

Abstract
Ionotropic glutamate receptors (iGluRs) are the major excitatory neurotransmitter receptor in the vertebrate CNS and, as a result, their activation properties lie at the heart of much of the neuronal network activity observed in the developing and adult brain. iGluRs have also been implicated in many nervous system disorders associated with postnatal development (e.g. autism, schizophrenia), cerebral insult (e.g. stroke, epilepsy), and disorders of the ageing brain (e.g. Alzheimer's disease, Parkinsonism). In view of this, an emphasis has been placed on understanding how iGluRs activate and desensitize in functional and structural terms. Early structural models of iGluRs suggested that the strength of the agonist response was primarily governed by the degree of closure induced in the ligand-binding domain (LBD). However, recent studies have suggested a more nuanced role for the LBD with current evidence identifying the iGluR LBD interface as a “hotspot” regulating agonist behaviour. Such ideas remain to be consolidated with recently solved structures of full-length iGluRs to account for the global changes that underlie channel activation and desensitization.
Introduction
Fast excitatory neurotransmission in the vertebrate CNS is mediated by a class of plasma membrane-bound, multimeric proteins called ionotropic glutamate receptors (iGluRs). iGluRs assemble as tetramers from subunits encoded by eighteen genes that are divided into three major subgroups, based on their differential sensitivities to the agonists N-methyl d-aspartate (NMDAs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPAs) and kainate (KAs) (Nakanishi, 1992; Seeburg, 1993; Hollmann & Heinemann, 1994; Collingridge et al. 2009), while a fourth, orphan group, comprising the δ subunits, does not appear to form functional ion-conducting channels (Yuzaki, 2003).
At central synapses, iGluRs respond to the transient presence of the neurotransmitter l-glutamate (l-Glu) in the synaptic cleft through a series of binding and conformational steps that lead to membrane depolarization by the opening of a cation-permeable transmembrane pore (Dingledine et al. 1999; Traynelis et al. 2010; Huettner, 2014). Although iGluRs are often found together in the postsynaptic membrane, their global distribution and specific functions differ. The rapid millisecond activation of AMPA receptors (AMPARs) allows them to facilitate postsynaptic depolarization, which is also a necessary first step in relieving tonic Mg2+ block of NMDA receptors (NMDARs) (MacDonald & Nowak, 1990). Once unblocked, NMDARs affect neuronal signalling by transporting extracellular Ca2+ into the cytoplasm, a process prolonged by the slow intrinsic gating properties of these receptors (Qian & Johnson, 2002; Paoletti et al. 2013). In contrast, kainate receptors (KARs) are thought to fulfil more of a modulatory role on synaptic transmission, acting from both pre- and postsynaptic locales via ionotropic and metabotropic signalling mechanisms (Huettner, 2003; Contractor et al. 2011; Lerma & Marques, 2013). Curiously, the orphan-class δ iGluRs exhibit a much restricted expression pattern in the CNS with their most notable role in cerebellar Purkinje cells, where they regulate synaptic plasticity and synaptogenesis via a non-ionotropic pathway (Yuzaki, 2012).
Over the past decade, a wealth of structural information on iGluRs has emerged, describing the topology and assembly of domains (Gouaux, 2004; Mayer, 2011; Sobolevsky, 2013). Importantly, insight into their structural properties is expected to have a significant impact in the long term, as we start to unravel how iGluRs are implicated in several prominent CNS disorders (Bowie, 2008) and continue the rational design of therapeutic compounds (Lipton, 2006; Sanacora et al. 2008; Santangelo et al. 2012; Pirotte et al. 2013). Despite this, our understanding of the basic events that lead to iGluR activation is still emerging. In this review, we re-examine this issue and highlight recent studies that place a greater emphasis on the iGluR ligand binding domain (LBD) dimer interface as a ‘hotspot’ of channel activation. Particular emphasis will be placed on AMPARs and KARs, as more structural information is available for these families than NMDARs. With the recent elucidation of (near) full-length, tetrameric AMPAR structures in several conformations (Dürr et al. 2014; Meyerson et al. 2014; Yelshanskaya et al. 2014), a more complete understanding of iGluRs will require a multidisciplinary approach that brings together functional and structural data with dynamic simulations of protein movement.
iGluR pharmacology and channel activation
By the mid-1980s it was accepted that iGluRs exist as pharmacologically distinct receptor families (for reviews see Watkins & Evans, 1981; McLennan, 1983; Mayer & Westbrook, 1987; Collingridge & Lester, 1989), though their rapid, millisecond time course of activation and inactivation was only fully appreciated with the application of the concentration-clamp technique (reviewed in Akaike, 1995). The use of the concentration-clamp permitted the first detailed kinetic analysis of iGluR channel kinetics in response to rapid agonist application. Typical exchange rates in whole-cell recording conditions were about 10 ms using stepper motors (Vyklicky et al. 1990), while sub-millisecond (300–500 μs) exchange was achieved in outside-out patch recordings with the help of a piezo translator (Jonas, 1995). Since exchange rates in whole-cell recordings are markedly slower than AMPAR or KAR gating kinetics, peak agonist responses can only be measured with accuracy in excised patches (see discussion in Bowie et al. 2003).
Using concentration-clamp recordings, it was demonstrated that native AMPAR/KARs expressed by rodent hippocampal neurons exhibited two distinct kinetic phenotypes in response to agonists: rapid desensitization with the neurotransmitter l-Glu or quisqualate (QA) and a non-desensitizing or non-decaying profile with kainate (KA) (Kiskin et al. 1986). Kinetic models developed to explain this behaviour proposed that KA binds to the desensitized state of AMPAR/KARs with lower affinity than l-Glu, QA or AMPA (Patneau & Mayer, 1991) (Fig. 1A and B). In keeping with this, the use of the benzothiadiazine diuretic, cyclothiazide, to attenuate receptor desensitization potentiated equilibrium responses elicited by KA much less than l-Glu (Patneau et al. 1993). Analysis of a more extensive range of receptor ligands, including sulfur-containing amino acids and willardiine derivatives, further established that neuronal AMPAR/KARs responded in an agonist-dependent manner (Patneau & Mayer, 1990; Patneau et al. 1992), presumably due to the varying degrees of desensitization (Fig. 1C). To complicate matters further, single-channel recordings demonstrated that KA primarily activated a low conductance open state, whereas l-Glu or QA gated channels with a much larger unitary conductance(s) (Ascher & Nowak, 1988; Cull-Candy & Usowicz, 1989; Tang et al. 1989). Although it was debated whether KA may activate distinct receptor families from l-Glu and QA (Cull-Candy & Usowicz, 1987; Jahr & Stevens, 1987), cross-desensitization experiments on native receptors (Kiskin et al. 1986; Patneau & Mayer, 1991) and the cloning of AMPAR receptor subunits (Hollmann et al. 1989; Keinanen et al. 1990) confirmed that different agonists gate the AMPAR channel pore in quite distinct ways. At NMDARs, gating behaviour was revealed to be more complicated in that the binding of not one but two neurotransmitters, namely l-Glu as well as the inhibitory transmitter glycine, were required for activation (Johnson & Ascher, 1987; Kleckner & Dingledine, 1988). In this regard, KARs were more comparable to AMPARs, though studies of native receptors (Huettner, 1990; Lerma et al. 1993) and receptor clones (Egebjerg et al. 1991) revealed that KA elicited a strongly desensitizing response. As discussed below, an ongoing challenge has been to develop structural models of iGluR activation that can account for many of these complications. The first steps in this process began with the study of recombinant iGluRs.
Figure 1. A cyclic gating model recreates AMPAR responses to willardiine series agonists.
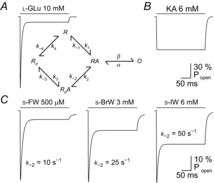
A, simulated response of AMPARs to l-Glu, generated from a five-state cyclic gating model (Patneau & Mayer, 1991). R, RA, Rd, and O represent resting, agonist-bound, desensitized, and open-channel states of the receptor, respectively. The rate constants for state transitions with l-Glu are adapted from values in another study (Vyklicky et al. 1991). B and C, simulated responses of AMPARs to KA (B), as well as to the willardiine series agonists, S-5-fluorowillardiine (s-FW), S-5-bromowillardiine (s-BrW), and S-5-iodowillardiine (s-IW) (C). As the halogen substituent increases in size, the equilibrium to peak current ratio recorded from AMPAR/KARs in hippocampal neurons also increases. To recapitulate this effect, the rate constant k–2 was increased, while k–1 and k–3 were adjusted to maintain microscopic reversibility. The rate constants are adapted from published values (Patneau et al. 1992), except for the s-BrW simulation, in which values were assigned to reproduce experimental observations.
Early insights into the topology of the agonist binding site
The cloning of the many genes that encode iGluR subunits (Nakanishi, 1992; Seeburg, 1993; Hollmann & Heinemann, 1994) permitted a detailed characterization not only of their functional properties, but also led the way to obtaining structural information (Madden, 2002; Gouaux, 2004; Mayer & Armstrong, 2004). Sequence analysis of the earliest cloned iGluRs revealed a large (approximately 500 amino acid) extracellular domain (ED), while overall amino acid sequence identity ranged from about 70% between AMPARs (Keinanen et al. 1990), down to 40% and 25% when they were compared to KARs and NMDARs, respectively (Bettler et al. 1990; Moriyoshi et al. 1991). Interestingly, a 100 amino acid segment of the ED just prior to the first predicted transmembrane domain (TM 1) shared 30% sequence identity with the bacterial periplasmic glutamine binding protein (GlnBP) (Nakanishi et al. 1990; Moriyoshi et al. 1991). The cloning of the first metabotropic glutamate receptors (mGluRs) revealed that these proteins also possess an approximately 500–600 residue ED (Masu et al. 1991; Tanabe et al. 1992), but retained significant homology within only two shorter, discontinuous segments (Masu et al. 1991). Through more refined sequence analysis techniques, it was later appreciated that iGluRs have two distinct EDs. The first, now referred to as the amino terminal domain (ATD), is homologous to the mGluR ED and the bacterial leucine-isoleucine-valine binding protein (LIVBP), while the second, now referred to as the ligand binding domain (LBD), is homologous to the GlnBP (O'Hara et al. 1993). At the time, X-ray crystal structural information was available for LIVBP (Adams & Oxender, 1989; Quiocho, 1990).
Drawing upon this structural information led research groups to use distinct, but complementary approaches to conclude that the agonist specificity of iGluRs and mGluRs is governed by two discontinuous segments of amino acid residues located in the extracellular domain (ED). For mGluRs, molecular modelling techniques were used to construct a homology model of mGluR1, which was then validated by assessing the impact of mutating conserved residues on agonist selectivity (O'Hara et al. 1993). For iGluRs, the replacement of native glycosylation sites with mutant sites was exploited to determine a topology of three membrane-spanning domains (TM 1, 3 and 4) with a re-entrant loop (TM 2) in between (Hollmann et al. 1994). From this framework, chimeric receptors (generated by swapping extracellular regions of GluA3 AMPARs and GluK2 KARs) were used to verify that two separate domains confer agonist selectivity (Stern-Bach et al. 1994). These domains were subsequently named S1 for the amino acid residues prior to transmembrane region 1 (TM 1) and S2 for residues between TM 3 and 4. The structural elucidation of an E. coli GlnBP (Hsiao et al. 1996) further established the unexpected link between the structural motifs of bacterial periplasmic transport proteins and neurotransmitter receptors expressed in the vertebrate CNS (Paas, 1998).
Two final observations led the way in permitting the resolution of the iGluR agonist/ligand binding site at atomic-level resolution. First, a truncated fusion protein consisting of the S1 and S2 domains of the GluA4 AMPAR, separated by a linker peptide, was shown to exhibit near-identical pharmacological properties to its wild-type counterpart (Kuusinen et al. 1995); a strategy that was exploited to obtain large quantities of the GluA2 AMPAR LBD for crystallographic analysis (Chen & Gouaux, 1997). Second, the upper and lower lobes were proposed to act in a ‘Venus flytrap’ arrangement (Mano et al. 1996), which helped further conceptualization of how the iGluR ligand binding cleft might trigger the process of channel activation.
A structural model of agonist efficacy
A new era in the study of iGluRs was heralded when Armstrong and colleagues solved the first high-resolution structure of a LBD; in this case it was the GluA2 AMPAR in complex with KA (Armstrong et al. 1998). As anticipated from earlier studies, the GluA2 structure was remarkably similar to that of the E. coli GlnBP (Sun et al. 1998), with the ligand binding cleft formed between two α-helix- and β-sheet-containing domains that were subsequently named D1 and D2 for the upper and lower lobes, respectively. Appreciating this structural similarity, the authors concluded that the GluA2–KA complex represented a partially closed structure, since the degree of domain closure was intermediate relative to GlnBP's open and closed conformations (Armstrong et al. 1998). Given this, they correctly predicted that further domain closure would be possible with different ligands, such as AMPA, with the rearrangement of a few key residues being required to accommodate the ligand. Interestingly, despite the overall low degree of sequence homology amongst iGluR family members, all seven residues that form direct contacts with KA in the structure were shown to be either identical or conservatively substituted, further underlining the importance of keeping these contact points as functionally diverse iGluR subtypes emerged during evolution.
Following this initial study, additional structures of the GluA2 AMPAR LBD in either apo, antagonist-bound (6,7-dinitroquinoxaline-2,3-dione, or DNQX), or agonist-bound (e.g. glutamate, AMPA and kainate) (Fig. 2A) conformations were described, which enabled comparisons to be made concerning what rearrangements of the LBD occur for different ligands (Armstrong & Gouaux, 2000; Hogner et al. 2002; Jin et al. 2002; Armstrong et al. 2003). Specifically, what distinguishes an agonist that opens the channel pore from an antagonist that does not? One measure that appeared to correlate well with agonist responsiveness was the angle of LBD closure formed by lobes D1 and D2 around the ligand binding cleft (relative to the apo structure), ranging from about 20 deg for the full agonist l-Glu to 5 deg for the antagonist DNQX (Armstrong & Gouaux, 2000). In support of the cleft closure hypothesis, a relationship between LBD closure, efficacy, and agonist response (in the presence of cyclothiazide, but see below for more details) held for a series of willardiine agonists, each with a different halogen-substituted group at the same position (Jin et al. 2003) (Fig. 2B and C). As predicted, the larger the halogen atom, the less closure around the agonist could be observed in structures, accounting for the lower weighted channel conductance and reduced efficacy seen in electrophysiological experiments (Jin et al. 2003). Taken together, all of these studies helped establish the idea that the degree of agonist-induced domain closure determines the extent of receptor activation and thus may represent the structural basis of agonist efficacy.
Figure 2. Agonist efficacy correlates with closure of the GluA2 ligand binding cleft.
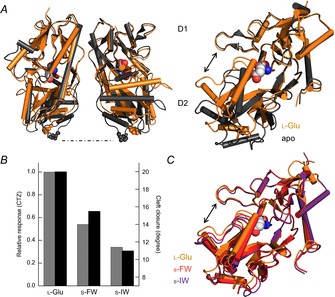
A, side view of the GluA2 LBD with apo (protein data bank (PDB) 1FTO) and l-Glu-bound (PDB 1FTJ) structures overlaid (left). The residue P632, found at the base of the D2 domain, is emphasized to illustrate how this region lifts up and separates upon agonist binding. Visualization of a single subunit highlights how the cleft between D1 and D2 is narrowed in the l-Glu-bound structure (right). B and C, in the presence of the allosteric modulator cyclothiazide (CTZ) to attenuate desensitization, agonist responsiveness correlates to the degree of closure between D1 and D2 at the ligand binding cleft. For example, willardiine series agonists with smaller halogen substituents were more efficacious and produced a greater degree of cleft closure. (Jin et al. 2003).
The need for a revised model of agonist efficacy
In subsequent years, high resolution structures of the LBD of KARs, NMDARs and orphan-class iGluRs were also described, in almost all cases, using the same approach as successfully applied to GluA2 AMPARs (Furukawa & Gouaux, 2003; Mayer, 2005; Naur et al. 2007) (though see Nanao et al. 2005). However, additional structural analyses of the three functional iGluR families has shown that full cleft closure can in fact be induced by partial agonists, and even antagonists. Structures of a GluA2 mutant for which AMPA becomes a partial agonist captured the AMPA-bound LBD cleft in intermediate, as well as fully closed conformations (Armstrong et al. 2003). Concurrently, the first structures of the GluN1 NMDAR subunit (Furukawa & Gouaux, 2003; Inanobe et al. 2005) indicated similar cleft closure for full and partial agonists, contrary to the model of agonist behaviour proposed for AMPARs. Moreover, although the initial structural analysis of GluK1 KARs suggested a close correlation between the degree of LBD closure and agonist behaviour (Mayer, 2005), subsequent work suggested that a more complicated relationship was at play (Fay et al. 2009; Frydenvang et al. 2009). This finding was not entirely surprising since earlier work had already established that external Na+ and Cl− ions profoundly affected the KAR agonist response (Bowie, 2002; Bowie & Lange, 2002; Wong et al. 2006) by binding to discrete sites at the interface between subunits (Plested & Mayer, 2007; Plested et al. 2008; Bowie, 2010) (see below). Taken together, these studies suggested that the structural basis of agonist behaviour was different between iGluR family members and/or that the initial structural model of agonist action at AMPARs needed further refinement.
On that note, Robert and colleagues addressed this issue through a series of structural and functional experiments that examined the impact of mutating a single amino-acid residue (i.e. T686A) that was expected to destabilize the closed conformation of the GluA2 AMPAR ligand binding cleft (Robert et al. 2005). Importantly, the authors were later able to more directly examine agonist efficacy by studying AMPARs at the single-channel level, taking into account sub-conductance levels. These studies suggested a model whereby more efficacious agonists can better stabilize the closed conformation of a dynamic ligand binding cleft (Zhang et al. 2008), an idea that has also been used to explain agonist behaviour at KARs (Maclean et al. 2011). In agreement, spectroscopic measurements of the willardiine-bound GluA2 S1S2 constructs pinpointed specific residues in the cleft that undergo dynamic motions correlated with agonist potency (Fenwick & Oswald, 2008). Thus, variations in cleft stability could potentially explain why crystal structures might capture this domain in multiple – and sometimes unexpected – conformations given the nature of the agonist or antagonist being examined. Such structures capture low energy states and poorly reflect stochastic processes that underlie channel gating (Lau & Roux, 2007). An alternative to the cleft closure paradigm proposed in recent studies is that other movements influence efficacy. In particular, the need to evaluate ligand-induced conformational changes in three dimensions has been emphasized (Birdsey-Benson et al. 2010) and at multiple locations, including the LBD dimer interface (Nayeem et al. 2011). As explained below, there has been a renewed focus on the dimer interface in determining agonist efficacy, particularly in KARs, where a novel role for the cation binding pocket has been identified.
Agonist efficacy, desensitization, and the LBD dimer interface
Although much emphasis was placed on relating conformations of iGluR LBDs to activation, it was also appreciated that subunits assemble back-to-back as a dimer of dimers (Armstrong & Gouaux, 2000; Tichelaar et al. 2004). Interactions between subunits along the LBD dimer interface are thought to remodel upon activation and desensitization (Fig. 3). Activation is proposed to lift apart the lower D2 lobes of the interface, while desensitization is proposed to reflect the rupture of cross-interface interactions as the upper D1 lobes separate (Sun et al. 2002) (Fig. 3). This structural arrangement is also significant from a functional perspective, since it helps explain the onset and recovery from AMPAR desensitization, which occurs in two sequential, kinetic steps (Robert et al. 2001; Bowie & Lange, 2002). This model emerged in part because the binding site for the cyclothiazide, and location of the leucine to tyrosine (L–Y) mutation, both of which strongly attenuate AMPAR desensitization (Patneau et al. 1993; Stern-Bach et al. 1998), are situated where subunits are predicted to come together. Accordingly, studies of the LBD dimer interface have often examined how individual residues affect the kinetics of desensitization (Horning & Mayer, 2004), with less attention paid to their role in agonist efficacy. However, other studies show that mutations along the KAR interface affect the relative efficacies of l-Glu and KA (Fleck et al. 2003; Zhang et al. 2006; Maclean et al. 2011), in addition to the time course of desensitization. Comparisons of agonist behaviour at GluA2 AMPARs in conditions with desensitization present or attenuated (in the presence of cyclothiazide or atop the L–Y mutation) even suggested that desensitization generally inverts the rank order of efficacy (Jin et al. 2003; Holm et al. 2005).
Figure 3. Early structural model of iGluR activation and desensitization.
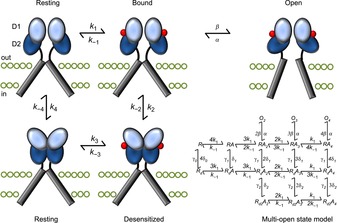
The binding of agonist molecules (red) to the LBD dimer permits subsequent rearrangement to one of two conformations: activated or desensitized. The activated state occurs when the two D2 lobes (dark blue) are lifted apart, generating forces on the transmembrane domains (grey bars) to open the channel pore. Alternatively, the desensitized state stems from the separation of the two D1 domains (light blue) at the dimer interface, relaxing the LBD such that the energy provided by ligand binding cannot open the pore. The structural states are arranged according to the cyclic gating model described in Fig. 1, although to better account for the complex functional behaviour of iGluRs, more advanced kinetic models (Robert & Howe, 2003) have been developed, into which the four agonist binding sites are incorporated (bottom right). Moreover, is not clear to what extent movements of LBD dimers occur in the absence of bound ligands. The ATD has also been excluded for simplicity.
In light of evidence linking the LBD dimer interface to desensitization, experiments were designed to directly test if it was possible to trap AMPARs or KARs into specific conformational states (i.e. active or desensitized) by introducing disulphide bonds via cysteine residues on opposing sides of the dimer interface to restrict protein movement (Armstrong et al. 2006; Priel et al. 2006; Weston et al. 2006) (Fig. 4A). As anticipated, the rapidly decaying response elicited by l-Glu was converted into a non-decaying phenotype via crosslinking of the D1 interface, suggesting that restricting dimer movement in this region locks the AMPAR or KAR into the activated state (Priel et al. 2006; Weston et al. 2006) (Fig. 4B). Despite this, it was surprising that responses elicited by the crosslinked GluA2 AMPAR were potentiated by cyclothiazide to a greater extent than the wild-type receptor (Weston et al. 2006). In principle, if desensitization is blocked, the agonist response would be expected to be unaffected by cyclothiazide unless a more complicated mechanism of cyclothiazide action is at play (see Mitchell & Fleck, 2007). Similarly, GluK2 KARs crosslinked to restrict dimer movement (Priel et al. 2006; Weston et al. 2006) elicited responses that were more consistent with the equilibrium response of wild-type receptors, where the occurrence of desensitization accounts for the slow decay kinetics and the higher apparent agonist affinity (Bowie et al. 2003). A clue to the unexpected behaviour of these cysteine mutants could be found in the GluK2 Y521C/L783C crystal structure (Weston et al. 2006), which showed the dimer interface to be in a relaxed conformation, in between an activated arrangement and the pseudo-desensitized arrangement of GluA2 S729C (Armstrong et al. 2006) (Fig. 5A). An important caveat in these studies is that the authors relied on macroscopic responses to infer the absence or presence of desensitization. However, given that desensitization represents long-lived inactive states of the channel (Sakmann et al. 1980), its occurrence can only be truly confirmed by single-channel analysis. As explained below, unitary measurements of crosslinked iGluRs revealed an unappreciated importance of the LBD dimer interface in governing the degree of activation.
Figure 4. Crosslinking of the GluK2 LBD dimer interface yields non-decaying current responses.
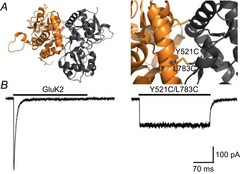
A, side views of the LBD dimer interface of GluK2 Y521C/L783C (PDB 2I0C), in its entirety (left) and close-up (right), detailing the inter-protomer disulphide bond (yellow). B, activation profiles of wild-type GluK2 and Y521C/L783C receptors in response to 10 mm l-Glu (holding potential –60 mV). Adapted from Daniels et al. (2013) with permission.
Figure 5. Cysteine crosslinking of the GluK2 LBD dimer interface disrupts activation.
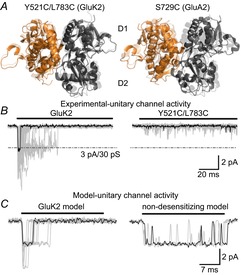
A, side views of the LBD dimer interface of GluK2 Y521C/L783C (left, PDB 2I0C) and GluA2 S729C (right, PDB 2I3W) in front of the corresponding l-Glu-bound wild-type receptors (transparent, PDB 3G3F and 1FTJ). For both mutants the distance across the interface between the D1 domains is increased. B, activation profiles of wild-type GluK2 and Y521C/L783C receptors at the single-channel level with a typical unitary response highlighted (black) for both receptors. C, simulations of GluK2 unitary channel activity in the presence (left) or absence (right) of desensitization, generated from a cyclic gating model (Bowie et al. 1998). When a single channel is simulated with desensitized states removed, the open channel probability is much greater than observed experimentally for the double cysteine mutant. Responses were recorded or simulated using 10 mm l-Glu and a holding potential of −100 mV, and experimental data were filtered at 1 kHz. Adapted from Daniels et al. (2013) with permission.
Single-channel measurements of the GluN1/GluN2A NMDAR crosslinked at equivalent positions to those in AMPARs and KARs demonstrated that restraining dimer movement had a profound effect on activation (i.e. Popen), whereas desensitization was still present (Borschel et al. 2011). The authors’ findings suggested two possibilities: (i) the structural basis of NMDAR desensitization is distinct from that of AMPARs and KARs and/or (ii) earlier studies implicating the dimer interface with AMPAR and KAR desensitization had overlooked the impact of crosslinking on the activation process. In keeping with the latter possibility, single-channel recordings comparing wild-type GluK2 receptor responses to the crosslinked receptor (i.e. GluK2 Y521C/L783C) showed that the mutant receptor activated poorly (Daniels et al. 2013) (Fig. 5B). Specifically, the crosslinked GluK2 receptor is effectively locked out of the main open-state, rather than having a preference for it, which would be expected if desensitization was abolished (Fig. 5C). Preliminary single-channel data on crosslinked GluA2 receptors (B. A. Daniels & D. Bowie, unpublished observations) reveals that channel activation is similarly disrupted. Taken together, these data suggest that the architecture of the LBD dimer interface of all iGluR subtypes is a key location for channel activation.
The cation binding pocket of KARs acts as an on/off switch
Although the strategy of covalent crosslinking did not lock KARs into the main activated state as expected, another GluK2 receptor mutant, namely GluK2 D776K, had also been proposed to eliminate macroscopic desensitization (Nayeem et al. 2009), but had yet to be studied at the single-channel level. Encouragingly, structural data showed that the positively charged Lys776 established a new inter-protomer contact across the dimer interface by tethering to the cation binding pocket (Nayeem et al. 2011) and thus affected KARs through a different mechanism from the Cys521/Cys783 disulphide bridge (Fig. 6A). This observation was also intriguing given the fact that earlier functional data had shown that occupancy of the cation binding pocket by external cations, such as Na+, was an absolute requirement for KAR activation (Wong et al. 2006; Bowie, 2010). Consequently, it was possible that Lys776 mimicked the effect of external cations and, by near-permanent occupancy of the cation binding pocket, was able to sustain activation and thus eliminate desensitization. In keeping with this, single channel recordings of individual GluK2 D776K receptors activated to the main conductance state of approximately 20 pS and remained there in the continued presence of the agonist (Dawe et al. 2013) (Fig. 6B and C). Using a combination of molecular dynamics simulations and electrophysiological recordings, it was shown that the cation binding pocket acts like an on/off switch with cation binding priming the KAR for activation, whereas desensitization proceeds when the cation site is unoccupied (Dawe et al. 2013). From this perspective, GluK2 D776K maximizes agonist efficacy and sustains the agonist response by keeping the KAR in the activated state rather than affecting the process of desensitization directly. Curiously, GluA1 AMPARs contain many of the residues that make up the cation binding pocket in KARs, but remain insensitive to modulation by external cations (Bowie, 2002). Previous studies have explained cation insensitivity of AMPARs by speculating that the Lys residue that lies in the pocket acts as a surrogate cation (Wong et al. 2006, 2007). This idea has yet to be tested experimentally; however, given that the Lys residue is conserved amongst some NMDARs, the KAR ‘cation binding pocket’ may prove to be a hotspot for activation of all iGluR families.
Figure 6. Occupancy of the GluK2 cation binding pocket sustains activation.
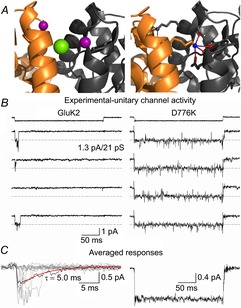
A, side views of the LBD dimer interface of wild-type GluK2 (left, PDB 3G3F) and the D776K mutant (right, PDB 2XXX). The former includes two allosteric sodium ions (purple) and a chloride ion (green) bound at the apex of the interface, while the latter possesses a charged amino group (blue) on residue 776 tethering into the electronegative pocket (red) normally occupied by sodium. B, representative single-channel responses of GluK2 (left) and D776K (right) to 10 mm l-Glu (holding potential −60 mV, filtered at 1 kHz). C, averaged responses of individual sweeps taken from the same patch recordings as shown in B, which mimic the phenotype exhibited by a large population of receptors. For wild-type GluK2 several individual responses (grey) are overlaid behind the average response, while a fit of the current decay (red) suggests the unitary events are representative of those that contribute to macroscopic decay kinetics, which occur over a similar time course. (Dawe et al. 2013).
New insights from full-length iGluR structures
Despite the identification of several discrete sites that regulate iGluR activation, the structural domains (ATD and LBD) to which they belong have been studied largely in isolation, making it difficult to ascribe a role to them during any global protein rearrangements that may accompany channel gating. Part of the problem has been the difficulty of obtaining full-length structures at atomic resolution (Mayer, 2011; Sobolevsky, 2013), though lower resolution, single particle electron microscopy (EM) images have actually been available for some time (Nakagawa, 2010). The first ‘image’ of a tetrameric iGluR was obtained following large-scale expression of recombinant GluA2 (Safferling et al. 2001), followed by a more refined three-dimensional reconstruction of the receptor at 20 Å resolution (Tichelaar et al. 2004). Native AMPAR complexes, with and without associated transmembrane AMPA receptor regulatory proteins (TARPs), were later imaged in multiple conformations at 30–40 Å using cryo-EM techniques (Nakagawa et al. 2005, 2006). The AMPARs were sorted into two major classes: a resting and/or activated class possessing a more compact extracellular region, and a desensitized class notable for separation of lobes at the ATD level (Nakagawa et al. 2005). This arrangement contrasts with an unliganded GluA2 receptor comprising a compact ATD and separate LBD lobes reported elsewhere (Midgett & Madden, 2008; Midgett et al. 2012), and suggests that protein purification conditions may heavily influence the resulting structure. Full-length tetrameric crystal structures have since supported the notion of a Y-shaped structure with a twofold axis of symmetry for apo and antagonist-bound GluA2 AMPARs (Sobolevsky et al. 2009; Dürr et al. 2014) (Fig. 7A). Meanwhile, crystallization in the presence of agonists and allosteric modulators or toxins that increase channel open probability suggest an activated state not too structurally dissimilar from the resting state: amongst many interesting movements, these structures reveal greater closure of the ligand binding cleft, opening between pairs of LBD dimers around the axis of symmetry, and an increase in inter-subunit LBD–TM linker distances (Fig. 7B), indicative of a pulling force that precedes channel opening (Chen et al. 2014; Dürr et al. 2014). These results were largely corroborated by cryo-EM data published simultaneously in which GluA2 is characterized in resting, activated and desensitized states, and contrasted with GluK2 in a desensitized conformation (Meyerson et al. 2014). Through improved image-processing techniques, resolution approaching 7 Å was achieved, in contrast to earlier images of apo and desensitized GluK2 receptors of around 20 Å (Schauder et al. 2013). Perhaps the most intriguing finding from the later EM study was an asymmetric rearrangement of the LBD during desensitization (Fig. 7C), in which the two subunits of each dimer rotate 125 deg and 13 deg in a horizontal plane (Meyerson et al. 2014), a movement captured to some extent in an fluorowillardiine-bound crystal structure (Dürr et al. 2014), but possibly constrained due to the extensive mutagenesis required for crystallization. GluA2 was also crystallized with nitrowillardiine in another study, but the LBD did not exhibit any drastic rearrangements relative to an antagonist-bound structure of the same, modified receptor (Yelshanskaya et al. 2014). Differences in agonist-bound structures are difficult to interpret, since it is unknown whether such structures represent closed or desensitized states, and to what extent mutagenesis of the GluA2 crystal construct or crystal lattice contacts affect the conformations adopted by these proteins (Yelshanskaya et al. 2014). Ultimately, the ideas presented in these structural studies remain to be consolidated, yet they will certainly generate many testable hypotheses concerning the mechanisms of AMPAR/KAR gating.
Figure 7. Structural rearrangements of full-length iGluRs during activation and desensitization.
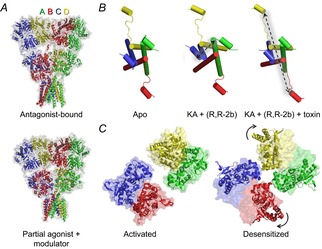
A, full-length GluA2 receptor bound by the competitive antagonist ZK200775 (top, PDB 3KG2) or KA and the allosteric modulator R,R-2b (bottom, PDB 4U1W), which potentiates GluA2 current responses in equilibrium conditions (Kaae et al. 2007). Both structures retain a twofold axis of symmetry, with the A/C and B/D subunits having distinct arrangements. The latter structure is thought to represent a ‘pre-open’ state of the receptor during the activation process. B, in contrast to the unliganded, apo state of GluA2 (left, PDB 4U2P), the binding of an agonist with positive modulator (centre, PDB 4U1W) causes separation between A/C subunits at the level of the LBD–TM 3 linker, generating forces that could open the pore. Addition of the con-ikot-ikot snail toxin further increases the B/D distance (right, PDB 4U5D). C, the tetrameric LBD of GluA2 bound by l-Glu and the allosteric modulator LY451646 (left, PDB 4UQK), a potentiator of AMPAR equilibrium currents (Gates et al. 2001), believed to be in an activated state. In contrast, the LBD of GluK2 bound by 2S,4R-4-methylglutamate (right, PDB 4UQQ) is believed to be in a desensitized state, characterized by large horizontal rotation of the B/D subunits.
Conclusion
Although we have witnessed great advances in our understanding of iGluR activation in the last three decades, the way forward faces two interrelated obstacles. First, we still do not have a detailed kinetic model of iGluRs that explains the relationship between different activated (i.e. channel conductance(s)) and desensitized states. This obstacle could be addressed with computational approaches that take into account the hierarchical structure of the iGluR (Ollivier et al. 2010). However, to provide an accurate starting template for the rule-based modelling of iGluRs, we require a clearer understanding of the stoichiometry of their activation and desensitization. The use of photo-switchable ligands tethered to the iGluR ligand binding cleft (Volgraf et al. 2006) could help address this problem, particularly if certain subunits are selectively stimulated (Reiner & Isacoff, 2014), although receptor modification and difficulty replicating response kinetics observed with rapid solution exchange will complicate interpretations made from this technique.
The second major roadblock to understanding iGluR activation is the scarcity of structural correlates for functional states. Despite the recent determination of the full-length structures of AMPARs in a range of agonist and antagonist-induced states (Dürr et al. 2014; Meyerson et al. 2014), it remains to be seen if these few ‘snapshots’ can account for the diversity of functional behaviour exhibited by iGluRs. It is also worth noting that no crystal structure has yet shown the pore in an open configuration. Consequently, there is still much to study regarding the signal transduction pathway that allows agonist binding to promote conformational changes that gate the channel pore. In NMDARs, where full-length structures are now available (Karakas & Furukawa, 2014; Lee et al. 2014), recent work has shown that the LBD–TM linker provides a mechanical force that increases the likelihood of channel opening (Kazi et al. 2014). In AMPARs and KARs, which have a low open probability in equilibrium conditions (e.g. Zhang et al. 2008), carrying out such investigations remains difficult. Molecular dynamics simulations of the GluA2 receptor have suggested that small increases in pore diameter can be achieved through rearrangement in the LBD (Dong & Zhou, 2011), but whether this mechanism can permit ion permeation is unclear. In the short term, full-length iGluR structures will provide more insight into which micro-domains may be critically linked to channel activation. But as an alternative approach, recent work on Cys-loop receptors suggests that insight into the allosteric nature of the ligand-gated ion channels may be achieved by mapping out the energy contributions of different structural domains during gating (Purohit et al. 2013). In either case, the identification of regions which contribute to the activation and desensitization of intact iGluRs opens up several new avenues of investigation. For instance, it is presently uncertain how dynamic the interactions are between auxiliary proteins (known to affect receptor trafficking and gating kinetics) and iGluRs at the synapse (Jackson & Nicoll, 2011), but having a three-dimensional model of AMPAR activation could shed light on state-dependent modulatory effects. In addition, such models might reveal previously unappreciated sites as new targets for therapeutic compounds. While it is clear that many secrets of glutamate receptor physiology remain to be uncovered, we have learned a great deal from 30 years of research on this neurotransmitter receptor.
Acknowledgments
We wish to thank members of the Bowie lab for their thoughtful comments on the manuscript.
Glossary
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- ATD
amino terminal domain
- ED
extracellular domain
- GlnBP
glutamine binding protein
- iGluR
ionotropic glutamate receptor
- KA
kainate
- KAR
kainate receptor
- LBD
ligand binding domain
- l-Glu
l-glutamate
- mGluR
metabotropic glutamate receptor
- NMDA
N-methyl d-aspartate
- NMDAR
NMDA receptor
- QA
quisqualate
- TARP
transmembrane AMPA receptor regulatory protein
- TM
transmembrane domain
Biographies
G. BrentDawe is a PhD candidate inMcGill's Integrated Program in Neuroscience and recipient of an NSERC doctoral scholarship. He obtained his BSc in Biology at the University of Ontario Institute of Technology, where he completed an Honours thesis project with Dr Sean Forrester. His research in the Bowie lab concerns the mechanisms of glutamate receptor activation and desensitization.
Derek Bowie is the Director of the FRQS (Fonds de recherche du Québec – Santé) research group GEPROM and has been a professor at McGill University in Montréal since 2002. His lab focuses on the structure–function properties of ionotropic glutamate receptors and GABAA receptors, as well as their role in neuronal circuit behaviour. Dr Bowie completed his doctoral work at the University of London followed by postdoctoral training in France (Université Louis Pasteur), Switzerland (University of Zurich) and the USA (National Institutes of Health) before holding a faculty position at Emory University in Atlanta.
Additional information
Competing interests
None declared.
Funding
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) as well as a personal award from the Canada Research Chair Program in Receptor Pharmacology. G.B.D. is funded by an Alexander Graham Bell Canada Graduate Scholarship (CGS-D) through the Natural Sciences and Engineering Research Council of Canada (NSERC). M.R.A. was funded through the Frederick Banting and Charles Best CGS-D through the CIHR. B.A.D. is funded through the CIHR/Fragile X Research Foundation of Canada (postdoctoral award).
References
- Adams MD. Oxender DL. Bacterial periplasmic binding protein tertiary structures. J Biol Chem. 1989;264:15739–15742. [PubMed] [Google Scholar]
- Akaike N. Concentration clamp technique. In: Walz W, editor; Boulton A, Baker G, editors. Neuromethods, Patch-Clamp Applications and Protocols. Humana Press Inc; 1995. pp. 141–154. [Google Scholar]
- Armstrong N. Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M. Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M. Gouaux E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci U S A. 2003;100:5736–5741. doi: 10.1073/pnas.1037393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ. Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- Ascher P. Nowak L. Quisqualate- and kainate-activated channels in mouse central neurones in culture. J Physiol. 1988;399:227–245. doi: 10.1113/jphysiol.1988.sp017077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M. Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-y. [DOI] [PubMed] [Google Scholar]
- Birdsey-Benson A, Gill A, Henderson LP. Madden DR. Enhanced efficacy without further cleft closure: reevaluating twist as a source of agonist efficacy in AMPA receptors. J Neurosci. 2010;30:1463–1470. doi: 10.1523/JNEUROSCI.4558-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borschel WF, Murthy SE, Kasperek EM. Popescu GK. NMDA receptor activation requires remodelling of intersubunit contacts within ligand-binding heterodimers. Nat Commun. 2011;2:498. doi: 10.1038/ncomms1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. External anions and cations distinguish between AMPA and kainate receptor gating mechanisms. J Physiol. 2002;539:725–733. doi: 10.1113/jphysiol.2001.013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Ionotropic glutamate receptors and CNS disorders. CNS Neurol Disord Drug Targets. 2008;7:129–143. doi: 10.2174/187152708784083821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Ion-dependent gating of kainate receptors. J Physiol. 2010;588:67–81. doi: 10.1113/jphysiol.2009.178863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Garcia EP, Marshall J, Traynelis SF. Lange GD. Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J Physiol. 2003;547:373–385. doi: 10.1113/jphysiol.2002.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Lange GD. Functional stoichiometry of glutamate receptor desensitization. J Neurosci. 2002;22:3392–3403. doi: 10.1523/JNEUROSCI.22-09-03392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD. Mayer ML. Activity-dependent modulation of glutamate receptors by polyamines. J Neurosci. 1998;18:8175–8185. doi: 10.1523/JNEUROSCI.18-20-08175.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GQ. Gouaux E. Overexpression of a glutamate receptor (GluR2) ligand binding domain in Escherichia coli: application of a novel protein folding screen. Proc Natl Acad Sci U S A. 1997;94:13431–13436. doi: 10.1073/pnas.94.25.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Dürr KL. Gouaux E. X-ray structures of AMPA receptor–cone snail toxin complexes illuminate activation mechanism. Science. 2014;345:1021–1026. doi: 10.1126/science.1258409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL. Lester RA. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev. 1989;41:143–210. [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J. Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Mulle C. Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG. Usowicz MM. Multiple-conductance channels activated by excitatory amino acids in cerebellar neurons. Nature. 1987;325:525–528. doi: 10.1038/325525a0. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG. Usowicz MM. On the multiple-conductance single channels activated by excitatory amino acids in large cerebellar neurones of the rat. J Physiol. 1989;415:555–582. doi: 10.1113/jphysiol.1989.sp017736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels BA, Andrews ED, Aurousseau MR, Accardi MV. Bowie D. Crosslinking the ligand-binding domain dimer interface locks kainate receptors out of the main open state. J Physiol. 2013;591:3873–3885. doi: 10.1113/jphysiol.2013.253666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe GB, Musgaard M, Andrews ED, Daniels BA, Aurousseau MR, Biggin PC. Bowie D. Defining the structural relationship between kainate-receptor deactivation and desensitization. Nat Struct Mol Biol. 2013;20:1054–1061. doi: 10.1038/nsmb.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D. Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Dong H. Zhou HX. Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat Commun. 2011;2:354. doi: 10.1038/ncomms1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr KL, Chen L, Stein RA, De Zorzi R, Folea IM, Walz T, McHaourab HS. Gouaux E. Structure and dynamics of AMPA receptor GluA2 in resting, pre-open, and desensitized states. Cell. 2014;158:778–792. doi: 10.1016/j.cell.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egebjerg J, Bettler B, Hermans-Borgmeyer I. Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- Fay AM, Corbeil CR, Brown P, Moitessier N. Bowie D. Functional characterization and in silico docking of full and partial GluK2 kainate receptor agonists. Mol Pharmacol. 2009;75:1096–1107. doi: 10.1124/mol.108.054254. [DOI] [PubMed] [Google Scholar]
- Fenwick MK. Oswald RE. NMR spectroscopy of the ligand-binding core of ionotropic glutamate receptor 2 bound to 5-substituted willardiine partial agonists. J Mol Biol. 2008;378:673–685. doi: 10.1016/j.jmb.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck MW, Cornell E. Mah SJ. Amino-acid residues involved in glutamate receptor 6 kainate receptor gating and desensitization. J Neurosci. 2003;23:1219–1227. doi: 10.1523/JNEUROSCI.23-04-01219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydenvang K, Lash LL, Naur P, Postila PA, Pickering DS, Smith CM, Gajhede M, Sasaki M, Sakai R, Pentikainen OT, Swanson GT. Kastrup JS. Full domain closure of the ligand-binding core of the ionotropic glutamate receptor iGluR5 induced by the high affinity agonist dysiherbaine and the functional antagonist 8,9-dideoxyneodysiherbaine. J Biol Chem. 2009;284:14219–14229. doi: 10.1074/jbc.M808547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H. Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates M, Ogden A. Bleakman D. Pharmacological effects of AMPA receptor potentiators LY392098 and LY404187 on rat neuronal AMPA receptors in vitro. Neuropharmacology. 2001;40:984–991. doi: 10.1016/s0028-3908(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Gouaux E. Structure and function of AMPA receptors. J Physiol. 2004;554:249–253. doi: 10.1113/jphysiol.2003.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogner A, Kastrup JS, Jin R, Liljefors T, Mayer ML, Egebjerg J, Larsen IK. Gouaux E. Structural basis for AMPA receptor activation and ligand selectivity: crystal structures of five agonist complexes with the GluR2 ligand-binding core. J Mol Biol. 2002;322:93–109. doi: 10.1016/s0022-2836(02)00650-2. [DOI] [PubMed] [Google Scholar]
- Hollmann M. Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Maron C. Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Hollmann M, O'Shea-Greenfield A, Rogers SW. Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature. 1989;342:643–648. doi: 10.1038/342643a0. [DOI] [PubMed] [Google Scholar]
- Holm MM, Naur P, Vestergaard B, Geballe MT, Gajhede M, Kastrup JS, Traynelis SF. Egebjerg J. A binding site tyrosine shapes desensitization kinetics and agonist potency at GluR2. A mutagenic, kinetic, and crystallographic study. J Biol Chem. 2005;280:35469–35476. doi: 10.1074/jbc.M507800200. [DOI] [PubMed] [Google Scholar]
- Horning MS. Mayer ML. Regulation of AMPA receptor gating by ligand binding core dimers. Neuron. 2004;41:379–388. doi: 10.1016/s0896-6273(04)00018-2. [DOI] [PubMed] [Google Scholar]
- Hsiao CD, Sun YJ, Rose J. Wang BC. The crystal structure of glutamine-binding protein from Escherichia coli. J Mol Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron. 1990;5:255–266. doi: 10.1016/0896-6273(90)90163-a. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Kainate receptors and synaptic transmission. Prog Neurobiol. 2003;70:387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Glutamate receptor pores. J Physiol. 2014 doi: 10.1113/jphysiol.2014.272724. (in press; DOI: 10.1113/jphysiol.2014.272724) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanobe A, Furukawa H. Gouaux E. Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron. 2005;47:71–84. doi: 10.1016/j.neuron.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Jackson AC. Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE. Stevens CF. Glutamate activates multiple single channel conductances in hippocampal neurons. Nature. 1987;325:522–525. doi: 10.1038/325522a0. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF. Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- Jin R, Horning M, Mayer ML. Gouaux E. Mechanism of activation and selectivity in a ligand-gated ion channel: structural and functional studies of GluR2 and quisqualate. Biochemistry. 2002;41:15635–15643. doi: 10.1021/bi020583k. [DOI] [PubMed] [Google Scholar]
- Johnson JW. Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Jonas P. Fast application of agonists to isolated membrane patches. In: Neher E, editor; Sakmann B, editor. Single-Channel Recording. New York: Plenum Press; 1995. pp. 231–243. [Google Scholar]
- Kaae BH, Harpsoe K, Kastrup JS, Sanz AC, Pickering DS, Metzler B, Clausen RP, Gajhede M, Sauerberg P, Liljefors T. Madsen U. Structural proof of a dimeric positive modulator bridging two identical AMPA receptor-binding sites. Chem Biol. 2007;14:1294–1303. doi: 10.1016/j.chembiol.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Karakas E. Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Dai J, Sweeney C, Zhou HX. Wollmuth LP. Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat Neurosci. 2014;17:914–922. doi: 10.1038/nn.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B. Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Kiskin NI, Krishtal OA. Tsyndrenko A. Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci Lett. 1986;63:225–230. doi: 10.1016/0304-3940(86)90360-5. [DOI] [PubMed] [Google Scholar]
- Kleckner NW. Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Kuusinen A, Arvola M. Keinanen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327–6332. doi: 10.1002/j.1460-2075.1995.tb00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AY. Roux B. The free energy landscapes governing conformational changes in a glutamate receptor ligand-binding domain. Structure. 2007;15:1203–1214. doi: 10.1016/j.str.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X. Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. Marques JM. Kainate receptors in health and disease. Neuron. 2013;80:292–311. doi: 10.1016/j.neuron.2013.09.045. [DOI] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Naranjo JR. Mellström B. Functional kainate-selective glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1993;90:11688–11692. doi: 10.1073/pnas.90.24.11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- MacDonald JF. Nowak LM. Mechanisms of blockade of excitatory amino acid receptor channels. Trends Pharmacol Sci. 1990;11:167–172. doi: 10.1016/0165-6147(90)90070-O. [DOI] [PubMed] [Google Scholar]
- Maclean DM, Wong AY, Fay AM. Bowie D. Cations but not anions regulate the responsiveness of kainate receptors. J Neurosci. 2011;31:2136–2144. doi: 10.1523/JNEUROSCI.4314-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan H( Receptors for the excitatory amino acids in the mammalian central nervous system. Prog Neurobiol. 1983;20:251–271. doi: 10.1016/0301-0082(83)90004-7. [DOI] [PubMed] [Google Scholar]
- Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- Mano I, Lamed Y. Teichberg VI. A venus flytrap mechanism for activation and desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. J Biol Chem. 1996;271:15299–15302. doi: 10.1074/jbc.271.26.15299. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R. Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Mayer ML. Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539–552. doi: 10.1016/j.neuron.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Mayer ML. Emerging models of glutamate receptor ion channel structure and function. Structure. 2011;19:1370–1380. doi: 10.1016/j.str.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Armstrong N. Structure and function of glutamate receptor ion channels. Annu Rev Physiol. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- Mayer ML. Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML. Subramaniam S. Structural mechanism of glutamate receptor activation and desensitization. Nature. 2014 doi: 10.1038/nature13603. (in press; DOI: 10.1038/nature13603) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett CR, Gill A. Madden DR. Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation. Front Mol Neurosci. 2012;4:56. doi: 10.3389/fnmol.2011.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett CR. Madden DR. The quaternary structure of a calcium-permeable AMPA receptor: conservation of shape and symmetry across functionally distinct subunit assemblies. J Mol Biol. 2008;382:578–584. doi: 10.1016/j.jmb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Mitchell NA. Fleck MW. Targeting AMPA receptor gating processes with allosteric modulators and mutations. Biophys J. 2007;92:2392–2402. doi: 10.1529/biophysj.106.095091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N. Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol Neurobiol. 2010;42:161–184. doi: 10.1007/s12035-010-8149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Cheng Y, Ramm E, Sheng M. Walz T. Structure and different conformational states of native AMPA receptor complexes. Nature. 2005;433:545–549. doi: 10.1038/nature03328. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Cheng Y, Sheng M. Walz T. Three-dimensional structure of an AMPA receptor without associated stargazin/TARP proteins. Biol Chem. 2006;387:179–187. doi: 10.1515/BC.2006.024. [DOI] [PubMed] [Google Scholar]
- Nakanishi N, Shneider NA. Axel R. A family of glutamate receptor genes: evidence for the formation of heteromultimeric receptors with distinct channel properties. Neuron. 1990;5:569–581. doi: 10.1016/0896-6273(90)90212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S( Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Green T, Stern-Bach Y, Heinemann SF. Choe S. Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci U S A. 2005;102:1708–1713. doi: 10.1073/pnas.0409573102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, Dravid SM, Pickering DS, Olsen L, Vestergaard B, Egebjerg J, Gajhede M, Traynelis SF. Kastrup JS. Ionotropic glutamate-like receptor δ2 binds d-serine and glycine. Proc Natl Acad Sci U S A. 2007;104:14116–14121. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Mayans O. Green T. Conformational flexibility of the ligand-binding domain dimer in kainate receptor gating and desensitization. J Neurosci. 2011;31:2916–2924. doi: 10.1523/JNEUROSCI.4771-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Zhang Y, Schweppe DK, Madden DR. Green T. A nondesensitizing kainate receptor point mutant. Mol Pharmacol. 2009;76:534–542. doi: 10.1124/mol.109.056598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL. Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- Ollivier JF, Shahrezaei V. Swain PS. Scalable rule-based modelling of allosteric proteins and biochemical networks. PLoS Comput Biol. 2010;6:e1000975. doi: 10.1371/journal.pcbi.1000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paas Y. The macro- and microarchitectures of the ligand-binding domain of glutamate receptors. Trends Neurosci. 1998;21:117–125. doi: 10.1016/s0166-2236(97)01184-3. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C. Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Patneau DK. Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK. Mayer ML. Kinetic analysis of interactions between kainate and AMPA: evidence for activation of a single receptor in mouse hippocampal neurons. Neuron. 1991;6:785–798. doi: 10.1016/0896-6273(91)90175-y. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML, Jane DE. Watkins JC. Activation and desensitization of AMPA/kainate receptors by novel derivatives of willardiine. J Neurosci. 1992;12:595–606. doi: 10.1523/JNEUROSCI.12-02-00595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L., Jr Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirotte B, Francotte P, Goffin E. de Tullio P. AMPA receptor positive allosteric modulators: a patent review. Expert Opin Ther Pat. 2013;23:615–628. doi: 10.1517/13543776.2013.770840. [DOI] [PubMed] [Google Scholar]
- Plested AJ. Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Vijayan R, Biggin PC. Mayer ML. Molecular basis of kainate receptor modulation by sodium. Neuron. 2008;58:720–735. doi: 10.1016/j.neuron.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Selak S, Lerma J. Stern-Bach Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron. 2006;52:1037–1046. doi: 10.1016/j.neuron.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Purohit P, Gupta S, Jadey S. Auerbach A. Functional anatomy of an allosteric protein. Nat Commun. 2013;4:2984. doi: 10.1038/ncomms3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian A. Johnson JW. Channel gating of NMDA receptors. Physiol Behav. 2002;77:577–582. doi: 10.1016/s0031-9384(02)00906-x. [DOI] [PubMed] [Google Scholar]
- Quiocho FA. Atomic structures of periplasmic binding proteins and the high-affinity active transport systems in bacteria. Philos Trans R Soc Lond B Biol Sci. 1990;326:341–351. doi: 10.1098/rstb.1990.0016. discussion 351–342. [DOI] [PubMed] [Google Scholar]
- Reiner A. Isacoff EY. Tethered ligands reveal glutamate receptor desensitization depends on subunit occupancy. Nat Chem Biol. 2014;10:273–280. doi: 10.1038/nchembio.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Armstrong N, Gouaux JE. Howe JR. AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J Neurosci. 2005;25:3752–3762. doi: 10.1523/JNEUROSCI.0188-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A. Howe JR. How AMPA receptor desensitization depends on receptor occupancy. J Neurosci. 2003;23:847–858. doi: 10.1523/JNEUROSCI.23-03-00847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Irizarry SN, Hughes TE. Howe JR. Subunit interactions and AMPA receptor desensitization. J Neurosci. 2001;21:5574–5586. doi: 10.1523/JNEUROSCI.21-15-05574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safferling M, Tichelaar W, Kummerle G, Jouppila A, Kuusinen A, Keinanen K. Madden DR. First images of a glutamate receptor ion channel: oligomeric state and molecular dimensions of GluRB homomers. Biochemistry. 2001;40:13948–13953. doi: 10.1021/bi011143g. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Patlak J. Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH. Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF. Liotta DC. Novel NMDA receptor modulators: an update. Expert Opin Ther Pat. 2012;22:1337–1352. doi: 10.1517/13543776.2012.728587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder DM, Kuybeda O, Zhang J, Klymko K, Bartesaghi A, Borgnia MJ, Mayer ML. Subramaniam S. Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain. Proc Natl Acad Sci U S A. 2013;110:5921–5926. doi: 10.1073/pnas.1217549110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH. The TINS/TiPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI. Structure and gating of tetrameric glutamate receptors. J Physiol. 2013 doi: 10.1113/jphysiol.2013.264911. (in press; DOI: 10.1113/jphysiol.2013.264911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP. Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O'Hara PJ. Heinemann SF. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M. Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M. Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Sun YJ, Rose J, Wang BC. Hsiao CD. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: comparisons with other amino acid binding proteins. J Mol Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Masu M, Ishii T, Shigemoto R. Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- Tang CM, Dichter M. Morad M. Quisqualate activates a rapidly inactivating high conductance ionic channel in hippocampal neurons. Science. 1989;243:1474–1477. doi: 10.1126/science.2467378. [DOI] [PubMed] [Google Scholar]
- Tichelaar W, Safferling M, Keinanen K, Stark H. Madden DR. The three-dimensional structure of an ionotropic glutamate receptor reveals a dimer-of-dimers assembly. J Mol Biol. 2004;344:435–442. doi: 10.1016/j.jmb.2004.09.048. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ. Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgraf M, Gorostiza P, Numano R, Kramer RH, Isacoff EY. Trauner D. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat Chem Biol. 2006;2:47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L, Jr, Benveniste M. Mayer ML. Modulation of N-methyl-d-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J Physiol. 1990;428:313–331. doi: 10.1113/jphysiol.1990.sp018214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L, Jr, Patneau DK. Mayer ML. Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron. 1991;7:971–984. doi: 10.1016/0896-6273(91)90342-w. [DOI] [PubMed] [Google Scholar]
- Watkins JC. Evans RH. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol. 1981;21:165–204. doi: 10.1146/annurev.pa.21.040181.001121. [DOI] [PubMed] [Google Scholar]
- Weston MC, Schuck P, Ghosal A, Rosenmund C. Mayer ML. Conformational restriction blocks glutamate receptor desensitization. Nat Struct Mol Biol. 2006;13:1120–1127. doi: 10.1038/nsmb1178. [DOI] [PubMed] [Google Scholar]
- Wong AY, Fay AM. Bowie D. External ions are coactivators of kainate receptors. J Neurosci. 2006;26:5750–5755. doi: 10.1523/JNEUROSCI.0301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, MacLean DM. Bowie D. Na+/Cl– dipole couples agonist binding to kainate receptor activation. J Neurosci. 2007;27:6800–6809. doi: 10.1523/JNEUROSCI.0284-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya MV, Li M. Sobolevsky AI. Structure of an agonist-bound ionotropic glutamate receptor. Science. 2014;345:1070–1074. doi: 10.1126/science.1256508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M. The δ2 glutamate receptor: 10 years later. Neurosci Res. 2003;46:11–22. doi: 10.1016/s0168-0102(03)00036-1. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. The ins and outs of GluD2–why and how Purkinje cells use the special glutamate receptor. Cerebellum. 2012;11:438–439. doi: 10.1007/s12311-011-0328-4. [DOI] [PubMed] [Google Scholar]
- Zhang W, Cho Y, Lolis E. Howe JR. Structural and single-channel results indicate that the rates of ligand binding domain closing and opening directly impact AMPA receptor gating. J Neurosci. 2008;28:932–943. doi: 10.1523/JNEUROSCI.3309-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nayeem N, Nanao MH. Green T. Interface interactions modulating desensitization of the kainate-selective ionotropic glutamate receptor subunit GluR6. J Neurosci. 2006;26:10033–10042. doi: 10.1523/JNEUROSCI.2750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]