

Abstract
Noradrenaline (NA)-releasing neurons in the locus coeruleus (LC) provide NA to the forebrain. Their activity is believed to be a key factor regulating the wakefulness/arousal level of the brain. In this study, we found that the activity of NA-releasing neurons in the LC (LC neurons) was subject to γ-aminobutyric acid (GABA) tonic inhibition through GABAB receptors (GABABRs), but not GABAA receptors. The intensity of GABABR tonic inhibition was found to depend on ambient GABA levels, as it was dramatically increased by blockade of GABA reuptake. It also varied with the function of GABABRs. The GABABR activity on LC neurons was found to increase with postnatal age up to postnatal days 8–10, resulting in increased tonic inhibition. Interestingly, there was no significant difference in the spontaneous activity of LC neurons at different postnatal ages unless GABABR tonic inhibition was blocked. These results show that, during postnatal development, there is a continuous increase in GABABR tonic inhibition that maintains the activity of LC neurons at a proper level. In male, but not female, rats, chronic perinatal treatment with citalopram, a selective serotonin reuptake inhibitor, reduced GABABR activity and tonic inhibition, which might result in the significantly higher spontaneous activity of LC neurons seen in these animals. In conclusion, our results show that GABABR-mediated tonic inhibition has a direct impact on the spontaneous activity of LC neurons and that the extent of the effect varies with ambient GABA levels and functionality of GABABR signalling.
Key points.
Noradrenaline (NA)-releasing neurons in the locus coeruleus (LC) provide NA to the forebrain and play important roles in regulating many brain functions.
LC neurons are subject to tonic inhibition mediated by GABAB receptors (GABABRs) and that the extent of the effect varies with ambient GABA levels.
GABABR-mediated tonic inhibition can effectively tune the spontaneous firing rate (SFR) of LC neurons; it is developmentally regulated and is responsible for maintaining a constant SFR of LC neurons during development.
In male, but not female rats, chronic perinatal treatment with citalopram, a selective serotonin reuptake inhibitor, results in downregulation of GABABR-mediated tonic inhibition of LC neurons that partially accounts for increased SFR in male, but not female, rats receiving such treatment.
Our results show that GABABR-mediated tonic inhibition could be an important player in the development of normal and abnormal behaviours/brain functions associated with the LC–NA system.
Introduction
Noradrenaline (NA) plays important roles in regulating many brain functions (Aston-Jones & Cohen, 2005; Tully & Bolshakov, 2010; Berridge et al. 2012). The major NA supply to the forebrain is from NA-releasing neurons in the locus coeruleus (LC), which is located in the dorsal pontine area. The NA-releasing neurons in the LC (referred to as LC neurons hereafter) spontaneously fire action potentials (APs) in a brain state-dependent manner (Aston-Jones & Cohen, 2005), displaying active firing during wakefulness and a decreased discharge rate during non-rapid eye movement (non-REM) sleep, and being silent during REM sleep. Since pharmacological activation or inhibition of their activity is always followed by a change in forebrain electroencephalographic activity, LC neurons are believed to control the wakefulness/arousal level of the brain (Adam & Foote, 1988; Berridge & Foote, 1991).
Several lines of evidence suggest that γ-aminobutyric acid (GABA), the principal inhibitory neurotransmitter in the brain, is involved in regulating LC neuron activity. Somogyi & Llewellyn-Smith (2001) showed that about two-thirds of synapses on LC neurons are GABAergic, while other studies showed that a substantial proportion of these GABAergic inputs come from the sleep-promoting nuclei, such as the ventrolateral preoptic nucleus (Lu et al. 2002; Luppi et al. 2006). Moreover, ambient GABA levels in the LC are significantly higher during REM/non-REM sleep than during wakefulness (Nitz & Siegel, 1997; Nelson et al. 2002). GABA, which acts on ionotropic GABAA receptors (GABAARs) and metabotropic GABAB receptors (GABABRs), provides important inhibitory control of neuronal circuits in the brain. In addition to classic phasic inhibition, GABAARs with a high affinity for GABA and extrasynaptic or perisynaptic location, are continuously activated by ambient GABA, thereby mediating a tonic inhibition that allows the long-term regulation of neuronal excitability (Farrant & Nusser, 2005). Tonic inhibition mediated by GABAARs has been reported throughout the brain and identified as an important player in both physiological and pathophysiological processes (Glykys & Mody, 2007a; Belelli et al. 2009).
Interestingly, recent reports have shown that GABA tonic inhibition in A6 (the LC) and A7 catecholamine cells in the pons (Wu et al. 2011; Wang et al. 2012) and layer 2/3 pyramidal neurons in the medial prefrontal cortex (Wang et al. 2010) is mediated by GABABRs, but not GABAARs. Since GABA is important for regulating LC neuron activity, the aims of the present study were to fully characterize the GABABR-mediated tonic inhibition of LC neurons and determine the functional outcome of manipulations that increase or decrease it. In brainstem neurons GABABR activity is reported to increase during development (Pfeiffer & Zhang, 2007) and to be reduced in animals receiving chronic treatment with a selective serotonin reuptake inhibitor (SSRI) (Cornelisse et al. 2007). In our study, normal postnatal development and chronic perinatal treatment with citalopram (CTM), a SSRI, were therefore used as study models for an increase or reduction in GABABR tonic inhibition, respectively. Our results showed that GABABR-mediated GABA tonic inhibition is developmentally regulated and is responsible for maintaining a constant spontaneous firing rate (SFR) of LC neurons. In male, but not female rats, chronic perinatal treatment with CTM resulted in downregulation of GABABR-mediated tonic inhibition of LC neurons, which partially accounts for the phenomenon reported by Darling et al. (2011), who used the same chronic perinatal CTM treatment procedures as ourselves and showed that the SFR of LC neurons is increased in male, but not female, rats receiving such treatment.
Methods
Animals
All animal experiments complied with the European Communities Council Directive (24 November 1986), and the use of animals in this study was approved by the Ethics Committee for Animal Research of National Taiwan University and Chung Shan Medical University. Every effort was made to minimize the number of animals used and their suffering. Sprague–Dawley rat pups of both sexes were used and were killed for slice preparation on postnatal day (PN) 8–10 in all electrophysiology experiments except the developmental study and the CTM treatment study. In the developmental study, rats were killed on PN2–4, PN8–10 and PN18–20, while, in the CTM treatment study, they were injected subcutaneously with CTM (10 mg (kg body weight)−1) or 0.9% NaCl (controls) twice a day for 7 days, starting on PN5, and were killed on PN18–20. For characterization of GABABR-mediated tonic inhibition (Figs 1, 2, 4 and 5) and the developmental study (Fig. 6), 14 PN2–4, 29 PN8–10 and 17 PN18–20 rats were used, and for the CTM treatment study (Figs 7 and 8) 21 male and 19 female rats were used. Another three naive rats (Fig. 3), and three saline-treated and three CTM-treated male rats (Fig. 9) were used for electron microscopy (EM) study.
Figure 1. Recordings from LC neurons.

A, identification of LC neurons in a sagittal brainstem slice. Aa and Ad are bright field images showing a paraformaldehyde-fixed slice at low magnification (Aa) and a non-fixed slice at high magnification (Ad). Ab and Ac are fluorescence images of slices stained with anti-TH antibody alone (Ab) or the merged image for Aa and Ab (Ac). Note the dense TH-ir neurons in the LC. Ae–g are fluorescence images of slices stained with anti-TH antibody (Ae) or biocytin (Af) and the merged images (Ag). The TH-ir neurons marked with white arrows in Ae are those marked with black arrows in Ad; the upper neuron was recorded and filled with biocytin (Af and g). Abbreviations: 4 V, 4th ventricle; 7n, 7th nerve; Me5, mesencephalic trigeminal nucleus; scp, superior cerebellar peduncle. The large arrows in Aa indicate slice orientation (D: dorsal; R: rostral) and the scale (200 μm). B, representative current recording from a TH-ir neuron showing spontaneous firing of APs and voltage oscillations (asterisks). C, voltage-clamp recordings from the same neuron as in B showing that the membrane currents underlying the voltage oscillations (asterisks in top trace) are blocked by application of 100 μm CBX, a gap junction blocker (bottom trace). D, representative current-clamp recording showing membrane voltage (Vm) responses (top trace) to the current injection protocol shown in the bottom trace (Da) and the summarized results (Db). Note that the delay in the onset of the action potential after the end of the hyperpolarizing current pulse (see arrow in Da) is Vm dependent (Db).
Figure 2. GABABR-GIRK currents in LC neurons.
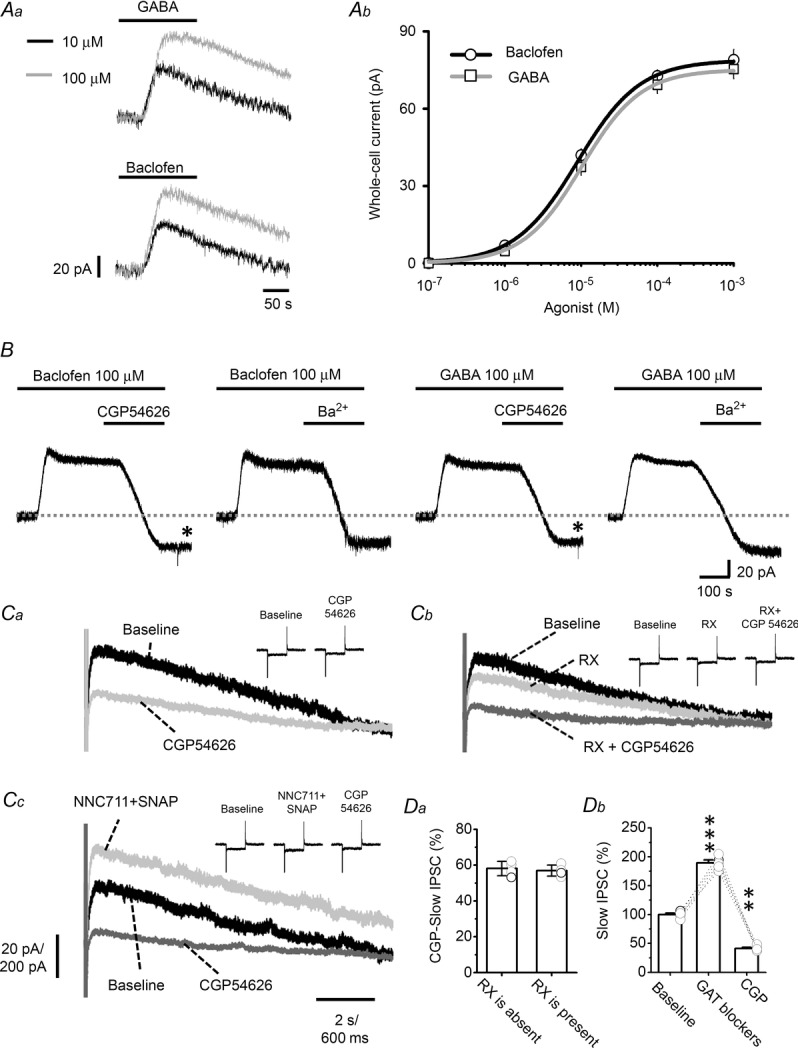
A, the traces in Aa are representative voltage-clamp recordings of the outward current induced by 10 or 100 μm baclofen or GABA. Ab shows summarized results using baclofen and GABA at concentrations of 0.1, 1, 10 and 100 μm and 1 mm and the fitting of the Hill equation to the data. B, representative recordings showing that the baclofen- or GABA-induced outward current is blocked by subsequent application of 10 μm CGP54626 or 1 mm Ba2+. C, GABABR-mediated slow-IPSCs in LC neurons. Ca, superimposed representative recordings showing the slow-IPSCs evoked in LC neurons by a 5-pulse stimulating train at 50 Hz under baseline conditions and in the presence of 10 μm CGP54626. Cb shows representative recordings of baseline, after addition of 100 nm RX781094 (RX), and after further addition of 10 μm CGP54626. Cc shows representative recording under baseline conditions after addition of two GAT blockers (50 μm NNC711 + 10 μm SNAP), and after further addition of 10 μm CGP54626. Inset traces in Ca–c show monitoring of series resistance by applying 5 mV voltage pulses throughout recording. Note no significant variance of series resistance. The vertical scale is 20 pA and 200 pA for recording of slow-IPSCs and series resistance monitoring, respectively, and the horizontal scale is 2 s and 600 ms for recording of slow-IPSCs and series resistance monitoring, respectively. D, summarized results of recording shown in C. Da shows the percentage blocking by CGP54626 of the slow-IPSCs (CGP slow-IPSC) in normal medium or the addition of the α2 blocker RX. Db shows the effect of GAT blockers on slow-IPSCs. The circles show results of individual experiments and the error bars show the mean and SEM for 4 cells (Da) or 8 cells (Db). The asterisks indicate a significant difference compared to the baseline value at the level of P < 0.01 (**) or P < 0.001 (***) using a paired t test. CGP, CGP54626.
Figure 4. GABABR-IStand in LC neurons.
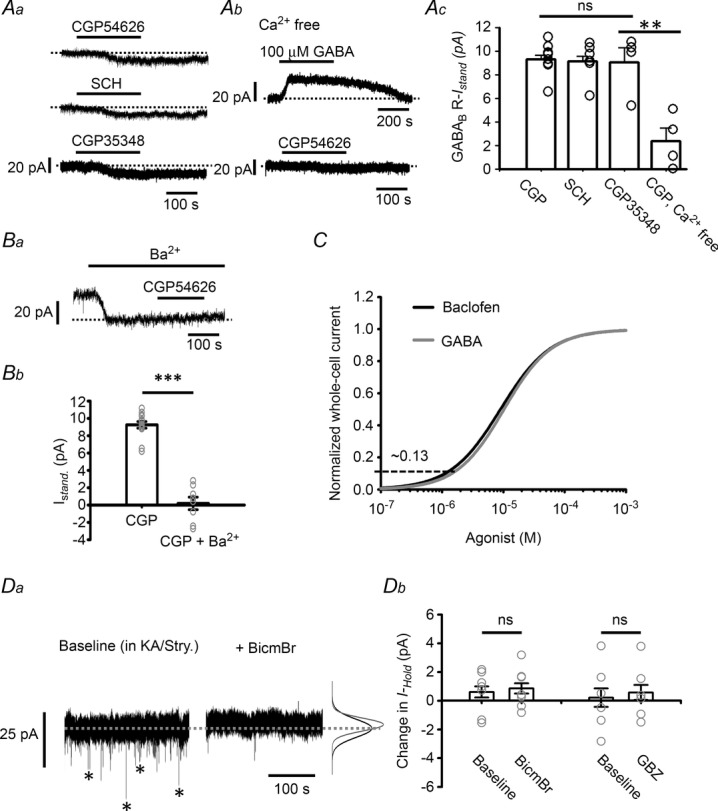
A, representative recordings showing the GABABR-IStand induced by application of two inverse agonists, CGP54626 and SCH50911, and a neutral antagonist CGP35348 (Aa), the IGABA induced by 100 μm GABA and the GABABR-IStand induced by 10 μm CGP54626 in Ca2+-free medium (Ab), and the summarized results (Ac). CGP, CGP54626. B, induction of the GABABR-IStand by 10 μm CGP54626 in the presence of 1 mm Ba2+; Ba shows a representative recording and Bb the summarized results compared to the values in the absence of Ba2+ (pooled data from CGP and SCH in panel Ac). C, normalization of the whole cell current versus concentration curve induced by GABA (grey) to that for baclofen (black) (data from Fig. 2Ab). The horizontal dashed line indicates the peak amplitude of the GABABR-IStand, which yields an estimate of the percentage of the GABABR-IStand to the GABABR-mediated total whole-cell current. Da, representative recordings of GABAergic mIPSCs (asterisks in the left trace), which are blocked by application of 20 μm BicmBr (right trace). Note that the inhibition of the mIPSC is not associated with a change in the holding current (dashed line) or noise level (indicated by the Gaussian curves on the right; black baseline, grey after BicmBr application). Db, summarized results for the effects of BicmBr and GBZ on the holding current. In all plots, the circles show results of individual experiments, while the error bars are the mean and SEM. ns in Ac and Db indicates no significant difference using one-way ANOVA or the paired t test, respectively, while ** in Ac and *** in Bb indicate, respectively, a statistically significant difference at the P < 0.01 and P < 0.001 level using Student's t test.
Figure 5. Tonic inhibition tunes the SFR of LC neurons and the effect of GABA reuptake.
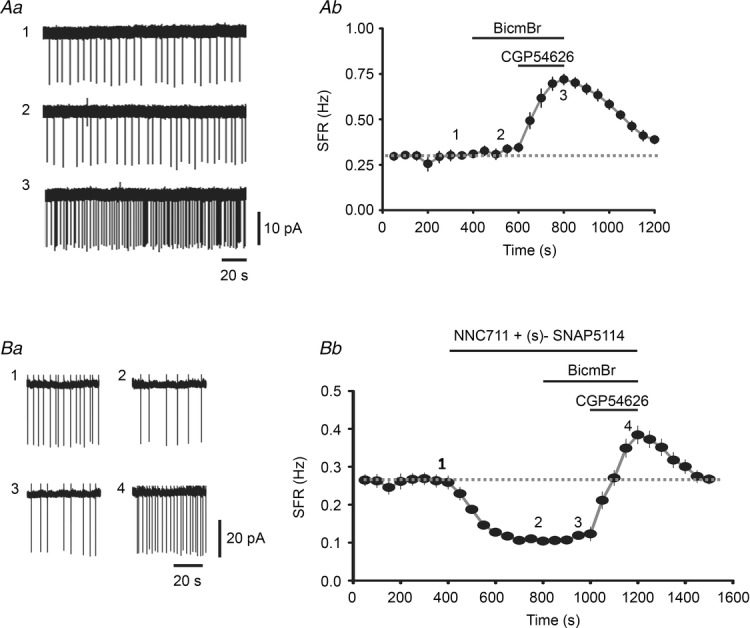
A, representative cell-attached recordings of the SFR of an LC neuron (Aa) at the times indicated by 1, 2 and 3 in the summarized results (Ab) showing the entire time-course of the recordings. Note the baseline SFR (trace 1) is not affected by 20 μm BicmBr (trace 2), but is dramatically increased by 10 μm CGP54626 (trace 3). In Ab, each point (circle plus vertical line) represents the mean and SEM for the instant firing rate averaged over a recording period of 50 s. B, representative recordings (Ba) and summarized results (Bb) showing the effect of prior application of the GAT1 blocker NNC711 (50 μm) and the GAT2/3 blocker (s)-SNAP5114 (10 μm).
Figure 6. Increased GABABR activity in LC neurons enhances tonic inhibition: developmental study.

A, representative recordings showing the IBac-Max (upper traces) and IGABA-Max (lower traces) induced by 1 mm baclofen and 1 mm GABA, respectively, in LC neurons in slices prepared at PN2–4 (Aa), PN8–10 (Ab), or PN18–20 (Ac). Note that the IBac-Max and IGABA-Max are blocked by subsequent application of 1 mm Ba2+ or 10 μm CGP54626. B, representative recordings showing the GABABR-IStand induced by 10 μm CGP54626 under normal conditions (upper traces) and in the presence of 1 mm Ba2+ (lower traces) in slices taken at PN2–4 (Ba) or PN18–20 (Bb). C and D, summarized results showing the developmental increase in the IBac-Max and IGABA-Max (C) and the GABABR-IStand (D). * and ** indicate P < 0.05 or < 0.01, respectively, between different developmental groups using one-way ANOVA, and ns indicates no significant difference between the indicated groups using the same test. †† indicates a difference at the level of P < 0.01 between the Ba2+ group and control group within a developmental stage using Student's t test. Ea, representative recording from an LC neuron at different developmental stages showing the SFR at baseline (trace 1) and after application of 20 μm BicmBr (trace 2) followed by 10 μm CGP54626 (trace 3) at the time points indicated in Eb. Eb, summarized results showing the entire time-course of the recordings for all 3 developmental stages. Note there is no difference in baseline SFR among the three developmental groups (shaded rectangle) and that, after CGP application, the SFR is significantly lower at PN2–4 than the other developmental stages (dashed rectangle).
Figure 7. Chronic perinatal CTM exposure reduces GABABR activity on LC neurons in male, but not female, rats.
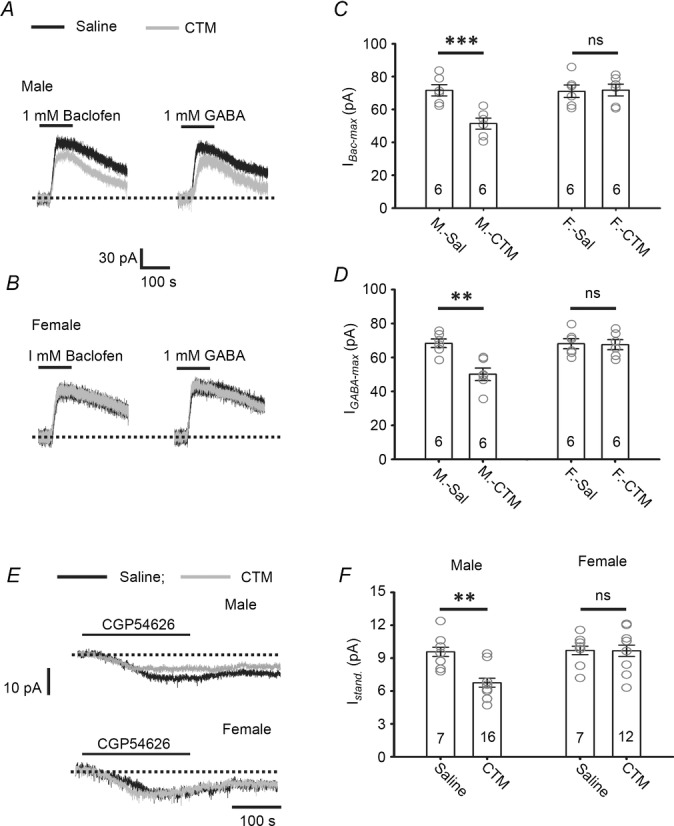
A–D, representative recordings showing the IBac-Max and IGABA-Max induced in LC neurons by 1 mm baclofen or 1 mm GABA in CTM- or saline-treated male (A) or female (B) rats, and the summarized results for the IBac-Max (C) and the IGABA-Max (D). E and F, representative recordings showing the GABABR-IStand induced by 10 μm CGP54626 in LC neurons from CTM-treated and saline-treated male rats and female rats (E) and the summarized results (F). In C, D and F, the circles are the results of individual experiments, while the error bars are the mean and SEM; the number of cells tested is indicated in each column. ** indicates a difference between the CTM-treated male rats (M-CTM) and saline-treated male rats (M-Sal) group at the level of P < 0.01 using two-way repeated-observation ANOVA. ns indicates no significant differences between the CTM-treated female rats (F-CTM) and saline-treated female rats (F-Sal) or between the M-Sal and F-Sal groups using the same test.
Figure 8. A reduction in GABABR activity on LC neurons decreases tonic inhibition: chronic perinatal CTM exposure study.
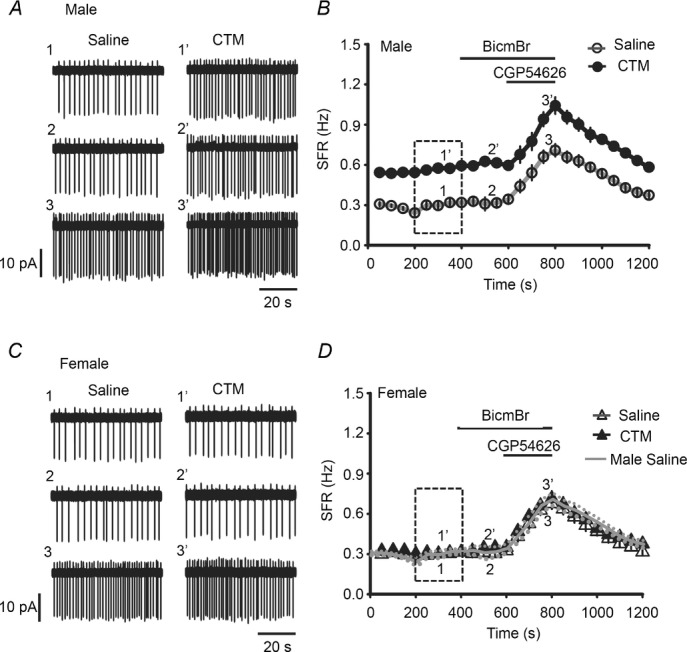
A, representative recordings from an LC neuron in a saline-treated male rat (1–3) and a CTM-treated male rat (1′–3′) showing the SFR at baseline (traces 1 and 1′) and after application of 20 μm BicmBr (traces 2 and 2′) followed by 10 μm CGP54626 (traces 3 and 3′); the numbers refer to the times in the time-course shown in B. B, summarized results showing the entire time-course of the recordings. Note the significant difference in the baseline SFR between the saline- and CTM-treated groups (dashed rectangle). C and D, representative recordings in saline- or CTM-treated female rats (C) and the summarized results (black lines and symbols) (D). Note CTM has no effect on the baseline SFR (dashed rectangle). The continuous and dotted lines (grey) in D are the results (mean ± SEM) for the saline-treated male rats taken from panel B and superimposed for comparison.
Figure 3. Subcellular distribution of GABABRs in the LC.
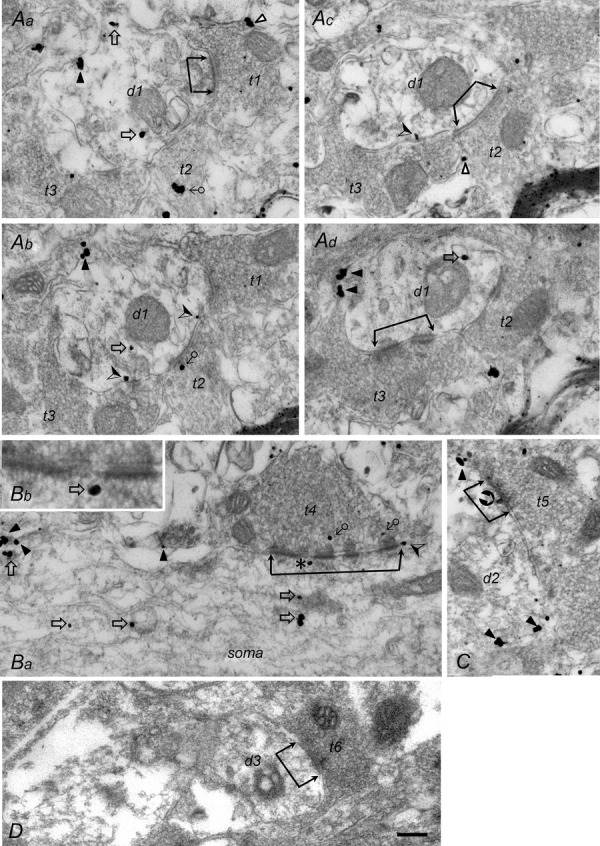
A, EM photographs of 4 serial ultrathin sections showing a dendrite (d1) receiving 3 axon terminals (t1–t3), with the contacts of the t1 and t3 terminal being asymmetric (Aa and d) and those of the t2 terminal (Ac) being symmetric. B, EM photograph showing an axon terminal (t4) making multiple asymmetric contacts with a soma at low (Ba) and high (Bb) magnification. C, EM photograph showing an axon terminal (t5) making symmetric contact on a dendrite (d2). D, EM photographs showing an axon terminal (t6) making asymmetric contact on a dendrite (d3) in a control section. All photographs, except that in Bb, are at the same magnification; the scale in D is 0.2 μm, which also applies to A, Ba and C; it indicates 0.08 μm for Bb. The active zones of the synaptic junctions are labelled with 2 connected arrows. GABABR-ir gold particles located on the plasma membrane of presynaptic terminals are indicated with the symbol Δ (Aa and c), those located on the membrane of cytosolic organelles in axonal terminals with ♂ (Ab and Ba), those located at the plasma membrane of the postsynaptic dendrite/soma with ▴ (extrasynaptic site; Aa, b and d, Ba and C),(perisynaptic site; Ab and Ba), or (synaptic site; see t5 in C), and those located on the membrane of cytosolic organelles in the dendrite/soma with ♂ (Aa, b and d, Ba and b). Note that Bb shows part of the active zone t4 (indicated by the asterisk in Ba) at high magnification with a GABABR-ir gold particle on a cytosolic organelle located beneath the active zone.
Figure 9. CTM treatment does not change the distribution and density of GABABRs in the LC.
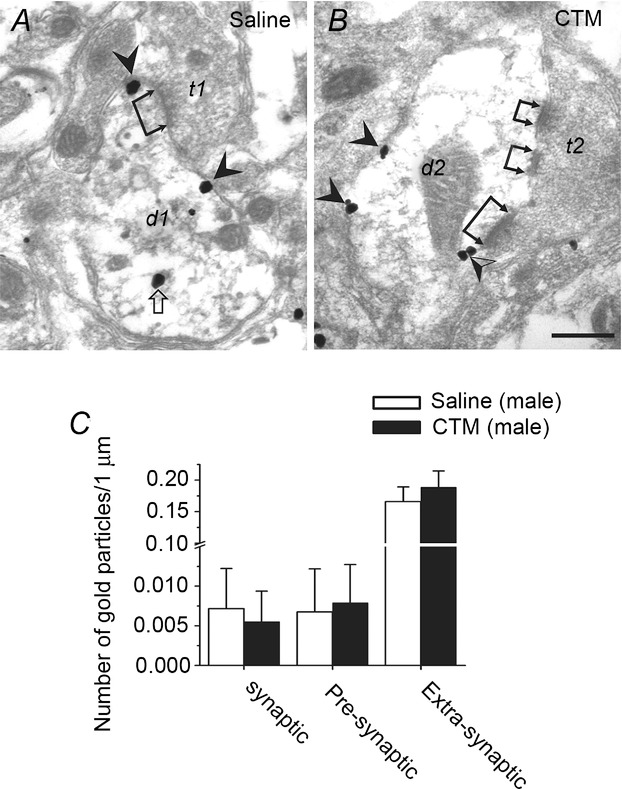
A, EM photograph of a section from a saline-treated male rat showing an axon terminal (t1) making an asymmetric contact (see dual-headed arrow) on a dendrite (d1) in the LC. GABABR-ir gold particles can be seen at extrasynaptic sites and on a cytosolic organelle. B, EM photographs of sections from CTM-treated male rats showing axon terminals (t2) making asymmetric contacts (dual-headed arrows) on a dendrite (d2) in the LC. GABABR-ir gold particles can be seen at extrasynaptic sites and perisynaptic sites. C, distribution of GABABR-ir gold particles at synaptic sites, and perisynaptic and extrasynaptic sites in the LC at 89 synapses on slides prepared from 3 saline-treated or CTM-treated males. Note that most GABABR-ir gold particles are found at extrasynaptic sites in both the saline- and CTM-treated groups. The scale bar is 0.2 μm in A and B.
Preparation of brainstem slices and electrophysiology
The animals were anaesthetized with 5% isoflurane in O2 and decapitated, then their brains were rapidly exposed and chilled with ice-cold artificial cerebrospinal fluid (ACSF) consisting of (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 26.2 NaHCO3, 1 NaH2PO4, 2.5 CaCl2 and 11 glucose, oxygenated with 95% O2 and 5% CO2, pH 7.4. Sagittal brainstem slices (300 μm) containing the LC were prepared using a vibroslicer (D.S.K. Super Microslicer Zero 1, Dosaka EM, Kyoto, Japan) and were maintained in a moist air–liquid (ACSF) interface chamber and allowed to recover for at least 90 min before being transferred to an immersion-type chamber mounted on an upright microscope (BX51WI, Olympus Optical Co., Ltd, Tokyo, Japan) for recording. Throughout the recording period, they were perfused at 2–3 ml min−1 with oxygenated ACSF containing 5 mm kynurenic acid (or a combination of 10 μM DNQX and 50 μM APV) and 1 μm strychnine to block glutamatergic and glycinergic synaptic transmission, respectively. The ‘baseline recordings’ described in this paper refer to the recordings made under these conditions unless noted otherwise.
Neurons were viewed using Nomarski optics. Patch pipettes were pulled from borosilicate glass tubing (1.5 mm outer diameter, 0.32 mm wall thickness; Warner Instruments Corp., Hamden, CT, USA) and had a resistance of about 3–5 MΩ when filled with the pipette solutions. To record the GABABR-mediated current, the pipette solution consisted of (in mm): 131 potassium gluconate, 20 KCl, 10 Hepes, 2 EGTA, 8 NaCl, 2 ATP, and 0.3 GTP, pH adjusted to 7.2 with KOH. To record the GABAAR-mediated current, the potassium gluconate was replaced with an equimolar concentration of CsCl. To record the inhibitory postsynaptic currents (IPSCs), the potassium gluconate was replaced with an equimolar concentration of KCl. Recordings were made at 33°C in either the cell-attached or whole-cell configuration with a patch amplifier (Multiclamp 700B; Axon Instruments Inc., Union City, CA, USA). For current-clamp recording, the bridge was balanced and recordings were only accepted if the recorded neuron had a membrane potential (Vm) of at least –45 mV without applying a holding current and if the AP was able to overshoot 0 mV. For voltage-clamp recording in the whole-cell configuration, neurons were clamped at –70 mV unless specified otherwise. The series resistance was monitored throughout recording and the data discarded if the values varied by more than 20% of the original value, which was usually less than 20 MΩ. For recording in the cell-attached configuration, the holding current was set to 0 pA so that the recorded neurons were at their resting membrane potential during recording. Extracellular stimulation was performed by delivering a stimulating train through a bipolar stainless steel electrode (FHC, Bowdoinham, ME, USA) positioned in the LC. The stimulating train consisted of five constant-current pulses (50–250 μA; 100 μs) at 50 Hz and was delivered every 30 s. Signals were low-pass filtered at a corner frequency of 2 kHz and digitized at 10 kHz using a Micro 1401 interface running Signal software for episode-based capture or Spike2 software for continuous recording (Cambridge Electronic Design, Cambridge, UK). The miniature IPSCs were recorded with the addition of 1 μm tetrodotoxin (TTX) into the bath and were measured and analysed using the Mini-Analysis program (Synaptosoft Inc., Fort Lee, NJ, USA). The measurement of GABAAR tonic current or GABABR standing current was modified from that described by Glykys & Mody (2007b). Briefly, the entire digitized recording was loaded and seal tests were removed from the traces. An all-point histogram was plotted for every 10,000 points. A Gaussian was fitted to the distribution, and the mean and standard deviation of the fitted Gaussian were considered to be the mean holding current and the noise level, respectively. This process was repeated for the entire recording and all baseline mean values were obtained. A segment of the baseline data was plotted and a linear fit was obtained and extended to the whole recording to correct for potential steady shifts during the recordings. A period of 30 s was used for the baseline and in the presence of drugs (bicuculline methiodide or CGP54626). The magnitude of tonic or standing current was obtained by subtracting the baseline current recorded in the presence of the drugs. All data are presented as the mean ± standard error of the mean (SEM) and were compared using Student's t test, the paired t test, one-way ANOVA, or two-way ANOVA. Bonferroni's post hoc test was used for selected pairwise comparisons when significant group-dependent differences were observed. The criterion for significance was a P value < 0.05.
All chemicals used to prepare the ACSF and pipette solution, and BaCl2 and paraformaldehyde, were from Merck (Frankfurt, Germany), baclofen, biocytin, carbenoxolone (CBX), kynurenic acid, and strychnine were from Sigma (St Louis, MO, USA), and dl-2-amino-5-phosphonopentanoic acid (APV), bicuculline methobromine (BicmBr), CGP35348, CGP54626 hydrochloride, 6,7-dinitroquinoxaline-2,3-dione (DNQX), gabazine (GBZ), NNC711, SCH50911, (s)-SNAP 5114, and tetrodotoxin (TTX) were from Tocris Cookson (Bristol, UK).
Biocytin histochemistry and immunohistochemistry (IHC)
In some experiments, 6.7 mm biocytin was included in the internal solution to fill the recorded neurons. The detailed procedures for viewing the biocytin-filled neurons and post hoc immunohistochemistry (IHC) for cell-type identification have been described previously (Min et al. 2008). Briefly, after recording, the slices were fixed overnight at 4°C in 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, then were subjected to biocytin histochemistry and IHC procedures without further sectioning. The slices were incubated for 1 h at room temperature in phosphate-buffered saline (PBS) containing 0.03% Triton X-100 (PBST), 2% bovine serum albumin, and 10% normal goat serum, then were incubated overnight at 4°C in PBST containing a 1/1300 dilution of rabbit antibodies against tyrosine hydroxylase (TH) (Merck Millipore, cat. no. AB5986P, Darmstadt, Germany) and a 1/200 dilution of avidin-AMCA (Vector Laboratories, Burlingame, CA, USA). After PBST rinses, they were incubated for 2 h with a 1/50 dilution in PBST of tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit IgG antibodies (Jackson ImmunoResearch, West Grove, PA, USA), then observed under a fluorescence microscope (Axioplan 2, Zeiss, Oberkochen, Germany) or a confocal microscope (Leica TCS SP5, Hamburger, Germany).
GABABR immunohistochemistry for electron microscopy (EM)
Animals were deeply anaesthetized with sodium pentobarbitone and perfused via the cardiovascular system with normal saline, followed by 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m PB. The pre-embedding IHC method was used. Sagittal LC sections (70 μm thick) were cut with a Vibratome (Ted Pella, Inc., Redding, CA, USA) and equilibrated in cryoprotectant solution consisting of 25% sucrose and 10% glycerol in 0.05 m PB. To enhance the tissue penetration of reagents, the sections were freeze-thawed by freezing in liquid nitrogen-cooled isopentane, followed by liquid nitrogen, and thawed in PBS. The sections were preincubated for 1 h in PBS containing 2% bovine serum albumin and 10% normal goat serum, then for 48 h at 4°C with guinea pig anti-GABABR1A/B antibodies (1/1000 dilution in PBS; Merck Millipore, cat. no. AB1531). The specificity of the GABABR antibodies has been described previously (Gassmann et al. 2004; Lacey et al. 2005), and the results of staining being specific to the use of the primary antibodies was demonstrated by omitting them during the staining procedures. After rinses in PBS, the sections were incubated for 16–18 h with gold (1.4 nm)-conjugated goat anti-guinea pig IgG antibodies (Nanoprobes Inc., Stony Brook, NY, USA) diluted 1/100 in PBS, and then, after further rinses in PBS, they were postfixed for 10 min with 2% glutaraldehyde in 0.1 m PB, followed by silver enhancement of the gold particles with an HQ Silver kit (Nanoprobes Inc.). The sections were then treated with 1% osmium tetraoxide in 0.1 m PB, dehydrated in a graded series of ethanol, and flat embedded on glass slides in Durcupan (Fluka) resin. Serial ultrathin sections (60 nm) of the LC region were cut and collected on single-slot copper grids coated with 1.5% Formvar in chloroform, stained with uranyl acetate followed by lead citrate, and examined on a JOEL JEM-2000 EXII electron microscope.
Synaptic structures were identified by the presence of a restricted zone of parallel pre- and postsynaptic membrane specializations with slight enlargement of the intercellular space, i.e. a visible synaptic cleft, and/or associated postsynaptic thickening, and by the accumulation of synaptic vesicles in the presynaptic profile (Peters et al. 1991). Synaptic junctions were classified as asymmetrical when a prominent plaque of dense material was seen on the cytoplasmic face of the postsynaptic membrane and as symmetrical when a less prominent density was seen on the postsynaptic membrane (Peters et al. 1991).
Results
Recording from LC neurons
In all electrophysiology experiments except the developmental study and the CTM treatment study, animals were killed for slice preparation on postnatal day (PN) 8–10. On a Nomarski microscopy video at low magnification, the LC was identified as a transparent, long, oval-shaped area located rostral to the floor of the 4th ventricle and beneath the superior cerebellar peduncle (Fig. 1Aa–c). At high magnification, numerous large neurons with a soma diameter of ∼25 μm could be identified for recording (Fig. 1Ad). Initially, we filled the recorded neurons with biocytin to confirm that these cells were immunoreactive (ir) with anti-TH antibody (Fig. 1Ae–g), but found that almost all were TH-ir and discontinued this practice when we started making cell-attached recordings. A feature of the current-clamp recordings from these TH-ir neurons was that the cells were able to spontaneously fire APs at ∼0.3 Hz and these were usually associated with membrane-voltage oscillations at 3.5 ± 0.7 Hz (asterisks in Fig. 1B). In voltage-clamp recordings, these events appeared as repeating biphasic currents (see asterisks in the upper trace in Fig. 1C) at the same frequency as the voltage oscillations seen in the current-clamp recordings and were blocked by CBX, a gap junction blocker (Fig. 1C). As it has been shown that neurons in the LC are electrically coupled (Ishimatsu & Williams, 1996; Ballantyne et al. 2004), these events could be due to the flow of AP currents through gap junctions. As seen previously in recordings from other pontine NA neurons (Min et al. 2008, 2010), on injection of hyperpolarizing current pulses, the TH-ir neurons in the LC showed a delay in the firing of APs, the duration of which was voltage dependent (Fig. 1Da and b), possibly caused by A-type potassium currents (Burdakov & Ashcroft, 2002). Taking all these features together, we considered that the recorded TH-ir neurons were indeed LC neurons.
Functional extrasynaptic GABAB receptors on LC neurons
Having confirmed that we were recording from LC neurons, the activity of their GABABRs was characterized. Using a potassium gluconate-containing pipette solution and blocking GABAARs by addition to the bath of 20 μm BicmBr, a selective GABAAR antagonist, an outward current was induced by bath application of 10 or 100 μm GABA (IGABA) or baclofen (IBac), a selective GABABR agonist (Fig. 2Aa), and this was shown to be dose dependent (Fig. 2Ab). As shown in Fig. 2B, the IGABA and IBac induced by 100 μm baclofen (left panels) or GABA (right panels) were blocked by subsequent addition to the bath of 10 μm CGP54626, a GABABR inverse agonist, or 1 mm Ba2+, a G protein-coupled inwardly rectifying potassium (GIRK) channel blocker, showing that they were GABABR-mediated currents resulting from the opening of GIRKs. The EC50 and the maximum amplitude of the IGABA were 9.55 μm and 75.36 ± 3.69 pA (n = 8 cells), respectively, while the corresponding values for the IBac were 9.18 μm and 78.93 ± 4.07 pA (n = 8 cells). Interestingly, CGP reduced IGABA and IBac to a level below baseline (see asterisks in Fig. 2B), indicating basal tonic activation of GABABRs in LC neurons (see below).
We next examined whether GABABR–GIRK signalling could be evoked by extracellular stimulation. As shown in Fig. 2C, outward currents with slow kinetics, referred to as slow-IPSCs and measured by charge transfer, were evoked on delivery of a stimulating train consisting of five pulses at 50 Hz, and subsequent bath application of 10 μm CGP54626 reduced these to 42 ± 4% of the baseline value (n = 4 cells; P < 0.01, paired t test), showing that GABABRs were responsible for 58 ± 4% of the slow-IPSCs (left panel in Fig. 2Da). LC neurons are hyperpolarized by an autoreceptor feedback mechanism mediated by α2-adrenoreceptors (α2ARs), which also couple to GIRK channels (William et al. 1985). Since both GABABRs and α2ARs are coupled to GIRK channels, we examined whether the slow-IPSCs of the GABABR-mediated component were occluded by α2AR activation. As shown in Fig. 2Cb, application of 100 nm RX781094, an α2AR antagonist, reduced the slow-IPSC to 70 ± 3% of the total slow-IPSCs of the baseline value (n = 4 cells; P < 0.01, paired t test), showing that α2AR–GIRK signalling also contributed to the slow-IPSC, and subsequent application of 10 μm CGP54626 in the presence of RX781094 reduced the residual slow-IPSCs by 57 ± 3% (n = 4 cells; P < 0.01, paired t test; Fig. 2Cb and Da), showing that contribution of GABABRs to slow-IPSCs in the presence of RX781094 was similar to that without RX781094 application (Fig. 2Da). These results show minor occlusion of GABABR-mediated activity by α2AR-mediated activity and suggest that the majority of these two receptors couple to different populations of GIRK channels in LC neurons. Application of a combination of 10 μm (s)-SNAP5114, a selective GABA transporter 1 (GAT1) blocker, and 50 μm NNC711, a selective GAT2/3 blocker, resulted in a significant increase in the slow-IPSCs to 219.7 ± 18.6% of the baseline value (Fig. 2Cc and Db, n = 8 cells) and subsequent application of CGP54626 in the presence of these GAT blockers dramatically reduced the slow-IPSCs to a level significantly below baseline (40.9 ± 4.9% of baseline, Fig. 2Db). These results show that the GABABR-mediated component of slow-IPSCs was enhanced by inhibition of GABA reuptake.
The dependence of the GABABR-mediated slow-IPSC on GABA reuptake suggests that it was mediated by GABABRs at peri-/extrasynaptic sites. To confirm this, the subcellular distribution of GABABRs was examined using a pre-embedding EM method. EM observations revealed that the GABA-ir gold particles were localized at the presynaptic terminals of both asymmetric (Fig. 3Aa and d) and symmetric (Fig. 3Ac) synapses. GABABR-ir gold particles were also found at the postsynaptic elements of these two types of synapse at both synaptic (Fig. 3C) and peri-/extrasynaptic sites (Fig. 3A–C). In addition, they were also found on the membrane of cytosolic organelles, some of which were located beneath the synaptic or perisynaptic active zone (Fig. 3B). Of the GABABR-ir gold particles found near synaptic structures, 39.8% were seen at presynaptic terminals (25% on the plasma membrane and 14.8% on the membranes of cytosolic organelles) and 60.2% at postsynaptic elements (24.2% on the plasma membrane and 36% on the membranes of cytosolic organelles). Of the 24.2% on the postsynaptic membrane, 0.8% were at synaptic sites, 5.4% at perisynaptic sites, and 18% at extrasynaptic sites. No GABABR-ir gold particles were seen in control sections in which primary antibody was omitted during staining (Fig. 3D). The EM observations show that most functional (surface) postsynaptic GABABRs were at peri-/extrasynaptic sites, consistent with the slow-IPSC recording results (Fig. 2Cc and Db).
GABABRs, but not GABAARs, mediate tonic inhibition of LC neurons
The peri-/extrasynaptic location of GABABRs on LC neurons suggested that they might be activated by ambient GABA and mediate tonic inhibition. We therefore tested whether a GABABR-mediated standing current was present in LC neurons. In line with the observation shown in Fig. 2B, application of 10 μm CGP54626 induced an inward current with an amplitude of 9.3 ± 0.3 pA (n = 8 cells) under conditions when GABAARs were blocked, and similar results were obtained using another selective GABABR inverse agonist, SCH50911 (20 μm) (inward current of 9.16 ± 0.43 pA, n = 7 cells) (Fig. 4Aa and c). We refer to this GABABR antagonist-induced current as the GABABR-mediated standing current (GABABR-IStand) (see Wu et al. 2011). Since CGP54626 and SCH50911 both function as inverse agonists of GABABRs (Grünewald et al. 2002), a neutral antagonist, CGP35348, was used to examine whether the GABABR-IStand resulted from activation of GABABRs by ambient GABA or from constitutive activity of the receptor. Application of 100 μm CGP35348 induced a GABABR-IStand with a similar amplitude (9.1 ± 1.3 pA, n = 4 cells) to those induced by 10 μm CGP54626 or 20 μm SCH50911 (Fig. 4Aa and c). GABABRs are shown to subject to allosteric modulation by Ca2+, and Ca2+ concentrations in the physiological range can potentiate the ability of GABA to activate GABABRs (Galvez et al. 2000). In LC neurons, the IGABA induced by the application of 100 μm GABA in the absence of extracellular Ca2+ was 27.4 ± 8.6 pA (n = 4 cells) (Fig. 4Ab, top trace), corresponding to 34.7% of the amplitude induced in normal extracellular Ca2+ levels (2.5 mm) (see Fig. 2Aa and b), the difference between the two conditions being significant (P < 0.001, F(1,11) = 47.63, one-way ANOVA test). The GABABR-IStand induced by 10 μm CGP54626 was also reduced (2.3 ± 1.3 pA; n = 4 cells) under Ca2+-free conditions (Fig. 4Ab and c) to 24.7% of the amplitude under normal conditions (Fig. 4Aa and c) and, again, the difference was significant (P < 0.001, F(1,11) = 45.37, one-way ANOVA test). These results show that GABABRs on LC neurons are also subject to allosteric modulation by Ca2+, and that the amplitude of the GABABR-IStand is dependent on GABA binding to GABABRs, i.e. was not due to constitutive activity of GABABRs. Like IGABA and IBac, the induction of the GABABR-IStand by 10 μm CGP54626 failed (Fig. 4B) when GIRKs were blocked by the addition of 1 mm Ba2+ to the bath medium. Together, these results show that a GABABR-IStand of ∼10 pA peak, accounting for ∼13% of the maximal GABABR-mediated current (see dashed line in Fig. 4C), in LC neurons resulted from continuous activation of peri-/extrasynaptic GABABRs by ambient GABA, rather than a constitutive activity of the receptors.
Since tonic inhibition mediated by extrasynaptic GABAARs has been demonstrated in many CNS neurons (Farrant & Nusser, 2005), we examined whether this was also the case in LC neurons. Using a CsCl-based pipette solution and 1 μm TTX in the bath to block voltage-dependent Na+ channels, many miniature IPSCs (mIPSCs) were seen, and these were blocked by subsequent application of 20 μm BicmBr (Fig. 4Da) or 20 μm GBZ (raw data not shown), showing that they were mediated by GABAARs. Blockade of mIPSCs using BicmBr (Fig. 4Da and Db) or GBZ (Fig. 4Db) was not associated with a change in the holding current from baseline or the level of background noise (Fig. 4Da), indicating that there was only a minor, or no, GABAAR-mediated tonic current. Together, these results show that the tonic inhibition of LC neurons is mediated by GABABRs, and not GABAARs.
Tonic inhibition can tune the SFR of LC neurons: role of GABA reuptake
The physiological role of GABABR tonic inhibition was next explored by examining its effect on the spontaneous firing rate (SFR) of LC neurons. To avoid a change in the intracellular ion composition, the cell-attached configuration was used. As with current-clamp recording (Fig 1B), LC neurons recorded using the cell-attached method showed an SFR of ∼0.3 Hz in the baseline recording (Fig. 5Aa, trace 1, and b), which was not affected by bath application of 20 μm BicmBr (Fig. 5Aa, trace 2, and b; n = 10 cells, paired t test, P = 0.158), but was significantly increased from 0.31 ± 0.02 to 0.72 ± 0.03 Hz by the addition of 10 μm CGP54626 (Fig. 5Aa, trace 3, and b, paired t test, P < 0.001). In addition, increasing the extent of tonic inhibition by increasing ambient GABA levels to activate more peri-/extrasynaptic GABABRs resulted in a significant decrease in the SFR of LC neurons. As shown in Fig. 5B, after a stable period of baseline recording (Fig. 5Ba, trace 1, and b), bath application of a combination of 50 μm NNC711 and 10 μm (s)-SNAP5114 to inhibit GABA reuptake resulted in a dramatic decrease in the SFR from 0.27 ± 0.02 Hz to 0.11 ± 0.01 Hz (Fig. 5Ba, trace 2 and b, n = 8 cells; P < 0.001, paired t test) and, while subsequent application of BicmBr had no effect (Fig. 5Ba, trace 3 and b), application of CGP54626 not only reversed the effect of the GAT1/2 blockers, but also increased the SFR to 0.38 ± 0.02 Hz (Fig. 5Ba, trace 4, and b, P < 0.001, paired t test, compared to baseline). In summary, these results show that varying the extent of GABABR tonic inhibition can effectively tune the SFR of LC neurons. Interestingly, there was no significant effect of BicmBr application on the SFR even when GABA reuptake was blocked (Fig. 5B). These results show that no GABAAR-mediated tonic inhibition was seen in LC neurons.
An increase in GABABR activity increases tonic inhibition and maintains the SFR of LC neurons constant in developing rats
In addition to ambient GABA levels, the effect of manipulating GABABR activity on the tonic inhibition and thus the SFR of LC neurons was also tested. Postnatal development was used as the study model to examine the effect of increasing GABABR activity. In addition to PN8–10, two additional PN ages, PN2–4 and PN18–20, were examined. As shown in Fig. 6A and C, on going from PN2–4 to PN8–10, an increase in the maximal IBac (IBac-Max) and IGABA (IGABA-Max) induced by 1 mm baclofen or 1 mm GABA was seen in LC neurons. The peak amplitudes of the IBac-Max and IGABA-Max were, respectively, 40.8 ± 2.2 pA (n = 7 cells) and 39.8 ± 2.7 pA (n = 7 cells) at PN2–4 and 79.2 ± 3.6 pA (n = 8 cells) and 78.9 ± 4.1 pA (n = 8 cells) at PN8–10 (Fig. 6Aa and b, and C; one-way ANOVA, P < 0.01, F(1,14) = 42.65 for Bac, F(1,14) = 39.56 for GABA); the IBac-Max and IGABA-Max at PN18–20 did not differ from those induced at PN8–10 (Fig. 6Ab and c, and C; one-way ANOVA, P = 0.971, F(1,15) = 0.02 for Bac, P = 0.864, F(1,15) = 0.03 for GABA). Consistent with the lower IBac-Max and IGABA-Max seen at PN2–4, as shown in Fig. 6B and D, the GABABR-IStand recorded at PN2–4 was 4.6 ± 0.8 pA (n = 8 cells), significantly lower than that recorded at PN8–10 (9.2 ± 0.4 pA, n = 15 cells, P < 0.01, F(1,22) = 27.05, one-way ANOVA) or PN18–20 (9.5 ± 0.5 pA, n = 8 cells, P < 0.05, F(1,15) = 11.46, one-way ANOVA). Induction of the GABABR-IStand was blocked at all developmental stages when CGP or Ba2+ was added to the bath (Fig. 6B and D). Interestingly, no significant difference in the baseline SFR was observed among the three developmental stages (∼0.25 Hz, shaded area in Fig. 6Eb) unless the GABABR-IStand was abolished by addition of CGP54626 to the bath (dashed area in Fig. 6Eb). At all three developmental stages, the SFR was significantly increased after blockade of GABABRs (Fig. 6Ea and b; P < 0.001 in all cases, paired t test), but the increase was significantly lower at PN2–4 than at PN8–10 or PN18–20. After removal of tonic inhibition, the SFR of LC neurons at PD2–4 was 0.51 ± 0.03 Hz (n = 8 cells), significantly lower than that of 0.72 ± 0.03 Hz at PN8–10 (n = 10 cells, P < 0.01, F(1,17) = 18.31, one-way ANOVA) and that of 0.71 ± 0.03 Hz at PN18–20 (n = 8 cells, P < 0.01, F(1,17) = 16.85, one-way ANOVA) (Fig. 6Ea and b). There was no difference in the resting Vm (measured with addition to the bath of 1 μm TTX) among the three groups (PN2–4: 45.2 ± 2.3 mV, n = 10 cells; PN8–10: 45.4 ± 2.1 mV, n = 12 cells; PN18–20: 44.9 ± 2.7 mV, n = 10 cells; P = 0.99, F(2,31) = 0.01, one-way ANOVA). These results show that, during development, the GABABR-IStand and tonic inhibition increase to maintain the SFR of LC neurons at a constant level of 0.3 Hz. As already seen for the PN8–10 stage (Fig. 5A and B), BicmBr had no effect on the SFR at the other developmental stages (Fig. 6E).
A decrease in GABABR functionality caused by chronic perinatal citalopram treatment inhibits tonic inhibition and results in abnormal SFRs of LC neurons
In addition to increasing GABABR activity and tonic inhibition, the functional outcome of a manipulation reducing these parameters was also investigated. In dorsal raphe neurons, a prolonged increase in serotonin agonist levels caused either by knockdown of the serotonin transporter (Mannoury la Cour et al. 2001) or by chronic treatment with an SSRI (fluoxetine) leads to reduced levels of somatodendritic GABABRs (Cornelisse et al. 2007). We therefore examined whether a similar downregulation of GABABR activity also occurred in LC neurons after chronic SSRI treatment and, if so, what effect it had on tonic inhibition. The model described by Darling et al. (2011) was used; male and female rats were injected subcutaneously with the SSRI CTM (10 mg (kg body weight)−1) or 0.9% NaCl twice a day from PN5 to PN12 and measurements were made at PN18–20. As shown in Fig. 7 (see also Table 1), the IBac-Max was significantly reduced by 28.3 ± 4.7% in CTM-treated male rats compared to saline-treated male rats (Fig. 7A and C; n = 6 cells, P < 0.01, F(1,23) = 12.84, two-way ANOVA) and the IGABA-Max was significantly reduced by 26.6 ± 5.4% (n = 6 cells, P < 0.01, F(1,23) = 14.68, two-way ANOVA) (Fig. 7A and D), but no effect was seen in female rats (Fig. 7B–D; Table 1). In addition, there was no difference between the IBac-Max and IGABA-Max recorded in LC neurons of saline-treated male or CTM-treated female rats (Fig. 7B–D; Table 1). Consistent with the reduction in the IBac-Max and IGABA-Max, the GABABR-IStand was significantly decreased in CTM-treated male rats compared to saline-treated male rats (29.5 ± 6.2%, n = 16 cells, P < 0.01, F(1,41) = 16.02, two-way ANOVA), but no effect was seen in female rats (Fig. 7E and F; Table 1). There was no difference in the resting Vm (measured with addition to the bath of 1 μm TTX) between the male and female groups or between treatment groups (two-way ANOVA, P = 0.57, F(1,24) = 1.25, see Table 1). These results show that CTM treatment results in reduced GABABR activity in LC neurons in male, but not female, rats.
Table 1.
Effect of chronic perinatal CTM exposure on the GABABRs on LC neurons
|
Male |
Female |
|||
|---|---|---|---|---|
| Saline | CTM | Saline | CTM | |
| Resting Vm (mV) | 44.9 ± 2.3 | 39.2 ± 2.4 | 45.1 ± 2.4 | 45.6 ± 2.6 |
| (n = 6) | (n = 7) | (n = 6) | (n = 6) | |
| IGABA-Max (pA) | 71.7 ± 3.4 | 57.4 ± 3.2† | 71.1 ± 3.8 | 71.8 ± 3.6 |
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| IBac-Max (pA) | 68.3 ± 2.5 | 55.3 ± 3.0† | 68.1 ± 3.0 | 67.5 ± 3.0 |
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| GABABR-IStand (pA) | 9.6 ± 0.6 | 7.0 ± 0.6† | 9.7 ± 0.6 | 9. 7 ± 0.7 |
| (n = 7) | (n = 16) | (n = 7) | (n = 12) | |
| SFR-baseline (Hz) | 0.32 ± 0.02 | 0.59 ± 0.04† | 0.31 ± 0.07 | 0.31 ± 0.01 |
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| SFR-BicmBr (Hz) | 0.34 ± 0.03* | 0.60 ± 0.03†ns | 0.33 ± 0.02ns | 0.35 ± 0.03ns |
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| SFR-CGP (Hz) | 0.71 ± 0.05* | 1.04 ± 0.06†* | 0.70 ± 0.04* | 0.71 ± 0.02* |
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
Significant difference at P < 0.01 level compared to Male-CTM group using two-way repeated-observation ANOVA. Compared to SFR-baseline group
indicates significant difference at P < 0.01 level and
indicates no significant difference, using paired t test.
In slices from CTM-treated male rats, the baseline SFR of the LC neurons was 0.56 ± 0.01 Hz (n = 6 cells), significantly higher than that of 0.29 ± 0.02 Hz in slices from saline-treated male rats (n = 6 cells; P < 0.001, F(1,23) = 37.51, two-way ANOVA) (compare traces 1 and 1′ in Fig. 8Aa and the dashed rectangle in Fig. 8B; Table 1). In contrast, there was no difference in the baseline SFR between CTM-treated and saline-treated female rats (compare traces 1 and 1′ in Fig. 8C and the dashed rectangle in Fig. 8D; Table 1). Could the decrease in the IBac-Max and IGABA-Max (activity of the GABABR) and the GABABR-IStand (tonic inhibition) caused by chronic perinatal CTM exposure result in the increased SFR of LC neurons seen in CTM-treated male rats? Support for this idea was obtained when removal of the GABABR-IStand by addition of CGP54626 to the bath reduced the difference in the SFR of LC neurons between CTM-treated and saline-treated male rats. As shown in Fig. 8, application of CGP54626 resulted in a significant increase in the SFR in all cases, regardless of sex or CTM treatment (P < 0.001 in all cases, paired t test; Table 1). However, the ratio of the baseline SFR recorded in saline-treated male rats to that in CTM-treated male rats was 1.84 and decreased to 1.46 after blockade of the GABABR-IStand (tonic inhibition), while that in females was ∼1 before and after application of CGP54626. Together, these results show that, in male rats subjected to chronic perinatal CTM exposure, there is less GABABR activity in LC neurons, and thus a lower GABABR-IStand and tonic inhibition, than in saline-treated rats. This lower GABABR functionality might partially account for the higher SFR of LC neurons seen in the CTM-treated male rats. Again, we did not observe any effect of BicmBr on the SFR (Fig. 8), regardless of sex or whether the animals received CTM or not. These results show that chronic perinatal CTM exposure does not alter the expression of extrasynaptic GABAARs on LC neurons.
Finally, we examined whether the reduced GABABR activity in CTM-treated male rats involved a decrease in surface GABABRs levels using a pre-embedding EM method. At stage PN20, 89 synapses were randomly sampled from three saline-treated and 89 from three CTM-treated male rats. There was no difference between the two groups in the pattern of surface GABABR distribution on postsynaptic membranes (Fig. 9A and B). As observed in the saline-treated group (Fig. 9A) and naive animals (Fig. 3), GABABR-ir gold particles were also found at postsynaptic locations at synaptic and peri- and extrasynaptic sites in the CTM-treated group (Fig. 9B). The density of GABABRs, expressed as the number of GABABR-ir gold particles per 1 μm length of dendritic membrane is much higher at extrasynaptic sites than at peri- and synaptic sites (P < 0.001, F(1,177) = 88.48, one-way ANOVA; Fig. 9C). For all three types of postsynaptic location, no difference in GABABR density was observed between the saline-treated and CTM-treated group (P = 0.85, F(1,177) = 0.61, two-way ANOVA; Fig. 9C). These results show that the reduction in GABABR activity in LC neurons from CTM-treated male rats did not involve a decrease in surface GABABR expression.
Discussion
Our argument for GABABR-mediated tonic inhibition of LC neurons is based on the observations that bath application of two different GABABR inverse agonists, CGP54626 and SCH50911, and a neutral antagonist, CGP35348, induced an inward standing current (GABABR-IStand) and increased the SFR in LC neurons. Since all three GABABR blockers produced the same results and since the induction of the GABABR-IStand was abolished by blockade of GIRK, a well-known effector molecule downstream of GABABR activation, the GABABR-mediated tonic inhibition of LC neurons was a specific, rather than a non-specific, effect of these drugs. In addition, as CGP35348, one of the drugs used, is a neutral antagonist and since the induction of the GABABR-IStand by CGP54626 (an inverse agonist) is seen at an extracellular calcium concentration in the physiological range (2.5 mm), but not in Ca2+-free medium, the tonic inhibition is mainly caused by activation of GABABRs by ambient GABA and is not a constitutive activity of the receptor.
Previous EM studies showed that, in all brain regions examined, most GABABRs are located at peri- and/or extrasynaptic sites, rather than synaptic sites (Boyes & Bolam, 2003; Kulik et al. 2003; Lacey, 2005), and an electrophysiology study showed that activation of these extrasynaptic GABABRs is critically dependent on GABA reuptake and increases when GABA transporters are blocked (Beenhakker & Huguenard, 2010). The activation of GABABRs on LC neurons is consistent with these features, as our EM results showed that very few GABABR-ir gold particles in postsynaptic locations were located at synaptic and perisynaptic sites and most were located at extrasynaptic sites; in addition, our electrophysiological studies showed a critical dependence of the activation of these extrasynaptic GABABRs on GABA uptake, as the GABABR-mediated slow-IPSCs and tonic inhibition were dramatically increased by blockade of GABA reuptake. The time course of GABABR-mediated slow-IPSCs in the present study is much slower than those reported in previous studies (Beenhakker & Huguenard, 2010). An explanation for this could be that there is a unique distribution pattern and activity of GABA transporters in the LC that results in an ambient GABA profile generating a slow GABABR-mediated response following electrical stimulation. In addition to GABABR, the evoked slow-IPSCs also contain an α2AR-mediated response with the α2AR-mediated component being smaller than that of GABABR. However, using stimulation parameters similar to the present study, Courtney & Ford (2014) report a robust α2AR-mediated IPSC in LC neurons. Since Kimura & Nakamura (1987) report that, although α2ARs already function in LC neurons at birth, the activation of these autoreceptors by NA released from the synaptic terminals does not occur until PN9 and becomes mature in PN24–34, the discrepancy between Courtney & Ford's results and ours regarding the amount of α2AR-mediated IPSC evoked in LC neurons might be due to the use of different ages of animals for brain slice preparation. Courtney & Ford (2014) used Sprague–Dawley rats aged 3–6 weeks, while PN8–10 rats of the same strain were used in the present study for recording slow-IPSCs. In addition, α2AR-mediated whole-cell current induced in LC neurons of PN8–10 rats by addition into the bath of 50 μm UK14304, a selective α2AR agonist, is 41.2 ± 7.1 pA (n = 6 cells; raw data not shown), which is smaller than GABABR-mediated current and further explains a smaller contribution of α2AR-mediated response to the slow-IPSCs. Although extrasynaptic GABAARs have been reported to be present in many brain regions examined (Farrant & Nusser, 2005; Glykys & Mody, 2007a,b; Belelli. 2009), they do not seem to play a role in mediating GABA tonic inhibition in LC neurons (also see Belujon et al. 2009), as no difference was seen in the holding current or the SFR when GABAARs was blocked in the normal condition or in the condition in which GABA reuptake by GATs was inhibited.
In addition to ambient GABA levels, tonic inhibition also varied with GABABR activity on LC neurons. We observed a developmental increase in GABABR activity on LC neurons and, consistent with this, an increase in both the GABABR-IStand and tonic inhibition. It has been reported that, in the cortex of neonatal rats, GABABR-mediated somatic responses to baclofen application increase until the middle of the second postnatal week, and then remain stable (Luhmann & Prince, 1991; for review see Le Magueresse & Monyer, 2013). The present results are in agreement with this observation, as the IBac and IGABA of LC neurons did not change after PN8–10. Interestingly, there was no significant difference in the SFR of LC neurons among the three developmental ages examined (see also Nakamura et al. 1987) unless GABABR-mediated tonic inhibition was blocked. It is possible that there is a developmental increase in the number of ion channels that are involved in generating spontaneous APs in LC neurons and that their function is counteracted by increased GABABR-mediated tonic inhibition, resulting in a constant level of SFR of LC neurons during postnatal developmental. The physiological significance of maintaining a constant SFR during development remains to be explored. Since NA plays many important roles in regulating development and cortical plasticity (Lipton & Kater, 1989; Kasamatsu, 1991) and since it has been suggested that abnormal behaviours could result from developmental dysregulation of the LC–NA system (for review see Mehler & Purpura, 2009), the maintenance of a stable SFR of LC neurons might ensure suitable ambient NA levels for the maturation of normal brain function.
SSRIs, including CTM, are preferred for antidepressant treatment because of their low toxicity and wide therapeutic index. The major pharmacological effect of SSRIs is inhibition of serotonin reuptake, increasing ambient serotonin levels. LC neurons are known to receive dense serotoninergic innervations from the dorsal raphe nucleus and pericoeruleus 5-HT neurons (Cedarbaum & Aghajanian, 1978; Aston-Jones et al. 1991; Kim et al. 2004) and their SFR has been shown to be modulated by serotonin (Haddjeri et al. 1997). Consistent with these facts, SSRI treatment has a profound effect on LC neuron activity. In mature rats, chronic SSRI exposure for 8–14 days leads to a reduction in the SFR of LC neurons (Nestler et al. 1990; Szabo et al. 1999; West et al. 2009). However, in contrast to these results, our results in juvenile rats and those of Darling et al. (2011) in mature rats show an increase in the SFR of LC neurons in male, but not female, rats receiving chronic perinatal CTM treatment. Although the cellular mechanisms underlying this observation remain to be explored, the present results are the first to show that downregulation of GABABR–GIRK activity and, thus, of tonic inhibition is involved.
In dorsal raphe neurons, both 5-HT1A receptor (5-HT1AR)- and GABABR-induced GIRK currents are reduced in rats after chronic treatment with fluoxetine, another SSRI (Cornelisse et al. 2007). Since 5-HT1ARs and GABABRs are coupled to the same population of GIRKs (Innis et al. 1987; Williams et al. 1988), and since there is a parallel reduction in 5-HT1AR–GIRK and GABABR–GIRK currents (Cornelisse et al. 2007), it has been previously proposed that GIRKs, rather than 5-HT1ARs and GABABRs, are desensitized after chronic fluoxetine exposure. In support of this, radioligand-binding results have shown that GIRKs, but not 5-HT1ARs, are desensitized after chronic fluoxetine exposure (Pejchal et al. 2002; Shen et al. 2002; Castro et al. 2003). In line with these observations, our EM study showed that chronic perinatal CTM exposure did not alter the distribution and density of GABABRs on the postsynaptic membrane in the LC, showing that the reduced GABABR–GIRK activity could not be ascribed to a decrease in surface GABABRs, but may be due to the GIRK desensitization described above and/or other mechanisms, such as change in GABABR phosphorylation, GABABR–GIRK coupling efficiency, regulators of G-protein signalling protein activity, and activity of the auxiliary GABAB receptor subunit, K+ channel tetramerization domain-containing protein (for review, see Padgett & Slesinger, 2010; Gassmann & Bettler, 2012).
In summary, our studies of chronic perinatal CTM exposure, development, and inhibition of GABA reuptake (activity of GATs) clearly showed that alteration of GABABR-mediated tonic inhibition can effectively tune the SFR of LC neurons and thus LC–NA-associated brain function. For example, since ambient GABA levels in the LC are significantly higher during REM/non-REM sleep than during wakefulness (Nitz & Siegel, 1997; Nelson et al. 2002), our results suggest that GABABR-mediated tonic inhibition may play an important role in regulating the SFR of LC neurons, and thus the wakefulness/arousal level of the brain. In addition to postsynaptic regulation, GABABRs at presynaptic sites have been shown to regulate glutamate and GABA release in many brain areas (Wu et al. 2011; Wang et al. 2013), perhaps including the LC neurons, as our EM study showed presynaptic location of GABABRs in the LC. As GABABR signalling and the activity of GABA transporters in many physiological and pathological processes can be regulated by various transmitter/receptor systems, such as the activation of 5-HT1A receptors during chronic exposure to SSRIs, our results suggest that tonic inhibition and presynaptic regulation of transmitter release mediated by peri/extrasynaptic GABABRs on LC neurons could be important players in the development of normal and abnormal behaviours/brain functions that are associated with the LC–NA system.
Acknowledgments
We thank Miss Y.-Y. Yu for EM support.
Glossary
- ACSF
artificial cerebrospinal fluid
- AP
action potential
- a2AR
a2-adrenoreceptor
- BicmBr
bicuculline methobromine
- CBX
carbenoxolone
- CTM
citalopram
- EM
electron microscopy
- GABA
γ-aminobutyric acid
- GABAAR
GABAA receptor
- GABABR
GABAB receptor
- GAT
GABA transporter
- GBZ
gabazine
- GIRK
G protein-coupled inwardly-rectifying potassium channel
- 5-HT1AR
5-HT1A receptor
- IGABA
GABA-induced whole-cell current
- IBac
baclofen-induced whole-cell current
- IBac-Max
the maximal IBac
- IGABA-Max
the maximal IGABA
- IHC
immunohistochemistry
- IPSCs
inhibitory postsynaptic currents
- ir
immunoreactive
- IStand
standing current
- LC
locus coeruleus
- mIPSCs
miniature IPSCs
- NA
noradrenaline
- PB
phosphate buffer
- PBS
phosphate-buffered saline
- PBST
PBS containing 0.03% Triton X-100
- PN
postnatal day
- REM
rapid eye movement
- SFR
spontaneous firing rate
- SSRI
selective serotonin reuptake inhibitor
- TH
hydroxylase
- TTX
tetrodotoxin
- Vm
membrane potential
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
H-Y.W. collected and analysed the electrophysiology data, and Z.-C.K. and Y.-S.F. collected and analysed the EM data. R.-F.C., M.-Y.M. and H.-W.Y. conceived and designed the experiments and wrote the first draft of the paper. The electrophysiological experiments were performed at NTU and CSMU, and EM experiments were performed at CHMU and NYMU. All authors have contributed to the writing of this paper and have approved its final version.
Funding
This work was supported by grants NSC-98-2311-B-002-003-MY3 (R.-F. Chen), NSC-99-2321-B-002-039 (M.-Y. Min) and NSC-100-2320-B-040-010-MY3 (H.-W. Yang) from the National Science Council, Taiwan.
References
- Adams LM. Foote SL. Effects of locally infused pharmacological agents on spontaneous and sensory-evoked activity of locus coeruleus neurons. Brain Res Bull. 1988;21:395–400. doi: 10.1016/0361-9230(88)90151-7. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G. Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Shipley MT, Chouvet G, Ennis M, van Bockstaele E, Pieribone V, Shiekhattar R, Akaoka H, Drolet G, Astier B, Charléty P, Valentino RJ. Williams JT. Afferent regulation of locus coeruleus neurons: anatomy, physiology and pharmacology. Prog Brain Res. 1991;88:47–75. doi: 10.1016/s0079-6123(08)63799-1. [DOI] [PubMed] [Google Scholar]
- Ballantyne D, Andrzejewski M, Muckenhoff K. Scheid P. Rhythms, synchrony and electrical coupling in the locus coeruleus. Respir Physiol Neurobiol. 2004;143:199–214. doi: 10.1016/j.resp.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP. Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC. Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci. 2009;29:12757–12763. doi: 10.1523/JNEUROSCI.3340-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belujon P, Baufreton J, Grandoso L, Boué-Grabot E, Batten TF, Ugedo L, Garret M. Taupignon AI. Inhibitory transmission in locus coeruleus neurons expressing GABAA receptor epsilon subunit has a number of unique properties. J Neurophysiol. 2009;102:2312–2325. doi: 10.1152/jn.00227.2009. [DOI] [PubMed] [Google Scholar]
- Berridge CW. Foote SL. Effects of locus coeruleus activation on electroencephalographic activity in neocortex and hippocampus. J Neurosci. 1991;11:3135–3145. doi: 10.1523/JNEUROSCI.11-10-03135.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Schmeichel BE. Espana RA. Noradrenergic modulation of wakefulness/arousal. Sleep Med Rev. 2012;16:187–197. doi: 10.1016/j.smrv.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J. Bolam JP. The subcellular localization of GABAB receptor subunits in the rat substantia nigra. Eur J Neurosci. 2003;18:3279–3293. doi: 10.1111/j.1460-9568.2003.03076.x. [DOI] [PubMed] [Google Scholar]
- Burdakov D. Ashcroft FM. Cholecystokinin tunes firng of an electrically distinct subset of arcuate nucleus neurons by activating A-type potassium channels. J Neurosci. 2002;22:6380–6387. doi: 10.1523/JNEUROSCI.22-15-06380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M, Diaz A, del Olmo E. Pazos A. Chronic fluoxetine induces opposite changes in G protein coupling at pre and postsynaptic 5-HT1A receptors in rat brain. Neuropharmacology. 2003;44:93–101. doi: 10.1016/s0028-3908(02)00340-4. [DOI] [PubMed] [Google Scholar]
- Cedarbaum JM. Aghajanian GK. Afferent projections to the rat locus coeruleus as determined by a retrograde tracing technique. J Comp Neurol. 1978;178:1–16. doi: 10.1002/cne.901780102. [DOI] [PubMed] [Google Scholar]
- Cornelisse LN, Van der Harst JE, Lodder JC, Baarendse PJ, Timmerman AJ, Mansvelder HD, Spruijt BM. Brussaard AB. Reduced 5-HT1A- and GABAB receptor function in dorsal raphe neurons upon chronic fluoxetine treatment of socially stressed rats. J Neurophysiol. 2007;98:196–204. doi: 10.1152/jn.00109.2007. [DOI] [PubMed] [Google Scholar]
- Courtney NA. Ford CP. The timing of dopamine- and noradrenaline-mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci. 2014;34:7645–7656. doi: 10.1523/JNEUROSCI.0166-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling RD, Alzghoul L, Zhang J, Khatri N, Paul IA, Simpson KL. Lin RC( Perinatal citalopram exposure selectively increases locus ceruleus circuit function in male rats. J Neurosci. 2011;31:16709–16715. doi: 10.1523/JNEUROSCI.3736-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M. Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, Malitschek B, Heid J, Brabet I, Froestl W, Bettler B, Kaupmann K. Pin J-P. Ca2+-requirement for high affinity γ-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol. 2000;57:419–426. doi: 10.1124/mol.57.3.419. [DOI] [PubMed] [Google Scholar]
- Gassmann M. Bettler B. Regulation of neuronal GABAB receptor functions by subunit composition. Nat Rev Neurosci. 2012;13:380–394. doi: 10.1038/nrn3249. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Shaban H, Vigot R, Sansig G, Haller C, Barbieri S, Humeau Y, Schuler V, Müller M, Kinzel B, Klebs K, Schmutz M, Froestl W, Heid J, Kelly PH, Gentry C, Jaton AL, Van der Putten H, Mombereau C, Lecourtier L, Mosbacher J, Cryan JF, Fritschy JM, Lüthi A, Kaupmann K. Bettler B. Redistribution of GABAB(1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J Neurosci. 2004;24:6086–6097. doi: 10.1523/JNEUROSCI.5635-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J. Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007a;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Glykys J. Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol. 2007b;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald S, Schupp BJ, Ikeda SR, Kuner R, Steigerwald F, Kornau HC. Köhr G. Importance of the γ-aminobutyric acidB receptor C-termini for G-protein coupling. Mol Pharmacol. 2002;61:1070–1080. doi: 10.1124/mol.61.5.1070. [DOI] [PubMed] [Google Scholar]
- Haddjeri N, de Montigny C. Blier P. Modulation of the firing activity of noradrenergic neurones in the rat locus coeruleus by the 5-hydroxtryptamine system. Br J Pharmacol. 1997;120:865–875. doi: 10.1038/sj.bjp.0700968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis RB. Aghajanian GK. Pertussis toxin blocks 5-HT1A and GABAB receptor-mediated inhibition of serotonergic neurons. Eur J Pharmacol. 1987;143:195–204. doi: 10.1016/0014-2999(87)90533-4. [DOI] [PubMed] [Google Scholar]
- Ishimatsu M. Williams JT. Synchronous activity in locus coeruleus results from dendritic interactions in pericoerulear regions. J Neurosci. 1996;16:5196–5204. doi: 10.1523/JNEUROSCI.16-16-05196.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasamatsu T. Adrenergic regulation of visuocortical plasticity: a role of the locus coeruleus system. Prog Brain Res. 1991;88:599–616. doi: 10.1016/s0079-6123(08)63837-6. [DOI] [PubMed] [Google Scholar]
- Kim MA, Lee HS, Lee BY. Waterhouse BD. Reciprocal connections between subdivisions of the dorsal raphe and the nuclear core of the locus coeruleus in the rat. Brain Res. 2004;1026:56–67. doi: 10.1016/j.brainres.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Kimura F. Nakamura S. Postnatal development of α-adrenoceptor-mediated autoinhibition in the locus coeruleus. Brain Res. 1987;432:21–26. doi: 10.1016/0165-3806(87)90004-6. [DOI] [PubMed] [Google Scholar]
- Kulik A, Vida I, Luján R, Haas CA, López-Bendito G, Shigemoto R. Frotscher M. Subcellular localization of metabotropic GABAB receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus. J Neurosci. 2003;23:11026–11035. doi: 10.1523/JNEUROSCI.23-35-11026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey CJ, Boyes J, Gerlach O, Chen L, Magill PJ. Bolam JP. GABAB receptors at glutamatergic synapses in the rat striatum. Neuroscience. 2005;136:1083–1095. doi: 10.1016/j.neuroscience.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C. Monyer H. GABAergic interneurons shape the functional maturation of the cortex. Neuron. 2013;77:388–405. doi: 10.1016/j.neuron.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- Lu J, Bjorkum AA, Xu M, Gaus SE, Shiromani PJ. Saper CB. Selective activation of the extended ventrolateral preoptic nucleus during rapid eye movement sleep. J Neurosci. 2002;22:4568–4576. doi: 10.1523/JNEUROSCI.22-11-04568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ. Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol. 1991;65:247–263. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Gervasoni D, Verret L, Goutagny R, Peyron C, Salvert D, Leger L. Fort P. Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. J Physiol Paris. 2006;100:271–283. doi: 10.1016/j.jphysparis.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Mannoury la Cour C, Boni C, Hanoun N, Lesch KP, Hamon M. Lanfumey L. Functional consequences of 5-HT transporter gene disruption on 5-HT1a receptor-mediated regulation of dorsal raphe and hippocampal cell activity. J Neurosci. 2001;21:2178–2185. doi: 10.1523/JNEUROSCI.21-06-02178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehler MF. Purpura DP. Autism, fever, epigenetics and the locus coeruleus. Brain Res Rev. 2009;59:388–392. doi: 10.1016/j.brainresrev.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Wu YW, Shih PY, Lu HW, Lin CC, Wu Y, Li MJ. Yang HW. Physiological and morphological properties of, and effect of substance P on, neurons in the A7 catecholamine cell group in rats. Neuroscience. 2008;153:1020–1033. doi: 10.1016/j.neuroscience.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Min MY, Wu YW, Shih PY, Lu HW, Wu Y, Hsu CL, Li MJ. Yang HW. Roles of A-type potassium currents in tuning spike frequency and integrating synaptic transmission in noradrenergic neurons of the A7 catecholamine cell group in rats. Neuroscience. 2010;168:633–645. doi: 10.1016/j.neuroscience.2010.03.063. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Kimura F. Sakaguchi T. Postnatal development of electrical activity in the locus ceruleus. J Neurophysiol. 1987;58:510–524. doi: 10.1152/jn.1987.58.3.510. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP. Maze M. The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci. 2002;5:979–984. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, McMahon A, Sabban EL, Tallman JF. Duman RS. Chronic antidepressant administration decreases the expression of tyrosine hydroxylase in the rat locus coeruleus. Proc Natl Acad Sci U S A. 1990;87:7522–7526. doi: 10.1073/pnas.87.19.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz D. Siegel JM. GABA release in the locus coeruleus as a function of sleep/wake state. Neuroscience. 1997;78:795–801. doi: 10.1016/s0306-4522(96)00549-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett CL. Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–147. doi: 10.1016/S1054-3589(10)58006-2. [DOI] [PubMed] [Google Scholar]
- Pejchal T, Foley MA, Kosofsky BE. Waeber C. Chronic fluoxetine treatment selectively uncouples raphe 5-HT1A receptors as measured by [35S]-GTPγS autoradiography. Br J Pharmacol. 2002;135:1115–1122. doi: 10.1038/sj.bjp.0704555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL. Webster H. The Fine Structure of the Nervous System. New York: Oxford University Press; 1991. [Google Scholar]
- Pfeiffer A. Zhang W. Postnatal development of GABAB-receptor-mediated modulation of potassium currents in brainstem respiratory network of mouse. Respir Physiol Neurobiol. 2007;158:22–29. doi: 10.1016/j.resp.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Shen C, Li H. Meller E. Repeated treatment with antidepressants differentially alters 5-HT1A agonist-stimulated [35S]GTPγS binding in rat brain regions. Neuropharmacology. 2002;42:1031–1038. doi: 10.1016/s0028-3908(02)00064-3. [DOI] [PubMed] [Google Scholar]
- Somogyi J. Llewellyn-Smith IJ. Patterns of colocalization of GABA, glutamate and glycine immunoreactivities in terminals that synapse on dendrites of noradrenergic neurons in rat locus coeruleus. Eur J Neurosci. 2001;14:219–228. doi: 10.1046/j.0953-816x.2001.01638.x. [DOI] [PubMed] [Google Scholar]
- Szabo ST, de Montigny C. Blier P. Modulation of noradrenergic neuronal firing by selective serotonin reuptake blockers. Br J Pharmacol. 1999;126:568–571. doi: 10.1038/sj.bjp.0702343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K. Bolshakov VY. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain. 2010;3:15. doi: 10.1186/1756-6606-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Min MY. Yang HW. GABAB. New Orleans: Society for Neuroscience; 2012. & receptor-mediated tonic inhibition of locus coeruleus neurons in developing rats . Program No. 537.07. 2012 Neuroscience Meeting Planner. , LA, USA. Online. [Google Scholar]
- Wang Y, Neubauer FB, Luscher HR. Thurley K. GABAB receptor-dependent modulation of network activity in the rat prefrontal cortex in vitro. Eur J Neurosci. 2010;31:1582–1594. doi: 10.1111/j.1460-9568.2010.07191.x. [DOI] [PubMed] [Google Scholar]
- Wang T, Rusu SI, Hruskova B, Turecek R. Borst JG. Modulation of synaptic depression of the calyx of Held synapse by GABAB receptors and spontaneous activity. J Physiol. 2013;591:4877–4894. doi: 10.1113/jphysiol.2013.256875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West CH, Ritchie JC, Boss-Williams KA. Weiss JM. Antidepressant drugs with differing pharmacological actions decrease activity of locus coeruleus neurons. Int J Neuropsychopharmacol. 2009;12:627–641. doi: 10.1017/S1461145708009474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Colmers WF. Pan ZZ. Voltage- and ligand-activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci. 1988;8:3499–3506. doi: 10.1523/JNEUROSCI.08-09-03499.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Henderson G. North RA. Characterization of alpha 2-adrenoceptors which increase potassium conductance in rat locus coeruleus neurones. Neuroscience. 1985;14:95–101. doi: 10.1016/0306-4522(85)90166-6. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang HY, Lin CC, Lu HC, Cheng SJ, Chen CC, Yang HW. Min MY. GABAB receptor-mediated tonic inhibition of noradrenergic A7 neurons in the rat. J Neurophysiol. 2011;105:2715–2728. doi: 10.1152/jn.00459.2010. [DOI] [PubMed] [Google Scholar]