

Abstract
The voltage-gated potassium channel KV10.1 (Eag1) is widely expressed in the mammalian brain, but its physiological function is not yet understood. Previous studies revealed highest expression levels in hippocampus and cerebellum and suggested a synaptic localization of the channel. The distinct activation kinetics of KV10.1 indicate a role during repetitive activity of the cell. Here, we confirm the synaptic localization of KV10.1 both biochemically and functionally and that the channel is sufficiently fast at physiological temperature to take part in repolarization of the action potential (AP). We studied the role of the channel in cerebellar physiology using patch clamp and two-photon Ca2+ imaging in KV10.1-deficient and wild-type mice. The excitability and action potential waveform recorded at granule cell somata was unchanged, while Ca2+ influx into axonal boutons was enhanced in mutants in response to stimulation with three APs, but not after a single AP. Furthermore, mutants exhibited a frequency-dependent increase in facilitation at the parallel fibre–Purkinje cell synapse at high firing rates. We propose that KV10.1 acts as a modulator of local AP shape specifically during high-frequency burst firing when other potassium channels suffer cumulative inactivation.
Key points.
Voltage-gated KV10.1 potassium channels are widely expressed in the mammalian brain but their function remains poorly understood.
We report that KV10.1 is enriched in the presynaptic terminals and does not take part in somatic action potentials.
In parallel fibre synapses in the cerebellar cortex, we find that KV10.1 regulates Ca2+ influx and neurotransmitter release during repetitive high-frequency activity.
Our results describe the physiological role of mammalian KV10.1 for the first time and help understand the fine-tuning of synaptic transmission.
Introduction
Neuronal potassium channels are involved in setting the resting membrane potential, influencing firing patterns, repolarizing the action potential (AP), and in controlling neurotransmitter release and synaptic plasticity. KV10.1 is the founding member of the eag (ether-à-go-go) family of voltage-gated potassium channels (Warmke & Ganetzky, 1994). In mammals, channel expression is restricted to the CNS (Ludwig et al. 1994; Saganich et al. 2001; Martin et al. 2008). While Drosophila eag is implicated in controlling neuronal excitability (Wu et al. 1983), little is known about the physiological role of KV10.1 in higher organisms. A biophysical hallmark of Kv10.1 is that it activates orders of magnitude faster at depolarized potentials than at hyperpolarized potentials (Ludwig et al. 1994); this provides the channel with a short-term molecular memory and could make its role dependent on the average potential previous to the AP and therefore on activity. Electron microscopy, single particle tracking (Gómez-Varela et al. 2010), and recent immunocytochemistry and biochemical data (Chuang et al. 2014) indicate a (pre)synaptic localization of KV10.1. As no specific pharmacological blockers for KV10.1 are available, KV10.1-deficient mice represent the best possibility to analyse its significance in neuronal function. KV10.1 knock-out (KO) mice are viable and show no obvious abnormal behaviour except increased spontaneous locomotor activity (Ufartes et al. 2013). We compared the synaptic transmission at the parallel fibre–Purkinje cell (PF–PC) synapse of wild-type (WT) and KV10.1 KO mice to study the physiological role of KV10.1. The PF–PC synapse has a moderate release probability (pr) (Dittman et al. 2000; Isope & Barbour, 2002; Sims & Hartell, 2005; Valera et al. 2012; Schmidt et al. 2013) and exhibits paired-pulse facilitation (Konnerth et al. 1990) that can be caused by several mechanisms including residual free Ca2+ (Cares), a facilitated release machinery, or buffer saturation (reviewed by Zucker and Regehr, 2002). In hippocampal mossy fibre boutons (Wheeler et al. 1996; Geiger & Jonas, 2000) and in the calyx of Held (Borst & Sakmann, 1999; Ishikawa et al. 2003), it has been shown that the width of an AP determines the duration of the Ca2+ nanodomain signal that triggers release (Bollmann & Sakmann, 2005) and subsequently influences synaptic plasticity. We found that single excitatory postsynaptic currents (EPSCs) recorded from Purkinje cells are unchanged in KV10.1 KO mice, but that they are differently affected by changes in the extracellular Ca2+ concentrations. By two-photon Ca2+ imaging we show that loss of KV10.1 causes a frequency- and pulse number-dependent increase in presynaptic Ca2+ signals. Additionally, facilitation is increased in KV10.1 KO mice at the PF–PC synapses. Somatic excitability of granule cells (GCs) is unchanged in KV10.1 KO mice, suggesting that the phenotype originates at the synapse. Our results suggest that KV10.1 is important for regulating AP width at high-frequency stimulus trains and thereby contributes to regulating synaptic strength.
Methods
Ethical statement
All experiments were done following the guidelines of the German law on animal protection.
HEK cell electrophysiology
Monoclonal HEK293 cells expressing hEag1 (García-Ferreiro et al. 2004) were grown for 24–72 h on poly-l-lysine-coated glass coverslips. Macroscopic currents were recorded in the whole-cell configuration of the patch-clamp technique using an EPC-9 amplifier and Pulse software (HEKA, Lambrecht/Pfalz, Germany). Patch pipettes with a tip resistance of 1.5–2 MΩ were made from Corning no. 0010 capillary glass (World Precision Instruments, Sarasota, FL, USA). The internal solution contained (in mm) 100 KCl, 45 N-methyl-d-glucamine, 10 BAPTA.K4 and 10 Hepes/HCl (pH 7.35). The control external recording solution contained (in mm) 160 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 8 glucose and 10 Hepes/NaOH (pH 7.4). Series resistance was determined using the automated capacity compensation of the amplifier and compensated for by 60–80%. Currents were elicited by injection of square voltage pulses, sampled at 20 kHz and filtered at 4 kHz. To determine the I–V relationship, cells were held at −60 mV and voltage pulses ranging from −60 to +80 mV were injected. To determine the dependence of activation kinetics on the prepulse potential, cells were stepped to +40 mV from a 5 s prepulse ranging from −120 to −70 mV and the rise time between 10 and 80% of the maximal current calculated. The Q10 value was calculated as
with R2 and R1 being the rise times, and T2 and T1 the temperature.
Data analysis was performed offline with PulseFit software (HEKA).
Slice preparation and electrophysiology
Slices were prepared as described previously (Bao et al. 2010). Briefly, KV10.1 KO mice and WT littermates (postnatal day (P)20–28) were decapitated under isoflurane (Essex Tierarznei, Munich, Germany) anaesthesia. The cerebellum was removed and placed in an ice-cold oxygenated solution containing (in mm) 60 NaCl, 120 sucrose, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 25 d-glucose, 0.1 CaCl2, 3 MgCl2, 3 myo-inositol, 2 sodium pyruvate and 0.4 ascorbic acid. Coronal slices (200 μm) of the cerebellar vermis were cut using a vibratome (VT1200S; Leica, Wetzlar, Germany). The slices were kept at 36°C for 45 min to 1 h in oxygenated artificial cerebrospinal fluid containing (in mm) 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 3 myo-inositol, 2 sodium pyruvate and 0.4 ascorbic acid.
Cells were visually identified and patched at 33 ± 1°C in oxygenated artificial cerebrospinal fluid using an EPC 10/2 amplifier controlled by Patchmaster software (HEKA). Thick-walled borosilicate pipettes were pulled to resistances of 3–4 MΩ for Purkinje cell recordings and 6–7 MΩ for granule cells when filled with intracellular solution containing (in mm) 135 potassium gluconate, 5 KCl, 10 Hepes, 5 MgATP, 0.5 NaGTP, 1 EGTA and 5 N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium chloride (QX-314; Tocris, Ellisville, MO, USA; a Na+ channel blocker to prevent clamp escape). Liquid junction potential was assumed to be 10 mV; the reported voltages were not corrected for liquid junction potential. For recording of evoked EPSCs, voltage pulses (20 μs, 1–5 V) were applied through an extracellular electrode placed in the GC layer and the GABAA receptor antagonist 2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)pyridazinium bromide (SR-95531; Tocris) was added. Series resistance was compensated for to leave ≤ 5 MΩ uncompensated resistance. QX-314 was omitted in current clamp recordings. Extracellular potentials were recorded in the molecular layer while voltage pulses were applied through an electrode placed 200–600 μm lateral to the recording site. Spontaneous miniature (m)EPSCs were recorded in Purkinje cells in sagittal slices in the presence of 1 μm TTX (Alomone, Jerusalem, Israel) and 100 μm picrotoxin (Tocris).
Data acquisition and analysis
Data were low-pass filtered at 5–10 kHz and sampled at 50–100 kHz. Offline analysis was performed with custom-written macros in Igor Pro (Wavemetrics, Lake Oswego, OR, USA). Trains of EPSCs were recorded 10–20 times and averaged per cell. Statistical analysis was performed with Prism software (GraphPad Software Inc., La Jolla, CA, USA). All data are presented as mean ± SEM and Student’s t test was used to test for statistical significance if not noted otherwise.
Variance mean analysis
Variance mean analysis for determination of synaptic parameters is an established tool for analysis of synaptic parameters (Silver et al. 1998; Reid & Clements, 1999; Clements & Silver, 2000; Humeau et al. 2001, 2002, 2007; Silver, 2003). It has been employed in experimental paradigms using extracellular stimulation at the PF–PC synapse previously (Sims & Hartell, 2005; Valera et al. 2012) Briefly, the binomial model of release describes the mean of the response
with N being a binominal parameter that can reflect the number of independent release sites under certain conditions (Meyer et al. 2001) with average release probability pr and quantal size Q. The variance of the response is given by
Modifying pr by changing the concentration of extracellular Ca2+, VarI = f(Imean) takes the form of a parabola:
| 4 |
where the initial slope of the parabola provides an estimate for Q and the greater x-axis intercept of the parabola for (N · Q). pr can be estimated from the parabola, as it will be 0.5 at the peak and 1 at the greater x-axis intercept. This model greatly simplifies the physiological situation, as it assumes uniformity of pr and Q across release sites and linear summation of individual quantal responses. Furthermore, N is merely a functional parameter, representing the number of release sites with release-ready vesicles at the time of stimulation, and excluding silent sites (Humeau et al. 2007; Valera et al. 2012).
In each cell, we recorded ≥ 50 EPSCs in 2, 4, 0.5 and again 2 mm Ca2+. Cells were only used for analysis if series resistance changed less than 10% during recording and if the average EPSC amplitude in the initial and final records in 2 mm Ca2+ did not differ. Variances of the average EPSC amplitudes under the different recording conditions were plotted against the respective means and fitted with eq.(1). To compare data from WT and KV10.1 KO cells, the data were normalized to the maximum variance and x-axis intercept of the parabolas.
Two-photon-Ca2+ imaging in axonal boutons
GCs were whole-cell patch-clamped and filled via the pipette with intracellular solution containing 0.1 mm Oregon Green 488 BAPTA-1 (OGB; Invitrogen, Carlsbad, CA, USA). The axon was visually identified and presumed synaptic boutons were visible as varicosities along the axon. For analysis, preference was given to boutons that were found distally to the bifurcation of the axon (see also Fig. 5). Current pulses of 1 ms duration were given to elicit 1–3 APs at 50–100 Hz 30–60 min after breaking into the cell. Fluorescence signals in the boutons were recorded at 33°C by performing a line scan across the bouton with a laser-scanning microscope (Olympus). Offline analysis was performed in Fluoview (Olympus) and Igor Pro with custom-written algorithms. Background fluorescence was corrected for and the change in intensity was divided by the baseline fluorescence resulting in ΔF/F0 values. After manual identification of a maximum, the signal decay was fitted with an exponential function. The amplitude parameter of the fit was used for statistical analysis.
Figure 5. Increased calcium influx into the presynaptic terminal.
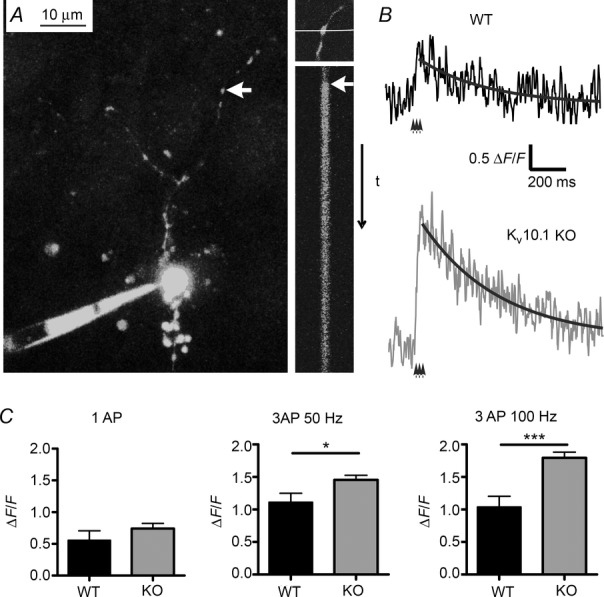
A, left, image of a granule cell filled with OBG-1 via the somatic patch pipette. The axon extends into the molecular layer, and putative synaptic boutons are visible as swellings (red arrow). Top right, one bouton was selected and a line scan performed across it while 1–3 APs were evoked in the soma (bottom right, arrow marks time of stimulation). B, representative traces measured in a bouton of a WT (top, black) and KV10.1 KO (bottom, grey) fibre in response to three APs at 100 Hz. The decay was fitted with an exponential function (black line). C, fluorescence ratio ΔF/F0 measured in response to a single AP (left, n = 7 in WT and n = 7 in KV10.1 KO), three APs at 50 Hz (middle, n = 7 and 16) and three APs at 100 Hz (right, n = 7 and 15). Ca2+ influx in the KO is increased in a frequency-dependent manner. *P < 0.05; ***P < 0.001; Student’s t test.
Subcellular fractioning and Western blotting
Synaptosomes were prepared as previously described (Fischer von Mollard et al. 1991). Briefly, 4-week-old mice (KV10.1 KO and WT littermates) were decapitated and their brains were removed. After homogenization in ice-cold homogenization buffer (320 mm sucrose, 5 mm Hepes, pH 7.4), cell debris was removed by centrifugation at 3000 g for 2 min at 4°C. Synaptosomes were then collected by re-centrifuging the supernatant for 12 min at 14,000 g, and laid on a three-step discontinuous Ficoll gradient consisting of 4 ml 13% Ficoll, 1 ml 9% Ficoll and 4 ml 6% Ficoll. After centrifugation of the gradients at 75,000 g for 35 min, the synaptosome band at the interface between 13% and 9% Ficoll was collected, diluted in homogenization buffer and again centrifuged for 12 min at 14,000 g to remove Ficoll. The isolated synaptosomes (5 mg) were resuspended in 20 ml sucrose buffer (320 mm sucrose, 5 mm Hepes, pH 8) and supplemented with 50 μg trypsin (Roche, Indianapolis, IN, USA) to give a final protein/protease ratio of 100:1. Proteolytic digestion of the post-synaptic membranes was performed by incubation of synaptosomes with the protease for 30 min at 30°C. Afterwards, the synaptosomes were pelleted by centrifugation at 22,500 r.p.m. in a Beckman SW41 swing-out rotor for 35 min and protease activity was blocked by addition of Pefabloc (Roche). Samples were separated by SDS-PAGE and proteins were subsequently transferred to a nitrocellulose membrane. The membrane was dried for 30 min at 37°C for Western blotting. After rehydration, the Quentix Western Blot Enhancer (Pierce, Rockford, IL, USA) was applied, the membrane was washed in H2O and incubated in 5% non-fat milk in Tris-buffered saline–Tween 20 (TBST) before application of primary antibodies against KV10.1 (Chen et al. 2011), AMPAR (Synaptic Systems, Göttingen, Germany), synaptophysin (Synaptic Systems) and synaptobrevin 2 (Synaptic Systems). After further washes with TBST, the membrane was incubated with horseradish peroxidase-coupled secondary antibodies for 1 h. The blot was developed using WesternLightening TMPlus-ECL (Perkin Elmer, Waltham, MA, USA) and protein bands were detected by a luminesce detector (Boehringer Mannheim, Mannheim Germany).
Freeze substitution and post-embedding immunogold labelling
Sprague-Dawley rats were transcardially perfused with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.15, for 1.5 h. Cerebellar regions were carefully dissected and processed for freeze substitution and low-temperature embedding as previously described (Douyard et al. 2007). For post-embedding immunocytochemistry, ultrathin sections (80 nm in thickness) on nickel grids were incubated in sodium borohydride and glycine in TBS solution with Triton X-100. After being preblocked with serum, the sections were incubated with affinity-purified primary antibody mAb62 (1 μg ml−1; 1:200 dilution). Primary antibody was detected with secondary antibodies conjugated to 5 nm gold particles (1:20; Amersham, Arlington Heights, IL, USA). The specificity of the antibody had been previously confirmed (Hemmerlein et al. 2006; Martin et al. 2008; Gómez-Varela et al. 2010). Controls included omitting mAb62 and preabsorption of mAb62 with the corresponding blocking protein (10 μg ml−1 final concentration). Ultrathin sections were counterstained with uranyl acetate and lead citrate and studied with a transmission electron microscope. Electron micrographs were taken at 30,000× magnification and scanned at a resolution of 3600 d.p.i. using a Linotype-Hell scanner (Heidelberg, Germany). Image processing was performed with Adobe Photoshop using only the brightness and contrast commands to enhance gold particles.
Results
KV10.1 activation kinetics at near physiological temperature
To date, all available studies on KV10.1 have been performed at room temperature and report a slow activation of the channel (e.g. Ludwig et al. 1994; Terlau et al. 1996). When we compared channel properties measured in stably transfected HEK-293 cells at room temperature (25 ± 1°C) and near physiological temperatures (30 ± 1°C), we found markedly accelerated activation kinetics (Fig. 1A, B; Q10 of approximately 1.4 at −90 mV holding potential), while the channel conductance remained unchanged (Fig. 1C). Interestingly, upon repeated brief depolarization, the amplitude of the current increased with each pulse (Fig. 1D). This suggests that due to the previously described two activity states of the channel (Ludwig et al. 1994; Terlau et al. 1996) it switches from a slow state at resting potential to a ‘fast mode’ during cell activity. This endows the channel with a short-lasting memory of the recent activity of the cell.
Figure 1. KV10.1 currents at near physiological temperature in HEK-293 cells.
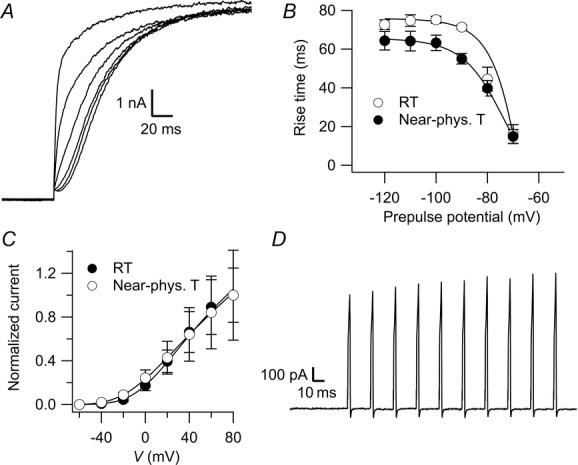
A, representative recording in an HEK-293 cell at near physiological temperature where a 2 s depolarization to 40 mV was preceded by 5 s prepulses ranging from −120 to −70 mV in steps of 10 mV. For clarity, only the first 200 ms are shown. The slowest trace corresponds to the −120 mV prepulse. B, 10–80% rise time of the currents measured with the protocol in A plotted against the prepulse potential at room temperature (open circles) and near physiological temperature (filled circles). C, I–V relationship as determined by measuring the whole-cell current elicited by stepping to potentials ranging from –60 to +80 mV in increments of 20 mV from a holding potential of –60 mV at room temperatures (open circles) and near physiological temperature (filled circles). D, currents recorded in response to a train of ten 2 ms depolarizations to 20 mV at 50 Hz.
Granule cell firing is unaffected in KV10.1 KO mice
In situ hybridization data suggest expression of KV10.1 in cerebellar GCs (Ludwig et al. 2000; Saganich et al. 2001). To test if KV10.1 plays a role in regulation of excitability and firing of GCs, we performed whole-cell patch clamp in GCs from WT and KV10.1 KO mice in acute cerebellar slices. The resting potential (under whole-cell current clamp) directly after breaking into the cell did not differ significantly between WT and KV10.1 KO cells (−79.3 ± 1.5 mV, n = 11 and −76.0 ± 1.8 mV, n = 5, respectively, P > 0.05). Neither WT nor KV10.1 KO cells showed spontaneous firing. Active membrane properties were evaluated by measuring the voltage response to injection of square current pulses ranging from −35 to +35 pA in steps of 5 pA into the soma (Fig. 2A). The resulting I–V relationship did not differ between genotypes (Fig. 2B). WT cells fired the first AP after injection of 6.4 ± 1.0 pA (n = 11), while KV10.1 KO cells started firing upon injection of 5.4 ± 1.5 pA (n = 10, P > 0.05). In accordance with previous studies (D'Angelo et al. 1998; Chadderton et al. 2004; Brickley et al. 2007), WT and KV10.1 KO cells displayed fast repetitive firing of APs with little or no adaptation during the depolarization (Fig. 2A). Evoked firing frequencies increased with the amplitude of the injected current (Fig. 2C). KV10.1 channels could be implicated in the repolarization of the AP. To test this possibility, the first 8–12 APs recorded just after reaching spike threshold were averaged, and undershoot, amplitude, half-width, rise time and de-/repolarization speeds were evaluated for both WT and KV10.1 KO. As these APs usually occurred at low frequencies, we did not expect KV10.1 kinetics to influence the results. None of the parameters tested resulted in significant differences between genotypes (Fig. 2D–F). In summary, we detected no difference in excitability or AP properties between GCs of WT and KV10.1 KO mice.
Figure 2. Somatic responses in granule cells are unaffected by deletion of KV10.1.
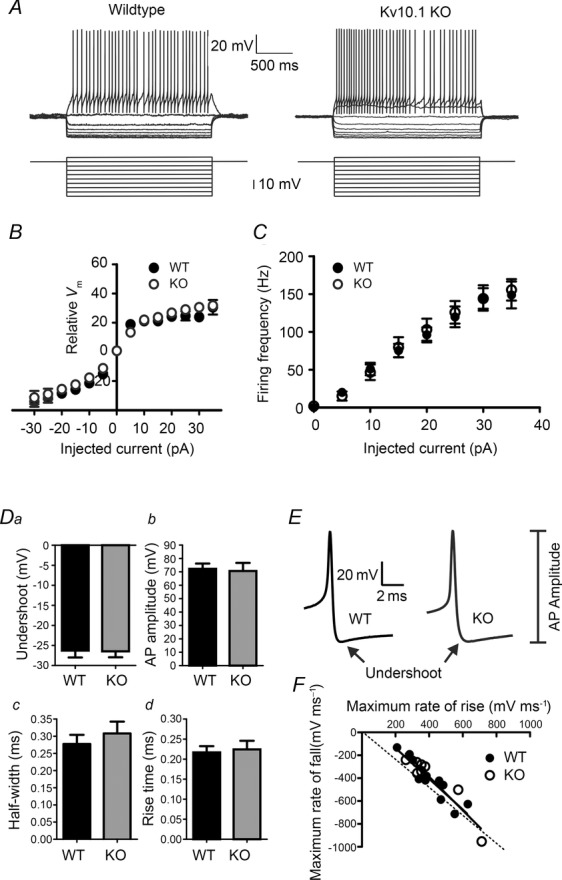
A–C, electroresponsiveness of cerebellar granule cells of WT and KV10.1 KO mice. A, representative traces recorded in granule cells from WT (left) and KV10.1 KO mice (right) in response to injection of square current pulses (bottom). Both genotypes showed inward rectification of the membrane potential during hyperpolarizing pulses and regular spiking after reaching a threshold potential. B, average membrane potential recorded in whole-cell current clamp mode in response to injection of square current pulses (2 s) in the presence of TTX. C, average firing frequency of WT and KV10.1 KO cells upon injection of 2 s square current pulses ranging between −35 and +35 pA in 5 pA increments. D–F, AP properties are not altered in KV10.1 KO mice. D, undershoot amplitudes measured from threshold (a), AP amplitude (b), half-width (c) and 10–90% rise time (d) are compared. Filled bars, WT, n = 11 cells; grey bars, KV10.1 KO, n = 8 cells. E, representative APs recorded in a WT (left) and KV10.1 KO (right) granule cell. F, maximum rate of rise of the AP plotted against the maximum rate of fall as determined by analysing the first differential. Filled circles, WT; open circles, KV10.1 KO. The data points were fitted with straight lines, which were not significantly different between genotypes. The dashed line represents unity.
Kv10.1 localizes to the presynaptic terminal
In immunoelectron microscopy images on rat cerebellum using a well-characterized anti-KV10.1 antibody (mAb62; Hemmerlein et al. 2006; Martin et al. 2008; Gómez-Varela et al. 2010) KV10.1 was preferentially localized to the synapses of the parallel fibres onto the Purkinje cell spines (Fig. 3A).
Figure 3. KV10.1 is enriched in the presynaptic compartment.
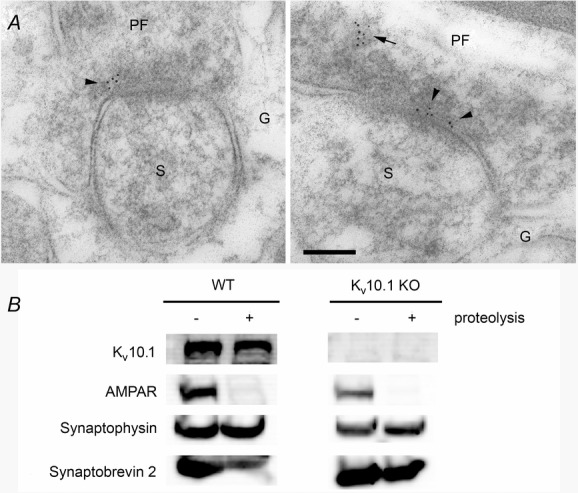
A, electron micrographs show post-embedding immunogold labelling for Kv10.1 in parallel fibre (PF) synapses on Purkinje cells spines (S) within the molecular layer of the cerebellum. Gold particles (5 nm in diameter) are localized in the presynaptic terminal close to the presynaptic plasma membrane (arrowhead) and within the cytoplasm of the PF synapse (arrow). G, Bergmann glial cells. Scale bar: 100 nm. B, immunoblot analysis of synaptosomes before and after treatment with protease reveals the presynaptic localization of KV10.1. The protein is detected before and after trypsination, as are the presynaptic markers synaptophysin and synaptobrevin 2. The AMPA receptor is not detected after protease treatment, indicating removal of the postsynaptic part. No signal for KV10.1 is detected in samples from KV10.1 KO mice.
To confirm presynaptic localization in the mouse, where mouse monoclonal antibodies are not optimal for morphological studies, we aimed at supporting the synaptic localization of KV10.1 biochemically. Subcellular fractionation by centrifugation through a Ficoll gradient has been shown to be effective in separating synaptosomes from cell bodies. The presynaptic compartment can subsequently be isolated by digestion of postsynaptic proteins by trypsin treatment, as described by Boyken et al. (2013).
Immunoblot analysis revealed that KV10.1 was present in the synaptosome preparation and protected from proteolysis along with the presynaptic markers synaptophysin and synaptobrevin 2. The signal for KV10.1 was slightly weaker after proteolysis, which might reflect some postsynaptic expression. The postsynaptic AMPA receptor was removed from the sample and was no longer detectable (Fig. 3B). This provides further evidence not only for the synaptic localization of KV10.1, but specifically for a presynaptic presence of the channel, as previously suggested by Gómez-Varela et al. (2010) and recently confirmed by others (Chuang et al. 2014).
Synaptic transmission at the PF–PC synapse
The lack of evidence for an involvement of KV10.1 in the regulation of the somatic membrane potential or AP firing despite the clear signal for KV10.1 mRNA expression (Ludwig et al. 2000; Saganich et al. 2001; M. Kuscher, personal communication), together with electron microscopy (Gómez-Varela et al. 2010), light microscopy (Chuang et al. 2014) and the described biochemical evidence, indicates presynaptic localization of the channel. In presynaptic terminals, potassium channels determine the duration of an AP, limit frequency of APs (Matsukawa et al. 2003) and set the resting potential (Huang & Trussell, 2011), thus controlling pr. To evaluate basic synaptic transmission, we measured spontaneously occurring mEPSCs in Purkinje cells of WT and KV10.1 KO mice. The inter-event interval was unchanged between WT and KO (207 ± 47 ms, n = 8 cells and 193 ± 57 ms, n = 7 cells, respectively), suggesting that the number of synapses formed onto Purkinje cells, the basal release probability and the resting Ca2+ levels are not affected in KV10.1 KO mice. Furthermore, the mEPSC amplitude was unchanged (19.5 ± 1 and 18.8 ± 1.4 pA, respectively), indicating that the postsynaptic compartment is functional and unaltered in KV10.1 KO cells.
We then recorded EPSCs in Purkinje cells evoked by extracellular stimulation of GCs (Fig. 4A, B). The kinetic parameters of EPSCs were identical in WT and KV10.1 KO cells (Fig. 4C). Due to the variability introduced by extracellular stimulation and dendritic filtering (Roth & Häusser, 2001), no conclusion can be drawn from comparing absolute EPSC amplitudes, but the change in amplitude as a reaction to altered external Ca2+ concentrations can provide information about synaptic properties (Foster et al. 2005). Figure 4D shows the relationship between EPSC amplitude and external Ca2+ concentration, normalized to the amplitude at 4 mm [Ca2+]o. In both genotypes, amplitude was strongly dependent on [Ca2+]o and the relationship could be fitted with a sigmoid function as described previously (Mintz et al. 1995; Foster et al. 2005). Neither the normalized amplitudes nor the parameters of the fit differed significantly between WT and KO (n = 12 and 13 cells, respectively). To further analyse synaptic parameters, we employed a variance mean analysis (Meyer et al. 2001). The variance of the EPSC amplitude during repetitive, low-frequency stimulation at different [Ca2+]o was plotted against the respective mean value and fitted with a parabolic function. For each cell, we determined N (64.0 ± 33.1 in WT and 44.7 ± 25.8 in KO, P ≥ 0.05) and Q (6.96 ± 3.23 pA in WT and 8.57 ± 4.18 pA in KO, P ≥ 0.05) values. To summarize data from various cells, the values were normalized to the maximum variance and N·Q, the (theoretical) maximum amplitude (Valera et al. 2012) (Fig. 4E). As pr is assumed to be 0.5 at the peak of the parabola, one can determine the release probability at the different Ca2+ concentrations. In 2 mm [Ca2+]o, pr was 0.24 ± 0.04 for the WT and 0.32 ± 0.03 for the KO (P > 0.05). At 0.5 mm [Ca2+]o, pr was 0.06 ± 0.01 and 0.06 ± 0.01; and at 4 mM pr was 0.63 ± 0.06 and 0.69 ± 0.06, respectively (n = 7 and 8, respectively, all P > 0.05). From these data, we conclude that the initial release probability is not significantly altered in KV10.1 KO cells.
Figure 4. EPSCs in Purkinje cells evoked by extracellular stimulation.
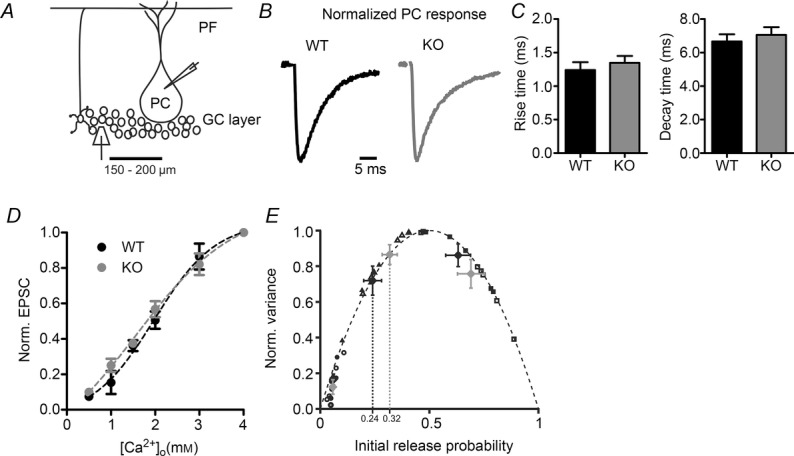
A, illustration showing the positions of the stimulation and measurement pipettes in the cerebellar cortex. PCs were whole-cell voltage clamped during extracellular stimulation in the granule layer. B, normalized sample traces in response to a single stimulus. Each trace is the average of 20 consecutive recordings. Black trace, WT; grey trace, KV10.1 KO. Stimulus artefacts are blanked for clarity. C, kinetics of EPSCs in Purkinje cells. Left, 10–90% rise time of EPSC in WT (black) and KV10.1 KO (grey). Right, decay time constant of EPSCs in WT (black) and KV10.1 KO (grey) Purkinje cells. D, EPSC amplitudes as a function of [Ca2+]o. Amplitudes are normalized to the EPSC at 4 mm and fitted with a sigmoid with the formula: base + max/(1+exp(([Ca2+]half – [Ca2+]o)/rate)). Parameters for WT fit were base = −0.05, max = 1.11, [Ca2+]half = 1.97, rate = 0.70. Parameters for KO fit were base = −0.31, max = 1.45, [Ca2+]half = 1.57, rate = 1.12. Black circles, WT, n = 6–12; grey circles, KV10.1 KO, n = 6–14. E, pooled results of the variance mean analysis for all cells normalized to the maximal variance and maximal amplitude (N · Q). Filled symbols (black, WT; grey, KO) represent initial pr and corresponding mean variance at 0.5 mm (circles), 2 mm (triangles) and 4 mm (diamonds) [Ca2+]o. Open symbols are values from individual cells (black, WT, n = 6; grey, KO, n = 8).
Two-photon Ca2+ imaging in the PF bouton
To address a possible increase in Ca2+ influx into presynaptic terminals of KV10.1 KO cells, we performed two-photon Ca2+ imaging in presynaptic boutons of PFs. After loading the GC with OGB, a line scan was performed across a putative bouton (Fig. 5A, left). One to three APs were elicited by injection of short current pulses into the soma, which led to a transient increase in fluorescence (Fig. 5B shows sample recordings in a KV10.1 KO cell and a WT cell in response to three APs at 100 Hz). Single APs evoked a slight but not significantly larger increase in the Ca2+ response in KV10.1 KO cells compared to WT (Fig. 5C). While increasing the number of APs to three nearly doubled the peak ΔF/F0 in both genotypes (Fig. 5C), an increase in frequency from 50 to 100 Hz did not cause a further rise in the WT (Fig. 5C). This is in line with earlier findings that the more intense facilitation at higher frequencies is not due to an increase in Ca2+ influx, for example by Ca2+-dependent facilitation of Ca2+ currents, but by downstream mechanisms such as increase in Cares, a facilitated release machinery or saturation of endogenous Ca2+ buffers (Atluri & Regehr, 1996; Naraghi & Neher, 1997; Kreitzer & Regehr, 2000; Blatow et al. 2003; Felmy et al. 2003; Brenowitz & Regehr, 2007; Bornschein et al. 2013). In the KO, however, doubling the frequency to 100 Hz further increased the peak ΔF/F0 by 23% (P < 0.01). This finding is compatible with the kinetic properties of KV10.1, which would take part in repolarization of the membrane depending on the frequency and pulse number (Fig. 1). Comparing the results from WT and KV10.1 KO boutons, the peak ΔF/F0 after three APs at 50 Hz was increased by 31% in the KO (P = 0.0226, Fig. 5C) and by 73% after three APs at 100 Hz (P = 0.0002, Fig. 4C). This suggests frequency-dependent increases in Ca2+ influx during high frequency bursts of APs characteristic for GC activity (Chadderton et al. 2004).
Increased facilitation at the PF–PC synapse in KV10.1 KO cells
We continued by investigating the consequences of loss of KV10.1 on short-term modulation of synaptic strength. When tested over a range of intervals, percentage facilitation (defined as (EPSC3 – EPSC1)/EPSC1) × 100) decays with a time constant of 99 ms in WT and 66 ms in KV10.1 KO cells (Fig. 6). Paired pulse ratio (PPR) at interstimulus intervals < 50 ms was significantly increased in KO cells. Thus, the frequency-dependent increase in Ca2+-influx observed by Ca2+-imaging correlates well with a frequency-dependent increase in facilitation at the PF–PC synapse.
Figure 6. Increased PPR in KV10.1 KO cells.
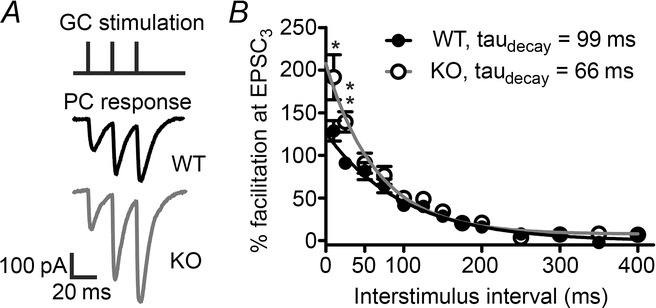
A, 50 Hz stimulus train (top) with representative recordings from a WT (middle) and KV10.1 KO (bottom) PC. The traces shown are the average of ten sweeps each, and stimulus artefacts have been blanked for clarity. B, facilitation of the third EPSC plotted against the interstimulus interval. The amplitude of facilitation decays exponentially with a time constant of 99 ms in the WT (filled circles) and 66 ms in the KO (open circles). Note the significant differences at intervals < 50 ms (P ≤ 0.05, Student’s t test).
Examining facilitation during train stimulations (Fig. 7) revealed that the effect of KV10.1 loss was again dependent both on the pulse number and on the stimulation frequency. In Fig. 7B–E, percentage facilitation is plotted against the stimulus number. A regular 10 Hz train evoked the same response in WT and KV10.1 KO cells (Fig. 7B). Upon 20 Hz stimulation (Fig. 7C), the responses reached a significant difference at pulse 10 (61 ± 8%, n = 8 in WT and 85 ± 7%, n = 10 in KV10.1 KO cells; P < 0.05). Stimulation with a 50 Hz train caused increased facilitation throughout the train in KV10.1 KO cells compared to the WT (Fig. 7A, D). The difference between KO and WT cells was largest after the third pulse. During a 100 Hz train (Fig. 7E), the difference between WT and KO increased until the fifth pulse. Facilitation in both genotypes declined after reaching the peak value. To control for the number of activated fibres during a train, we measured the presynaptic volley during stimulation at the frequencies mentioned (Sabatini & Regehr, 1997) (Fig. 7F). In both genotypes, the amplitude of the volley decreased by 15% in the second pulse, similar to the results of Kreitzer & Regehr (2000). This suggests that the number of activated fibres during the train was similar in both genotypes and no additional fibres were recruited in the KO during the train. When analysing the EPSC kinetics during the trains, it was noticeable that the 10–90% rise time of WT EPSCs did not change during a 50 Hz train, whereas it increased markedly in the KV10.1 KO (Fig. 7G). At the tenth pulse, the KO rise time was increased 29% in comparison to the WT rise time (n = 20 in WT, n = 14 in KO, P < 0.05). The rise time of an EPSC is related to the duration of the Ca2+ transient at the release site. It has been demonstrated that AP broadening causes a prolonged duration of the active zone Ca2+ signal, which results in a prolonged rise time (Borst & Sakmann, 1999; Bollmann & Sakmann, 2005). The large size of the Purkinje cell complicates space clamp quality, so kinetic values have to be interpreted with care. However, the rise time could serve as a clue towards pulse number-dependent AP broadening in the KV10.1 KO synapses. The decay time of the EPSC did not change during the train in either genotype (Fig. 7H), indicating that glutamate pooling and spillover does not contribute significantly to shaping the postsynaptic response (Takahashi et al. 1995). Interestingly, facilitation can be maintained during prolonged stimulation in both genotypes (Fig. 8A). This would only be possible if the readily releasable pool (RRP) is replenished quickly (Valera et al. 2012), which has been shown to be Ca2+-dependent (Dittman & Regehr, 1998; Dittman et al. 2000; Crowley et al. 2007). We assessed RRP replenishment by calculating the cumulative EPSC amplitude for responses normalized to the first value, fitting a line to the steady state region, and extrapolating it to the starting time (Fig. 8B) (Schneggenburger et al. 1999, 2002; Valera et al. 2012). We found the rate of vesicle replenishment in the KO to be enhanced compared to WT, which we attribute to the increased intracellular Ca2+ during repeated stimulation (Fig. 5). This can explain how the observed increased Ca2+ influx and transmitter release do not exhaust the synapse in a short time. These findings suggest that KV10.1 is involved in modulation of synaptic strength during high-frequency trains. We interpret these observations as reflecting the pulse number-dependent increase in Ca2+ observed in the imaging experiment at the electrophysiological level.
Figure 7. Facilitation of EPSCs during regular stimulus trains.
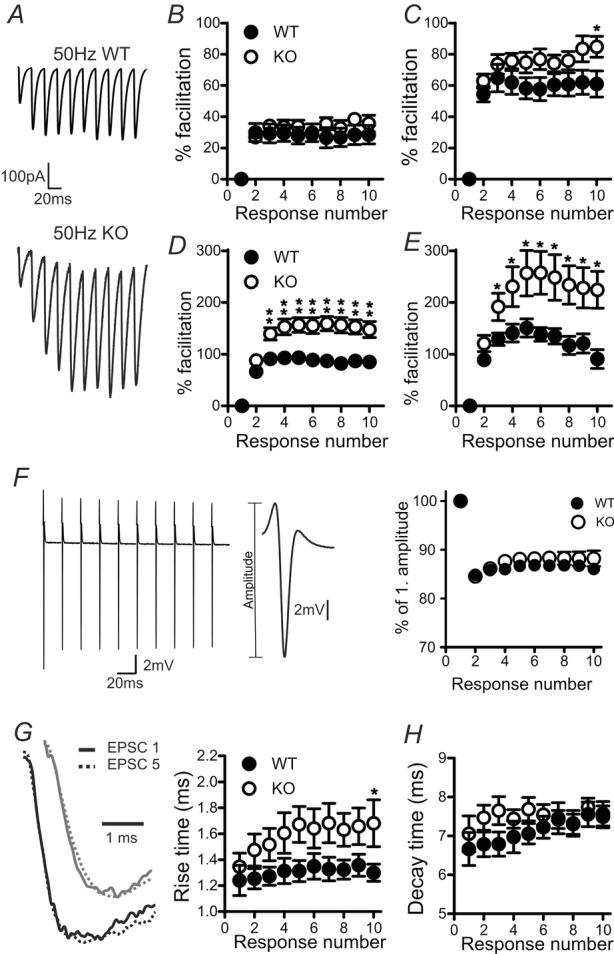
A, representative traces recorded in a WT (top) and a KO (bottom) Purkinje cell in response to a 50 Hz stimulus train. B–E, responses to train stimulations with 10 Hz (B), 20 Hz (C), 50 Hz (D) and 100 Hz (E). Percentage facilitation is defined as (EPSCn–EPSC1)/EPSC1×100 and plotted against the response number. F, left, presynaptic volley recorded in response to a 50 Hz stimulation. Stimulus artefacts have been blanked for clarity. Centre, the amplitude of the volley is a measure of the number of activated fibres. Right, relative change in the volley amplitude during a 50 Hz stimulation. The number of fibres activated during repetitive stimulation does not differ between WT and KO. G, left, representative traces of the first (continuous line) and fifth (dotted line) EPSC recorded in response to stimulation at 50 Hz in a WT (black) and KV10.1 KO (grey) Purkinje cell. Right, 10–90% rise time of the EPSCs measured in WT (filled circles) and KV10.1 KO (open circles) Purkinje cells during a 50 Hz stimulation plotted against the stimulus number. H, decay of the EPSCs during a 50 Hz stimulation was fitted with a single exponential and the time constant plotted against the stimulus number. In both genotypes, a slight increase in the decay time is observed, which is slightly more pronounced in the WT (filled circles) than in KV10.1 KO cells (open circles). *P < 0.05; **P < 0.01.
Figure 8. Facilitation during prolonged stimulation.
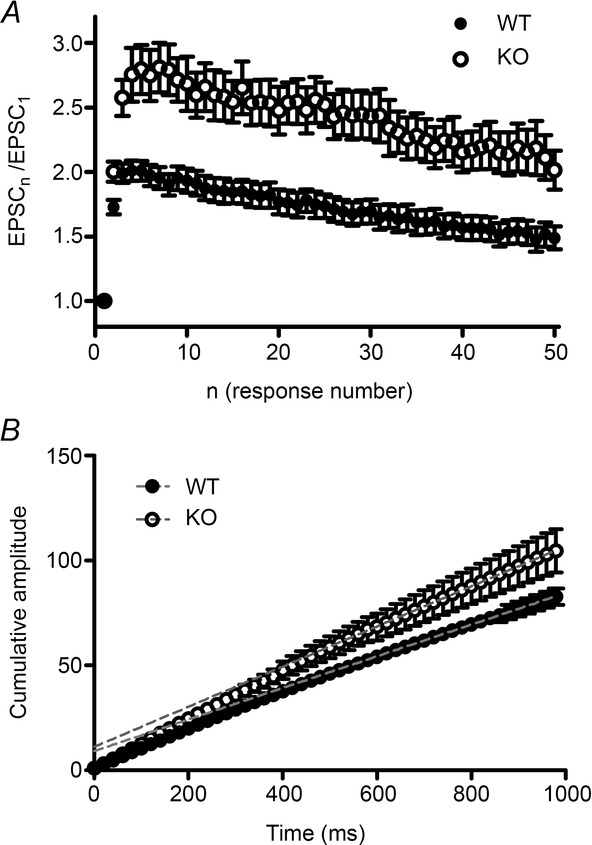
A, normalized EPSC amplitude (EPSCn/EPSC1) during 50 Hz stimulation at the PF–PC synapse plotted against response number. Facilitation decays only slowly in both genotypes (filled circles, WT; open circles, KO). B, cumulative plot of normalized EPSC amplitudes shown in A. A line is fitted to the steady-state region of the plot (dotted red line, WT; dotted blue line, KO) and extrapolated to intercept the y-axis at time 0. The slope represents the relative replenishment rate of the RRP (Schneggenburger et al., 1999, 2002). The fitting function is 0.076t + 9.06 for the WT, and 0.096t + 11.06 for the KV10.1. filled circles, WT (n = 24); open circles, KO (n = 19).
Changes in plasticity as a result of alterations in Ca2+ influx and dynamics
Decreasing [Ca2+]o causes less influx of Ca2+ into the terminal, reducing initial pr and decelerating build-up of Cares. By influencing pr, changes in external Ca2+ and Ca2+ influx have strong effects on the steady state amplitude of facilitation (Kreitzer & Regehr, 2000; Foster et al. 2005). In the WT, facilitation in 2 and 3 mm [Ca2+]o reached a plateau after 3–4 pulses. Lowering [Ca2+]o to 1 mm caused the facilitation in the WT to build up over 10 stimuli without reaching a stable plateau (Fig. 9A, filled circles). In KV10.1 KO cells, however, facilitation in 1 mm [Ca2+]o already reached a plateau after the 5th pulse (Fig. 9A, open circles). This indicates a faster build-up of intraterminal Ca2+ in the KV10.1 KO compared to WT. Plasticity under conditions where the release machinery (Valera et al. 2012) or postsynaptic receptors (Foster et al. 2005) are close to saturation should not differ between WT and KV10.1 KO. Increasing [Ca2+]o to 3 and 4 mm indeed decreased facilitation to the same extent in both genotypes (Fig. 9B). The steady state facilitations (defined as EPSC8/EPSC1) in different concentrations of [Ca2+]o for WT and KV10.1 KO cells are shown in Fig. 9B. In agreement with previous studies, facilitation decreased as [Ca2+]o increased (Kreitzer & Regehr, 2000; Foster et al. 2005) and vice versa. Taken together, loss of KV10.1 induced an effect only under conditions where the release machinery or the Ca2+ influx into the terminal is not saturated, i.e. where an increase in Ca2+ can translate into an increase in transmitter release.
Figure 9. The effects of altering extracellular or intracellular calcium on facilitation at the PF–PC synapse.
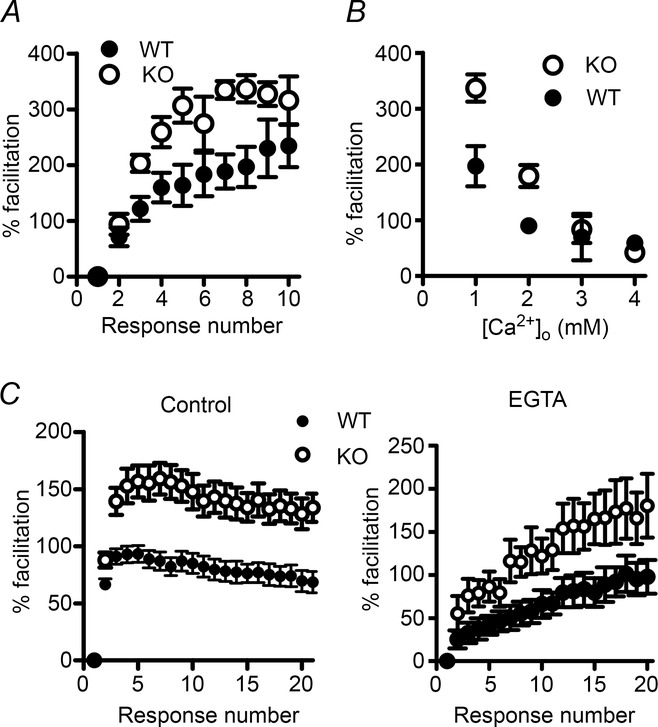
A, response to 50 Hz train stimulation in 1 mm external Ca2+. B, percentage facilitation at the 8th EPSC of a 50 Hz train, when facilitation reached a plateau. Open circles, KV10.1 KO; filled circles, WT. Each data point is the average of 5–10 cells. C, response to a 50 Hz stimulus under control conditions (left) and after incubation with 50 μM EGTA-AM (right). Both amplitude and time course are changed after incubation with EGTA-AM. Facilitation in the KO (open circles) was increased and rose faster than in the WT (filled circles).
If the increased facilitation observed in the KV10.1 KO is indeed due to a faster build-up of Cares in the presynaptic terminal, removal of residual free Ca2+ might rescue the phenotype. Introducing EGTA into the terminal reduces residual Ca2+ (Adler et al. 1991; Atluri & Regehr, 1996). Due to its buffering properties and the very tight influx–release coupling (Schmidt et al. 2013), moderate concentrations of EGTA will not significantly affect the peak Ca2+ transient at the active zone, while the decay of residual Ca2+ will be accelerated. Incubation with 50 μm EGTA-AM for 15 min reduced facilitation during a 50 Hz train in both genotypes (Fig. 9C, right) and showed a different time course than under control conditions (Fig. 9C, left). However, facilitation was still enhanced in the KV10.1 KO. Assuming the concentration of EGTA to be similar in terminals from both genotypes, this suggests first that more Ca2+ enters the terminal during each pulse and thus cannot be equally buffered by EGTA (see also Rozov et al. (2001), and secondly that this leads to faster build-up of residual Ca2+ and subsequently to enhanced facilitation even in the presence of EGTA.
Discussion
We analysed the consequences of loss of the KV10.1 K+ channel in the mouse cerebellum, providing for the first time information about the physiological function of the channel in the mammalian CNS. We found that loss of KV10.1 causes increased Ca2+ influx into presynaptic terminals. Furthermore, enhanced facilitation was observed at PF–PC synapses of KV10.1 KO mice. These effects were dependent on the stimulation frequency and number of pulses, which can be explained by the unique biophysical properties of KV10.1 activation. KV10.1 specifically regulates synaptic transmission at high-frequency AP trains and so acts at an intermediate between fast high-voltage activated (HVA) and slow low-voltage activated (LVA) channels.
KV10.1 in the CNS
Our data indicate an asymmetric distribution of KV10.1 between soma and terminal in GCs. Despite the high signal for KV10.1 mRNA in the GC somata (Ludwig et al. 2000; Saganich et al. 2001), no difference between WT and KO cells was found regarding excitability or AP shape at the soma (Fig. 2). This supports the described presynaptic localization of the channels (Gómez-Varela et al. 2010). Further biochemical and immunoelectron microscopy analysis showed an enrichment of KV10.1 in the presynaptic compartment (Fig. 3; see also Chuang et al. 2014). By selectively targeting ion channels to subcellular compartments, the cell can fine-tune the membrane properties to serve a specific purpose. In cerebellar basket cells and the calyx of Held for example, the asymmetric distribution of KV1 and KV3 channels ensures fast and reliable APs in the synaptic terminal, while the AP in the soma is slower and wider (Southan & Robertson, 1998; Ishikawa et al. 2003). The indicated presynaptic localization of KV10.1 suggests a role in regulating presynaptic excitability and/or transmitter release.
Synaptic transmission in KV10.1 KO mice
The frequency of mEPSCs was unaltered in KV10.1 KO mice, suggesting that basal release probability is not affected. Furthermore, one can assume that the resting Ca2+ is not elevated, as this would cause an increase of spontaneous vesicle release. The kinetics of single EPSCs were unchanged, as well as the dependence of the amplitude on the external Ca2+ concentration. In both genotypes, the relationship between [Ca2+]o and EPSC could be fitted with a sigmoidal function. Variance mean analysis has previously been used to characterize synaptic parameters of compound EPSCs at the PF–PC synapse (Sims & Hartell, 2005; Valera et al. 2012). This allowed us to determine pr at different [Ca2+]o in WT and KO cells. The values were in good agreement with previous studies (Sims & Hartell, 2005; Valera et al. 2012; Schmidt et al. 2013), and suggest that the initial pr is not altered in KV10.1 KO cells. These results indicate that Kv10.1 channels do not participate significantly in repolarizing the first AP of a train as the resulting changes in AP shape should have altered pr. As we observe an increase in KV10.1-mediated current amplitude in HEK cells upon repeated stimulation at 50 Hz, we think this indicates that the Kv10.1 channel only becomes active later in a train of APs (see below).
The average mEPSC amplitude was more than twice the quantal size yielded by variance mean analysis in both genotypes. However, one has to consider that mEPSCs from climbing fibre synapses will be included, as they cannot be distinguished from events originating in parallel fibres. Furthermore, only mEPSCs larger than a certain detection limit (∼10 pA) were measured. Small events, especially from distal parts of the dendritic arbour, can be lost in dendritic filtering and noise. Thus, our recordings systematically overestimated mEPSC amplitude.
Increased Ca2+ influx and facilitation in the absence of KV10.1
Interestingly, stimulation with a short burst of APs increased the Ca2+ influx to a greater extent in the KO than in the WT. This effect was stronger at 100 Hz than at 50 Hz (Fig. 5). The frequency- (and pulse number-) dependent increase in Ca2+ influx resulted in enhanced facilitation of the PF–PC synapse in the KV10.1 KO at input frequencies > 20 Hz, whose extent and time course was also frequency-dependent (Fig. 7). We can exclude significant additional fibre recruitment in the KO as the fibre volley amplitude was unchanged between WT and KO (see also Valera et al. 2012). We did not detect a broadening in the field potential recording corresponding to the increase in EPSC rise time (Fig. 7G), probably because the extracellular recording failed to resolve this quite small difference.
It is intriguing how efficiently the PF synapse can increase its output without depleting the RRP. This property indicates a constant, rapid replenishment of the RRP, which is a Ca2+-dependent process (Dittman & Regehr, 1998; Dittman et al. 2000; Crowley et al. 2007). We found increased RRP replenishment in the KO, which can explain that facilitation decays only slowly even during prolonged stimulation, with the KO values constantly well above the WT (Fig. 8). Previous studies have shown medium, heterogeneous pr at PF synapses (Valera et al. 2012; Schmidt et al. 2013). Valera et al. (2012) suggested that during tract stimulation high pr synapses respond to the first stimulus in a train whereas low pr synapses become subsequently recruited as intracellular Ca2+ rises. Our data support this notion, as the increased Ca2+ influx into KO synapses could promote the recruitment of low pr synapses during trains, further contributing to the activity-dependent increase in EPSC size. Even though a single synapse could suffer from rundown caused by increased transmitter release, the compound response would remain or increase in size as new synapses become recruited.
The finding that the rise time of the EPSC during a 50 Hz train also increased in the KO, but remained constant in the WT, corroborates our hypothesis of a use-dependent AP broadening in the KO, as AP shape, the time course of Ca2+ influx and EPSC rise time are tightly related (Borst & Sakmann, 1999; Bollmann & Sakmann, 2005). Our results are reminiscent of the use-dependent AP broadening observed in the hippocampal mossy fibre bouton. There, cumulative inactivation of KV1 channels during trains of stimuli causes a broadening of the presynaptic AP and subsequently facilitation of EPSCs (Geiger & Jonas, 2000; Alle et al. 2011). Use-dependent increase in Ca2+ influx has not been shown at the PF–PC synapse (Kreitzer & Regehr, 2000), indicating that either channels taking part in repolarization of the AP at this synapse do not inactivate or that cumulative inactivation is compensated for by other channels. To our knowledge, no functional study has been done on KV1 channels in PF–PC synapses, but mRNA for KV1.1, 1.3 and 1.5 was detected in GCs. Of these, KV1.3 shows cumulative inactivation, and, in the presence of regulatory subunits, also KV1.1 and 1.5 can show inactivation (Mathie et al. 2003). During repeated activity of the synapse, these channels will suffer increasing inactivation, while the depolarizations will accelerate the activation kinetics of KV10.1 (Fig. 1), allowing it to participate in AP repolarization specifically at trains of high-frequency activity and to ensure a constant AP waveform (Sabatini & Regehr, 1997) during the AP train.
Physiological implications of KV10.1 loss
The behavioural consequences of KV10.1 deletion are mild; KO mice show only slightly increased spontaneous locomotor activity and an increased sensitivity to Haloperidol-induced catalepsy, both of which have been related to alterations in neurotransmitter systems (Ufartes et al. 2013). Knock-down of KV10.1 in zebrafish has been shown to severely disrupt development of the CNS (Stengel et al. 2012). Note that comparing knock-down and knock-out approaches is not always straightforward, as has been shown recently by Yang et al. (2013). There, the phenotypes observed upon knocking down or knocking out the synaptic protein complexin were quite different, depending on the method employed.
The role of KV10.1 in mammalian development is unlikely to be as important as in zebrafish. KV10.1 expression starts after birth, and reaches a maximum only in adult animals. KV10.1 KO mice breed and develop normally, and show no micro- or macroanatomical abnormalities. No compensatory up-regulation of other ion channel genes could be detected in KO mice (Ufartes et al. 2013), and our mEPSC data further speak against alterations in synapse number or postsynaptic organization.
Our data assign a new role to KV10.1, different from both HVA and LVA channels. It acts as a specific regulator of high-frequency AP trains and prevents excessive neurotransmitter release from saturating and in the end even depleting the synapse and endanger faithful encoding of signals from GCs to Purkinje cells.
Acknowledgments
We thank G. Bethge, B. Heidrich, S. Schmidt and H. Widera for expert technical assistance.
Glossary
- AP
action potential
- Cares
residual calcium
- GC
granule cell
- HVA
high voltage activating
- LVA
low voltage activating
- (m)EPSC
(miniature) excitatory postsynaptic current
- OGB
Oregon Green BAPTA-1
- PC
Purkinje cell
- PF
parallel fibre
- pr
release probability
- RRP
readily releasable pool
- TBST
Tris-buffered saline–Tween 20
Additional information
Competing interest
The authors declare no competing financial interests.
Author contributions
L.S.M., T.S., H.S., J.E., W.S. and L.A.P. designed experiments. L.S.M., Z.F., M.E.R., H.S. and L.A.P. performed experiments. L.S.M., Z.F., H.S. and L.A.P. analysed data. L.S.M., H.S., J.E., A.B.F. and R.U. contributed analytical or experimental tools. L.S.M. wrote the manuscript with critical inputs from H.S., T.S., Z.F., A.B.F., R.U., J.E., W.S. and L.A.P. All authors approved the final version of the manuscript. The experiments were performed at the Max-Planck-Institute of Experimental Medicine, Göttingen, the Max-Planck-Institute of Biophysical Chemistry, Göttingen, and the Carl-Ludwig-Institute of Physiology, Leipzig.
Funding
This work was supported by an IMPRS fellowship to L.S.M., a GGNB Excellence Stipend to A.B.F., NIH grant 1R01DC013048-01 to M.E.R., the Japanese Society for the Promotion of Science, Core-to-Core Program A, Advance Research Networks to T.S., and DFG grant EI 342/4-1 to J.E. and H.S.
Authors' present addresses
Z. Farsi, Max Planck Institute for Biophysical Chemistry, 37077 Göttingen, Germany.
A. Barrantes-Freer, Department of Neuropathology, University Medical Center Göttingen, 37099 Göttingen, Germany.
M. E. Rubio, Department of Otolaryngology and Neurobiology, University of Pittsburgh Medical School, Pittsburgh, PA 15261, USA.
T. Sakaba, Doshisha Universtiy, Kyoto 619-0225, Japan.
References
- Adler E, Augustine G, Duffy S. Charlton M. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H, Kubota H. Geiger JRP. Sparse but highly efficient Kv3 outpace BKCa channels in action potential repolarization at hippocampal mossy fiber boutons. J Neurosci. 2011;31:8001–8012. doi: 10.1523/JNEUROSCI.0972-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP. Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Reim K. Sakaba T. Target-dependent feedforward inhibition mediated by short-term synaptic plasticity in the cerebellum. J Neurosci. 2010;30:8171–8179. doi: 10.1523/JNEUROSCI.0276-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H. Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Bollmann JH. Sakmann B. Control of synaptic strength and timing by the release-site Ca2+ signal. Nat Neurosci. 2005;8:426–434. doi: 10.1038/nn1417. [DOI] [PubMed] [Google Scholar]
- Bornschein G, Arendt O, Hallermann S, Brachtendorf S, Eilers J. Schmidt H. Paired-pulse facilitation at recurrent Purkinje neuron synapses is independent of calbindin and parvalbumin during high-frequency activation. J Physiol. 2013;591:3355–3370. doi: 10.1113/jphysiol.2013.254128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JGG. Sakmann B. Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Phil Trans R Soc B. 1999;354:347–355. doi: 10.1098/rstb.1999.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyken J, Grønborg M, Riedel D, Urlaub H, Jahn R. Chua JJ. Molecular profiling of synaptic vesicle docking sites reveals novel proteins but few differences between glutamatergic and GABAergic synapses. Neuron. 2013;78:285–297. doi: 10.1016/j.neuron.2013.02.027. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD. Regehr WG. Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J Neurosci. 2007;27:7888–7898. doi: 10.1523/JNEUROSCI.1064-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Aller MI, Sandu C, Veale EL, Alder FG, Sambi H, Mathie A. Wisden W. TASK-3 two-pore domain potassium channels enable sustained high-frequency firing in cerebellar granule neurons. J Neurosci. 2007;27:9329–9340. doi: 10.1523/JNEUROSCI.1427-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW. Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sánchez A, Rubio ME, Kohl T, Pardo LA. Stühmer W. Functional Kv10.1 channels localize to the inner nuclear membrane. PLoS ONE. 2011;6:e19257. doi: 10.1371/journal.pone.0019257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C-C, Jow G-M, Lin H-M, Weng Y-H, Hu J-H, Peng Y-J, Chiu Y-C, Chiu M-M. Jeng C-J. The punctate localization of rat Eag1 K+ channels is conferred by the proximal post-CNBHD region. BMC Neuroscience. 2014;15:23. doi: 10.1186/1471-2202-15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD. Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Carter AG. Regehr WG. Fast vesicle replenishment and rapid recovery from desensitization at a single synaptic release site. J Neurosci. 2007;27:5448–5460. doi: 10.1523/JNEUROSCI.1186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo E, Filippi GD, Rossi P. Taglietti V. Ionic mechanism of electroresponsiveness in cerebellar granule cells implicates the action of a persistent sodium current. J Neurophysiol. 1998;80:493–503. doi: 10.1152/jn.1998.80.2.493. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC. Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS. Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douyard J, Shen L, Huganir RL. Rubio ME. Differential neuronal and glial expression of GluR1 AMPA receptor subunit and the scaffolding proteins SAP97 and 4.1 N during rat cerebellar development. J Comp Neurol. 2007;502:141–156. doi: 10.1002/cne.21294. [DOI] [PubMed] [Google Scholar]
- Felmy F, Neher E. Schneggenburger R. Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron. 2003;37:801–811. doi: 10.1016/s0896-6273(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G, Südhof TC. Jahn R. A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature. 1991;349:79–81. doi: 10.1038/349079a0. [DOI] [PubMed] [Google Scholar]
- Foster KA, Crowley JJ. Regehr WG. The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci. 2005;25:11655–11665. doi: 10.1523/JNEUROSCI.4029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Ferreiro RE, Kerschensteiner D, Major F, Monje F, Stühmer W. Pardo LA. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J Gen Physiol. 2004;124:301–317. doi: 10.1085/jgp.200409041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JRP. Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Gómez-Varela D, Kohl T, Schmidt M, Rubio ME, Kawabe H, Nehring RB, Schäfer S, Stühmer W. Pardo LA. Characterization of Eag1 channel lateral mobility in rat hippocampal cultures by single-particle-tracking with quantum dots. PLoS ONE. 2010;5:e8858. doi: 10.1371/journal.pone.0008858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerlein B, Weseloh RM, Mello de Queiroz F, Knotgen H, Sanchez A, Rubio ME, Martin S, Schliephacke T, Jenke M, Heinz Joachim R, Stuhmer W. Pardo LA. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer. 2006;5:41. doi: 10.1186/1476-4598-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. Trussell LO. KCNQ5 channels control resting properties and release probability of a synapse. Nat Neurosci. 2011;14:840–847. doi: 10.1038/nn.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Popoff MR, Benfenati F. Poulain B. Fast changes in the functional status of release sites during short-term plasticity: involvement of a frequency-dependent bypass of Rac at Aplysia synapses. J Physiol. 2007;583:983–1004. doi: 10.1113/jphysiol.2007.139899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Popoff MR, Kojima H, Doussau F. Poulain B. Rac GTPase plays an essential role in exocytosis by controlling the fusion competence of release sites. J Neurosci. 2002;22:7968–7981. doi: 10.1523/JNEUROSCI.22-18-07968.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont J-L, Du G, Frohman MA, Bader M-F. Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci USA. 2001;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li W-B, Iwasaki S. Takahashi T. Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci. 2003;23:10445–10453. doi: 10.1523/JNEUROSCI.23-32-10445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isope P. Barbour B. Properties of unitary granule cell→Purkinje cell synapses in adult rat cerebellar slices. J Neurosci. 2002;22:9668–9678. doi: 10.1523/JNEUROSCI.22-22-09668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Llano I. Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Regehr WG. Modulation of transmission during trains at a cerebellar synapse. J Neurosci. 2000;20:1348–1357. doi: 10.1523/JNEUROSCI.20-04-01348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig J, Terlau H, Wunder F, Brüggemann A, Pardo LA, Marquardt A, Stühmer W. Pongs O. Functional expression of a rat homologue of the voltage gated either á go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 1994;13:4451–4458. doi: 10.1002/j.1460-2075.1994.tb06767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig J, Weseloh R, Karschin C, Liu Q, Netzer R, Engeland B, Stansfeld C. Pongs O. Cloning and functional expression of rat eag2, a new member of the Ether-à-go-go family of potassium channels and comparison of its distribution with that of eag1. Mol Cell Neurosci. 2000;16:59–70. doi: 10.1006/mcne.2000.0851. [DOI] [PubMed] [Google Scholar]
- Martin S, Lino de Oliveira C, Mello de Queiroz F, Pardo LA, Stühmer W. Del Bel E. Eag1 potassium channel immunohistochemistry in the CNS of adult rat and selected regions of human brain. Neuroscience. 2008;155:833–844. doi: 10.1016/j.neuroscience.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Mathie A, Clarke C, Ranatunga K. Veale E. What are the roles of the many different types of potassium channel expressed in cerebellar granule cells? Cerebellum. 2003;2:11–25. doi: 10.1080/14734220310015593. [DOI] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH. Knöpfel T. Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Neher E. Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL. Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Naraghi M. Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA. Clements JD. Postsynaptic expression of long-term potentiation in the rat dentate gyrus demonstrated by variance-mean analysis. J Physiol. 1999;518:121–130. doi: 10.1111/j.1469-7793.1999.0121r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A. Häusser M. Compartmental models of rat cerebellar Purkinje cells based on simultaneous somatic and dendritic patch-clamp recordings. J Physiol. 2001;535:445–472. doi: 10.1111/j.1469-7793.2001.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B. Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL. Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, Machado E. Rudy B. Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci. 2001;21:4609–4624. doi: 10.1523/JNEUROSCI.21-13-04609.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Brachtendorf S, Arendt O, Hallermann S, Ishiyama S, Bornschein G, Gall D, Schiffmann SN, Heckmann M. Eilers J. Nanodomain coupling at an excitatory cortical synapse. Curr Biol. 2013;23:244–249. doi: 10.1016/j.cub.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC. Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T. Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Meth. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A. Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RE. Hartell NA. Differences in transmission properties and susceptibility to long-term depression reveal functional specialization of ascending axon and parallel fiber synapses to Purkinje cells. J Neurosci. 2005;25:3246–3257. doi: 10.1523/JNEUROSCI.0073-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan AP. Robertson B. Patch-clamp recordings from cerebellar basket cell bodies and their presynaptic terminals reveal an asymmetric distribution of voltage-gated potassium channels. J Neurosci. 1998;18:948–955. doi: 10.1523/JNEUROSCI.18-03-00948.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel R, Rivera-Milla E, Sahoo N, Ebert C, Bollig F, Heinemann SH, Schonherr R. Englert C. Kcnh1 voltage-gated potassium channels are essential for early zebrafish development. J Biol Chem. 2012;287:35565–35575. doi: 10.1074/jbc.M112.363978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kovalchuk Y. Attwell D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J Neurosci. 1995;15:5693–5702. doi: 10.1523/JNEUROSCI.15-08-05693.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Ludwig J, Steffan R, Pongs O, Stühmer W. Heinemann S. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflugers Arch. 1996;432:301–312. doi: 10.1007/s004240050137. [DOI] [PubMed] [Google Scholar]
- Ufartes R, Schneider T, Mortensen LS, de Juan Romero C, Hentrich K, Knoetgen H, Beilinson V, Moebius W, Tarabykin V, Alves F, Pardo LA, Rawlins JNP. Stuehmer W. Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum Mol Genet. 2013;22:2247–2262. doi: 10.1093/hmg/ddt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera AM, Doussau F, Poulain B, Barbour B. Isope P. Adaptation of granule cell to Purkinje cell synapses to high-frequency transmission. J Neurosci. 2012;32:3267–3280. doi: 10.1523/JNEUROSCI.3175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke JW. Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Randall A. Tsien R. Changes in action potential duration alter reliance of excitatory synaptic transmission on multiple types of Ca2+ channels in rat hippocampus. J Neurosci. 1996;16:2226–2237. doi: 10.1523/JNEUROSCI.16-07-02226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B, Haugland FN. Liu AX. Potassium currents in Drosophila: different components affected by mutations of two genes. Science. 1983;220:1076–1078. doi: 10.1126/science.6302847. [DOI] [PubMed] [Google Scholar]
- Yang X, Cao P. Südhof TC. Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proc Natl Acad Sci USA. 2013;110:20777–20782. doi: 10.1073/pnas.1321367110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]