, Abrahamsen B, Nielsen B, Gajhede M, Kastrup JS, Jensen AA, Pickering DS, Frydenvang K, Gefflaut T. Bunch L. Chemoenzymatic synthesis of new 2,4-syn-functionalized (S)-glutamate analogues and structure–activity relationship studies at ionotropic glutamate receptors and excitatory amino acid transporters. J Med Chem. 2013;56:1614–1628. doi: 10.1021/jm301433m. [DOI] [PubMed] [Google Scholar]
- Balannik V, Menniti FS, Paternain AV, Lerma J. Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron. 2005;48:279–288. doi: 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Balasuriya D, Goetze TA, Barrera NP, Stewart AP, Suzuki Y. Edwardson JM. α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors adopt different subunit arrangements. J Biol Chem. 2013;288:21987–21998. doi: 10.1074/jbc.M113.469205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets. 2008;7:129–143. doi: 10.2174/187152708784083821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Ion-dependent gating of kainate receptors. J Physiol. 2010;588:67–81. doi: 10.1113/jphysiol.2009.178863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry C, Plested AJ, Schuck P. Mayer ML. Energetics of glutamate receptor ligand binding domain dimer assembly are modulated by allosteric ions. Proc Natl Acad Sci U S A. 2009a;106:12329–12334. doi: 10.1073/pnas.0904175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry C, Weston MC, Schuck P, Rosenmund C. Mayer ML. Stability of ligand-binding domain dimer assembly controls kainate receptor desensitization. EMBO J. 2009b;28:1518–1530. doi: 10.1038/emboj.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copits BA. Swanson GT. Dancing partners at the synapse: auxiliary subunits that shape kainate receptor function. Nat Rev Neurosci. 2012;13:675–686. doi: 10.1038/nrn3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels BA, Andrews ED, Aurousseau MR, Accardi MV. Bowie D. Crosslinking the ligand-binding domain dimer interface locks kainate receptors out of the main open state. J Physiol. 2013;591:3873–3885. doi: 10.1113/jphysiol.2013.253666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Kumar J, Mayer ML. Plested AJ. Domain organization and function in GluK2 subtype kainate receptors. Proc Natl Acad Sci U S A. 2010;107:8463–8468. doi: 10.1073/pnas.1000838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe GB, Musgaard M, Andrews ED, Daniels BA, Aurousseau MR, Biggin PC. Bowie D. Defining the structural relationship between kainate-receptor deactivation and desensitization. Nat Struct Mol Biol. 2013;20:1054–1061. doi: 10.1038/nsmb.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H. Structure and function of glutamate receptor amino terminal domains. J Physiol. 2012;590:63–72. doi: 10.1113/jphysiol.2011.213850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R. Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- Gonzalez J, Du M, Parameshwaran K, Suppiramaniam V. Jayaraman V. Role of dimer interface in activation and desensitization in AMPA receptors. Proc Natl Acad Sci U S A. 2010;107:9891–9896. doi: 10.1073/pnas.0911854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H. Traynelis SF. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol. 2010;78:535–549. doi: 10.1124/mol.110.067157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Naur P, Kurtkaya NL, Kristensen AS, Gajhede M, Kastrup JS. Traynelis SF. Modulation of the dimer interface at ionotropic glutamate-like receptor δ2 by d-serine and extracellular calcium. J Neurosci. 2009;29:907–917. doi: 10.1523/JNEUROSCI.4081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB. Traynelis SF. Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors. J Neurosci. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms JE, Benveniste M, Maclean JK, Partin KM. Jamieson C. Functional analysis of a novel positive allosteric modulator of AMPA receptors derived from a structure-based drug design strategy. Neuropharmacology. 2013;64:45–52. doi: 10.1016/j.neuropharm.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Maron C. Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y. Gouaux E. Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J. 2009;28:1812–1823. doi: 10.1038/emboj.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N. Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N. Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A. Jones TA. The Uppsala Electron-Density Server. Acta Crystallogr D Biol Crystallogr. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- Krissinel E. Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kumar J. Mayer ML. Functional insights from glutamate receptor ion channel structures. Annu Rev Physiol. 2013;75:313–337. doi: 10.1146/annurev-physiol-030212-183711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P, Jin R. Mayer ML. The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat Struct Mol Biol. 2009;16:631–638. doi: 10.1038/nsmb.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P. Mayer ML. Structure and assembly mechanism for heteromeric kainate receptors. Neuron. 2011;71:319–331. doi: 10.1016/j.neuron.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH. Gouaux E. Amino terminal domains of the NMDA receptor are organized as local heterodimers. PLoS One. 2011;6:e19180. doi: 10.1371/journal.pone.0019180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS. Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour M, Nagarajan N, Nehring RB, Clements JD. Rosenmund C. Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron. 2001;32:841–853. doi: 10.1016/s0896-6273(01)00520-7. [DOI] [PubMed] [Google Scholar]
- Midgett CR, Gill A. Madden DR. Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation. Front Mol Neurosci. 2012;4:56. doi: 10.3389/fnmol.2011.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD. Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Zhu S, Carvalho S. Paoletti P. Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 2011;30:3134–3146. doi: 10.1038/emboj.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, Dravid SM, Pickering DS, Olsen L, Vestergaard B, Egebjerg J, Gajhede M, Traynelis SF. Kastrup JS. Ionotropic glutamate-like receptor δ2 binds d-serine and glycine. Proc Natl Acad Sci U S A. 2007;104:14116–14121. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Mayans O. Green T. Conformational flexibility of the ligand-binding domain dimer in kainate receptor gating and desensitization. J Neurosci. 2011;31:2916–2924. doi: 10.1523/JNEUROSCI.4771-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Mayans O. Green T. Correlating efficacy and desensitization with GluK2 ligand-binding domain movements. Open Biol. 2013;3:130051. doi: 10.1098/rsob.130051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Zhang Y, Schweppe DK, Madden DR. Green T. A non-desensitizing kainate receptor point mutant. Mol Pharmacol. 2009;76:534–542. doi: 10.1124/mol.109.056598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x. [DOI] [PubMed] [Google Scholar]
- Paoletti P. Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Cohen A, Stern-Bach Y. Lerma J. A role for extracellular Na+ in the channel gating of native and recombinant kainate receptors. J Neurosci. 2003;23:8641–8648. doi: 10.1523/JNEUROSCI.23-25-08641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJ. Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Vijayan R, Biggin PC. Mayer ML. Molecular basis of kainate receptor modulation by sodium. Neuron. 2008;58:720–735. doi: 10.1016/j.neuron.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Selak S, Lerma J. Stern-Bach Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron. 2006;52:1037–1046. doi: 10.1016/j.neuron.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Rambhadran A, Gonzalez J. Jayaraman V. Subunit arrangement in N-methyl-d-aspartate (NMDA) receptors. J Biol Chem. 2010;285:15296–15301. doi: 10.1074/jbc.M109.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou M, Stroebel D, Edwardson JM. Paoletti P. An alternating GluN1–2–1–2 subunit arrangement in mature NMDA receptors. PLoS One. 2012;7:e35134. doi: 10.1371/journal.pone.0035134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M, Sukumaran M, Penn AC, Veprintsev DB, Babu MM. Greger IH. Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J. 2011;30:959–971. doi: 10.1038/emboj.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salussolia CL, Prodromou ML, Borker P. Wollmuth LP. Arrangement of subunits in functional NMDA receptors. J Neurosci. 2011;31:11295–11304. doi: 10.1523/JNEUROSCI.5612-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder DM, Kuybeda O, Zhang J, Klymko K, Bartesaghi A, Borgnia MJ, Mayer ML. Subramaniam S. Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain. Proc Natl Acad Sci U S A. 2013;110:5921–5926. doi: 10.1073/pnas.1217549110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B. Klocker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Shanks NF, Maruo T, Farina AN, Ellisman MH. Nakagawa T. Contribution of the global subunit structure and stargazin on the maturation of AMPA receptors. J Neurosci. 2010;30:2728–2740. doi: 10.1523/JNEUROSCI.5146-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI. Structure and gating of tetrameric glutamate receptors. J Physiol. 2013 doi: 10.1113/jphysiol.2013.264911. doi: 10.1113/jphysiol.2013.264911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP. Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O'Hara PJ. Heinemann SF. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M. Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Straub C. Tomita S. The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr Opin Neurobiol. 2012;22:488–495. doi: 10.1016/j.conb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran M, Penn AC, Sala C, Greger IH, Kreutz MR. Synaptic Plasticity. Springer-Verlag; 2012. AMPA receptor assembly: atomic determinants and built-in modulators; pp. 241–264. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M. Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Timm DE, Benveniste M, Weeks AM, Nisenbaum ES, Partin KM. Structural and functional analysis of two new positive allosteric modulators of GluA2 desensitization and deactivation. Mol Pharmacol. 2011;80:267–280. doi: 10.1124/mol.110.070243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ. Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veran J, Kumar J, Pinheiro PS, Athane A, Mayer ML, Perrais D. Mulle C. Zinc potentiates GluK3 glutamate receptor function by stabilizing the ligand binding domain dimer interface. Neuron. 2012;76:565–578. doi: 10.1016/j.neuron.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SE, Harries M, Aldegheri L, Austin NE, Ballantine S, Ballini E, Bradley DM, Bax BD, Clarke BP, Harris AJ. Integration of lead optimization with crystallography for a membrane-bound ion channel target: discovery of a new class of AMPA receptor positive allosteric modulators. J Med Chem. 2011;54:78–94. doi: 10.1021/jm100679e. [DOI] [PubMed] [Google Scholar]
- Weston MC, Schuck P, Ghosal A, Rosenmund C. Mayer ML. Conformational restriction blocks glutamate receptor desensitization. Nat Struct Mol Biol. 2006;13:1120–1127. doi: 10.1038/nsmb1178. [DOI] [PubMed] [Google Scholar]
- Wong AY, MacLean DM. Bowie D. Na+/Cl− dipole couples agonist binding to kainate receptor activation. J Neurosci. 2007;27:6800–6809. doi: 10.1523/JNEUROSCI.0284-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nayeem N, Nanao MH. Green T. Interface interactions modulating desensitization of the kainate-selective ionotropic glutamate receptor subunit GluR6. J Neurosci. 2006;26:10033–10042. doi: 10.1523/JNEUROSCI.2750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]