

Abstract
The composition of brain extracellular fluid is shaped by a continuous exchange of substances between the cerebrospinal fluid (CSF) and interstitial fluid. The CSF is known to contain a wide range of endogenous neuromodulatory substances, but their collective influence on neuronal activity has been poorly investigated. We show here that replacing artificial CSF (aCSF), routinely used for perfusion of brain slices in vitro, with human CSF (hCSF) powerfully boosts spontaneous firing of CA1, CA3 and layer 5 pyramidal neurons in the rat brain slice. CA1 pyramidal neurons in hCSF display lowered firing thresholds, more depolarized resting membrane potentials and reduced input resistance, mimicking properties of pyramidal neurons recorded in vivo. The increased excitability of CA1 pyramidal neurons was completely occluded by intracellular application of GTPγS, suggesting that endogenous neuromodulators in hCSF act on G-protein coupled receptors to enhance excitability. We found no increase in spontaneous inhibitory synaptic transmission by hCSF, indicating a differential effect on glutamatergic and GABAergic neurons. Our findings highlight a previously unknown function of the CSF in promoting spontaneous excitatory activity, and may help to explain differences observed in the activity of pyramidal neurons recorded in vivo and in vitro.
Key points.
The cerebrospinal fluid contains numerous neuromodulators at ambient levels but whether, and how, they affect the activity of central neurons is unknown.
This study provides experimental evidence that human cerebrospinal fluid (hCSF) increases the excitability of hippocampal and neocortical pyramidal neurons.
Hippocampal CA1 pyramidal neurons in hCSF displayed lowered firing thresholds, depolarized resting membrane potentials and reduced input resistance, mimicking properties of pyramidal neurons recorded in vivo.
The excitability-increasing effect of hCSF on CA1 pyramidal neurons was entirely occluded by intracellular application of GTPγS, suggesting that neuromodulatory effects were mediated by G-protein coupled receptors.
These results indicate that the CSF promotes spontaneous excitatory neuronal activity, and may help to explain observed differences in the activity of pyramidal neurons recorded in vivo and in vitro.
Introduction
Central neurons are surrounded by an extracellular fluid whose composition results from the continuous mixing of cerebrospinal fluid (CSF), occupying the ventricles and subarachnoid space, with interstitial fluid of the parenchyma. In the brain, communication between these fluids occurs unrestrictedly (Brightman & Palay, 1963; Brightman & Reese, 1969; Smith et al. 2004) along local concentration gradients. The CSF is known to contain many different neuromodulatory substances (or neuromodulators) (Jackson, 1980; Post et al. 1988) that appear to derive from within the CNS. This raises the question of whether, and how, these substances influence the activity of central neurons in vivo.
Pyramidal neurons in vivo are bombarded with spontaneous excitatory synaptic activity and fire spontaneously at high rates (Steriade et al. 2001; Destexhe et al. 2003). In contrast, pyramidal neurons in brain slices (in vitro) are often quiescent in the sense that they rarely fire spontaneously and show much lower levels of excitatory synaptic activity. Moreover, pyramidal neurons in vivo are more depolarized, show prominent membrane potential (Vm) fluctuations and have lower input resistance (Rin) (Steriade et al. 2001; Destexhe et al. 2003). This discrepancy is typically attributed to the higher levels of neuronal connectivity existing in vivo. The relative quiescence of pyramidal neurons in brain slices could, however, also result from the fact that artificial CSF (aCSF) is routinely used in these recordings. aCSF lacks the organic content of physiological CSF (including neuromodulators), and contains simply electrolytes, glucose and water.
An intriguing finding suggesting that neuromodulators distributed in CSF could act to regulate neuronal activity is the presence of CSF-contacting neurons in vertebrate brains (Cupedo & de Weerd, 1980; Buma et al. 1989; Calle et al. 2005). Among these are neurons that send varicose neuromodulatory fibres that terminate directly in ventricular or subarachnoidal CSF (Parent, 1981; Vigh et al. 2004). This suggests that neuromodulators are actively released into the CSF, as diffusible signals, to influence the activity of recipient neurons. However, whether endogenous neuromodulators present in CSF can in fact influence neuronal activity has remained largely unknown, as direct physiological evidence has been scarce.
Here we directly examined the influence of human CSF (hCSF) on the activity of pyramidal neurons in brain slices, using a matched aCSF as control. We show that switching from aCSF to hCSF powerfully boosts spontaneous firing, and induces in vivo-like properties, in pyramidal neurons.
Methods
Artificial and human cerebrospinal fluid
hCSF was obtained via lumbar puncture by neurologists at the Sahlgrenska University Hospital in Gothenburg, Sweden. Samples were routinely centrifuged (2000 r.p.m., 10 min) within 30 min after lumbar puncture to remove cells, then transported to the lab on dry ice and stored at –80°C. The majority of samples came from patients diagnosed with normal pressure hydrocephalus (NPH), while a lesser amount was contributed by healthy volunteers. Patient samples were obtained from the clinical routine at the Department of Clinical Neuroscience, Sahlgrenska University Hospital. hCSF from healthy male and female volunteers was obtained specifically for the purpose of this study, with permission from the local ethical committee at the University of Gothenburg. hCSF samples were acquired from subjects after written informed consent and the clinical procedures conformed to the principles of the declaration of Helsinki. hCSF from patients and healthy volunteers had the same effects on pyramidal neurons and therefore data from these groups were combined. hCSF pools were made from 5–30 de-identified samples. The total concentration of electrolytes and glucose, as well as pH and osmolality, was measured in every pool. A matched aCSF was then designed based on measured variables in hCSF and used as a control (see Table 1). pH was measured in aCSF and hCSF while bubbling with a gas mixture of 95% O2 and 5% CO2. We estimated ionized Ca2+ and Mg2+ in hCSF to account for between 75 and 90% of measured total concentration (Goldstein & Massry, 1980; Joborn et al. 1991) in all experiments. Electrolyte and glucose concentrations were determined by board-certified laboratory technicians at the Clinical Chemistry Laboratory at Sahlgrenska University Hospital. Laboratory analyses were performed on Cobas instruments (Roche Diagnostics, Indianapolis, IN, USA) using accredited methods with inter-assay coefficients of variation (CVs) below 2%. Na+, K+ and Cl− were measured using ion selective electrodes. Total Ca2+ and Mg2+ concentrations were measured using colorimetric o-cresolphthalein and chlorophosphonazo III methods, respectively. Glucose concentration was determined using the hexokinase method. Only hCSF pools showing normal electrolyte (Na+, K+, Cl−, Ca2+, Mg2+) and glucose concentrations, pH, osmolality and colouring were used. Prior to experimentation, frozen hCSF was thawed to room temperature in a bowl of warm (37°C) water. Both the aCSF and the hCSF were bubbled with a gas mixture of 95% O2 and 5% CO2 during, and prior to, recordings. The amount of hCSF used in individual experiments ranged from 10 to 15 ml.
Table 1.
Measured variables in hCSF and aCSF
| pH | Osmolality (mosmol kg–1) | Na+ | K+ | Cl− | Ca2+ | Mg2+ | Glucose | |
|---|---|---|---|---|---|---|---|---|
| hCSF1 | 7.35 | 289 | 147.6 | 2.79 | 125.1 | 1.18 | 1.14 | 3.66 |
| aCSF1 | 7.39 ± 0.02 | 285 ± 0.7 | 147.3 ± 0.94 | 2.77 ± 0.03 | 124.3 ± 0.52 | 1.17 ± 0.02 | 1.12 ± 0.01 | 3.69 ± 0.03 |
| hCSF2 | 7.35 | 275 | 138 | 2.6 | 116 | 1.1 | 1.0 | 3.68 |
| aCSF2 | 7.41 ± 0.02 | 280 ± 0.9 | 138.3 ± 0.78 | 2.59 ± 0.012 | 115.1 ± 0.31 | 1.09 ± 0.008 | 1.01 ± 0.007 | 3.65 ± 0.02 |
Total concentrations of electrolytes and glucose are given in mmol l–1. hCSF1, CSF pool from healthy volunteers (n = 10 individuals); aCSF1, aCSF designed to match the healthy CSF pool (measurements were taken from aCSF solutions prepared from scratch on different experimental days) (n = 5); hCSF2, CSF pool from NPH patients (n = 10 individuals); aCSF2, aCSF designed to match the NPH pool (measurements were taken from aCSF solutions prepared from scratch on different experimental days) (n = 5).
Slice preparation
Experiments were carried out on acute hippocampal and neocortical brain slices from Wistar rats 10–25 days old (P10–25). Some experiments were performed on hippocampal slices from young adult rats 40–78 days old (P40–P78). All experiments were conducted in accordance with regulations of the Swedish Animal Welfare law and approved by the local ethical committee for animal research at the University of Gothenburg. Rats were anaesthetized via inhalation of isoflurane (Abbott, Chicago, IL, USA) and, following decapitation, the brain was removed and put in ice-cold (0–3°C) solution containing (in mm): 220 glycerol, 2.5 KCl, 1.2 CaCl2, 7 MgCl2, 26 NaHCO3, 1.2 NaH2PO4 and 11 d-glucose. Sagittal slices (300–400 μm) from the dorsal hippocampus and parietal cortex were cut using a vibratome (HM650V; Microm, Bicester, UK) and stored in aCSF containing (in mm): 129 NaCl, 3 KCl, 2 CaCl2, 4 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 0.5 ascorbic acid, 3 myo-inositol, 4 dl-lactic acid and 10 d-glucose. During slicing and storage, solutions were bubbled with a mixture of 95% O2 and 5% CO2. Following 1–5 h of storage at room temperature (22–25°C), a brain slice was transferred to a recording chamber perfused with aCSF at a rate of 3 ml min–1. The aCSF used during recordings contained 26 mm NaHCO3 and 1.2 mm NaH2PO4 in all experiments, whereas concentrations of Na+, K+, Cl−, Ca2+, Mg2+ and glucose varied with measured concentrations in different hCSF pools (see Table 1). All experiments were performed at room temperature.
Extracellular field recordings
Field recordings were performed in the hippocampal stratum radiatum using a borosilicate glass micropipette filled with 1 m NaCl (resistance 3–4 MΩ) mounted on a AgCl-coated electrode. Stimulation was delivered via a tungsten stimulation electrode (resistance ∼ 0.3–0.5 MΩ). Biphasic constant current pulses (200 + 200 μs, 15–40 μA) were applied to Schaffer collaterals/commissural fibres at a frequency of 0.2 Hz. Field excitatory postsynaptic potential (fEPSP) magnitude was measured from the first 0.8 ms (or 0.6 ms, see Results) of the initial slope. Ten paired pulses were applied (paired pulse interval 50 ms) following 15 min recording periods in aCSF and hCSF. Recordings were sampled at 10 kHz and filtered at 3 kHz, using an EPC-9 amplifier and Patchmaster software (HEKA Elektronik, Lambrecht/Pfalz, Germany). In field recordings, the aCSF and hCSF were supplemented with 100 μm picrotoxin (Sigma-Aldrich, St. Louis, MO, USA) to block GABAA receptor-mediated inhibition.
Whole-cell recordings
Whole-cell recordings from pyramidal neurons were acquired using a borosilicate glass micropipette with a resistance of 4–6 MΩ. Visual identification of neurons was obtained using differential interference contrast microscopy (Nikon E600FN) together with a CCD camera (Sony XC-73CE). Data were acquired with a sampling frequency of 10 kHz, and filtered at 3 kHz using an EPC-9 amplifier (HEKA Elektronik). After break-in, neurons were allowed to rest for at least 5 min before any current or voltage was applied. Series resistance was continuously monitored with a 20 ms/10 mV hyperpolarizing pulse and was not allowed to fluctuate more than 20% within recordings. The liquid junction potential was not corrected for. In voltage-clamp recordings, the intracellular solution contained (in mm): 130 caesium methanesulfonate, 2 NaCl, 20 Hepes, 0.2 EGTA, 5 QX-314, 4 Mg-ATP and 0.4 GTP (pH 7.2, 290 mosmol kg−1). Spontaneous and miniature excitatory postsynaptic currents (sEPSCs and mEPSCs) were recorded at –70 mV in the presence of 100 μm picrotoxin. In mEPSC recordings, the aCSF and hCSF were further supplemented with 0.5 μm TTX (Sigma-Aldrich). Spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded at 0 mV, close to the reversal potential of NMDA- and AMPA receptor-mediated synaptic currents, in the absence of NMDA/AMPA receptor blockers and picrotoxin. In current-clamp recordings, the intracellular solution contained (in mm): 127 K+-gluconate, 8 KCl, 10 Hepes, 15 phosphocreatine, 4 Mg-ATP and 0.3 Na-GTP (pH 7.2, 295 mosmol Kg−1). In current-clamp recordings where G-protein dependence of hCSF effects were evaluated, Na-GTP was replaced with 0.3 mm GTPγS (Sigma-Aldrich). Rin was calculated from the slope of the I–V relationship in response to step current injections (300 ms) ranging from 0 to –100 pA. Action potential (AP) threshold was defined as the voltage at which dV/dt exceeded 20 mV ms–1. AP amplitude was measured as the voltage difference between threshold and peak of overshoot. AP threshold and amplitude, Vmax and Vmin were extracted by plotting the time derivative of membrane voltage against membrane potential (phase plane plot). In frequency–current plots, AP firing frequency was calculated from the total number of APs produced during depolarizing current injections (300 ms) ranging from 0 to 390 pA.
Membrane dialysis
Two 50 ml batches of pooled hCSF (pH 7.33 and 7.36, 270 and 290 mosmol kg–1) were each divided into a 25 ml control aliquot and a 25 ml aliquot used for dialysis. In two separate experiments, hCSF was dialysed against an aCSF containing (in mm): 124 NaCl, 3 KCl, 0.87 CaCl2, 0.8 MgCl2, 26 NaHCO3, 1.2 NaH2PO4 and 4 d-glucose (pH 7.4, 280 mosmol kg–1). In each experiment, 25 ml hCSF was dialysed against 2×2 litres of aCSF over 10 h at room temperature. hCSF samples were bubbled to pH 7.33 and 7.36 (with a gas mixture of 95% O2, 5% CO2), respectively, prior to the start of dialysis. The aCSF was kept at pH 7.4 (by bubbling with a gas mixture of 95% O2 and 5% CO2) throughout the experiment. Using a cellulose membrane with a 6–8 kDa molecular weight cut-off (Spectra/Por, Spectrum Laboratories, Rancho Dominguez, CA, USA), constituents ≤ 8 kDa were removed from the hCSF. The aliquot acting as control was held at room temperature throughout the procedure and then frozen together with the dialysed hCSF at –80°C pending analysis. It was used as a matched control in experiments where the effect of dialysed hCSF was tested.
Human serum
Six healthy members of the research group (4 male, 2 female) volunteered to have 20 ml of venous blood drawn to examine the effects of human serum on hippocampal pyramidal neurons. Blood samples were collected at the Sahlgrenska University Hospital in Molndal, Sweden, and serum was isolated according to standard laboratory protocols. Samples were then pooled, transported to the lab on dry ice and subsequently stored at –80°C. Prior to experimentation, the serum was thawed to room temperature in a bowl of warm (37°C) water. A 1 ml aliquot human serum was added to 99 ml of aCSF, yielding a protein concentration similar to that of hCSF (Burtis et al. 2006), which was otherwise identical in composition to control.
Data analysis
Extracellular field- and whole-cell current-clamp recordings were analysed using the computer software Igor Pro (WaveMetrics, Lake Oswego, OR, USA). Field recordings were binned per minute so that an average wave (n = 12) was obtained. Spontaneous and miniature postsynaptic currents were analysed using the software MiniAnalysis (Synaptosoft, Decatur, GA, USA). The amplitude threshold for detection of postsynaptic currents was 4 pA. Statistical significance was determined using Student's paired t test. For unmatched comparisons statistical significance was determined using Student's unpaired t test. All data are presented as mean ± SEM.
Results
hCSF increases spontaneous AP firing in pyramidal neurons
To examine whether hCSF affects neuronal excitability, we performed current-clamp recordings from CA1 pyramidal neurons of P20–25 rats. CA1 pyramidal neurons displaying spontaneous firing ≥ 0.2 Hz (approximately 50% of cells examined) at resting membrane potential (Vrest) were chosen to evaluate the effect of hCSF on spontaneous firing frequency. hCSF profoundly increased the frequency of spontaneous APs (531 ± 154%, n = 9, P = 0.00009, Student's paired t test, Fig. 1A and B) and depolarized Vrest (2.9 ± 0.6 mV, n = 9, P = 0.0015, Student's paired t test, Fig. 1A and C). Control experiments in aCSF showed no depolarization of Vrest over the same time period (0.2 ± 1.3 mV, n = 6, P = 0.87, Student's paired t test). We also tested the effect of hCSF on spontaneous firing in parietal neocortical layer 5 pyramidal neurons. Similar to the effect in CA1 pyramidal neurons, hCSF produced a more than fivefold increase in spontaneous AP frequency in these neurons (586.4 ± 261.5%, n = 5, P = 0.003, Student's paired t test), which was largely reversed after 10 min of washout (154.2 ± 5.4%, n = 3).
Figure 1. hCSF increases spontaneous action potential firing in CA1 pyramidal neurons.
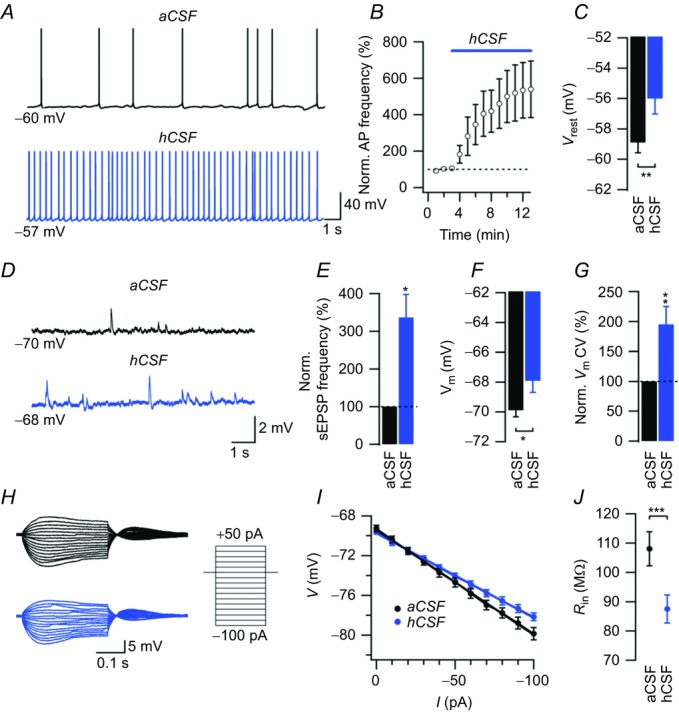
A, example sweeps of spontaneous APs recorded in aCSF and hCSF at Vrest. Note the depolarization of Vrest in hCSF. B, summary graph showing the effect of hCSF on normalized spontaneous AP frequency. C, summary bar graph showing Vrest in aCSF and hCSF. D, example sweeps of current-clamp recordings in aCSF and hCSF when starting at a Vm of –70 mV. E, summary bar graph showing the effect of hCSF on normalized sEPSP frequency. F, summary bar graph of Vm in aCSF and hCSF from recordings starting at –70 mV. G, the variability of voltage recorded at –70 mV in aCSF and hCSF. H, example sweeps of voltage responses to a series of current pulses (schematically illustrated to the right) in aCSF (black sweeps) and in hCSF (blue sweeps) from a Vm of –70 mV. I, summary graph showing the hyperpolarizing voltage response (from a Vm of –70 mV) to negative current injections in aCSF (black trace) and in hCSF (blue trace). J, summary graph of Rin in aCSF and hCSF. Rin was calculated from the slope of the I–V relationship (cf. I) in each experiment. Error bars represent SEM. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
In current-clamp recordings at –70 mV, hCSF increased the frequency of spontaneous excitatory postsynaptic potentials (sEPSPs) in CA1 pyramidal neurons (336.4 ± 61.4%, n = 4, P = 0.02, Student's paired t test, Fig. 1D and E), indicating that hCSF increased spontaneous firing in CA3 pyramidal neurons as well. The increased frequency of spontaneous excitatory input further increased the membrane potential CV to 195.2 ± 29.9% (n = 4, P = 0.04, Student's paired t test, Fig. 1G). We also found that Rin decreased in hCSF by approximately 20% (from 108.1 ± 5.8 to 87.5 ± 4.8 MΩ, n = 13, P = 0.00004, Student's paired t test, Fig. 1H and J). Control experiments in aCSF showed no significant change in Rin over the same time period (126.8 ± 24 vs. 124.1 ± 31.7 MΩ, n = 6, P = 0.78, Student's paired t test).
hCSF lowers the AP threshold in CA1 pyramidal neurons
We next examined the effect of hCSF on AP properties in current-clamp mode. hCSF shifted the threshold for AP firing towards more hyperpolarized potentials (from –50.7 ± 1.2 to –55.6 ± 1.4 mV, n = 13, P = 0.00005, Student's paired t test, Fig. 2A and C), probably contributing largely to the increased spontaneous excitatory activity observed in hCSF. Control experiments in aCSF showed no significant change in AP threshold over the same time period in whole-cell mode (–51 ± 1.5 vs. –51.6 ± 1.8 mV, n = 6, P = 0.7, Student's paired t test). We also found a decrease in AP amplitude in hCSF (106.8 ± 2.5 vs. 102.7 ± 2.1 mV, n = 13, P = 0.0001, Student's paired t test, Fig. 2D). Control experiments in aCSF showed no significant change in AP amplitude over the same time period in whole-cell mode (102.6 ± 5.3 vs. 100.7 ± 3.3 mV, n = 6, P = 0.44, Student's paired t test). The maximal AP upstroke velocity was decreased in hCSF, as indicated by a lower Vmax (243.04 ± 11.5 vs. 223.02 ± 8.5 mV ms–1, n = 13, P = 0.0011, Student's paired t test, Fig. 2B and E), whereas maximal downstroke velocity (Vmin) was increased (–46.1 ± 1.4 vs. –48.6 ± 1.4 mV ms–1, n = 13, P = 0.0012, Student's paired t test, Fig. 2B and F). Control experiments in aCSF showed no significant changes in Vmax (236.7 ± 19.5 vs. 225 ± 13.2 mV ms–1, n = 6, P = 0.31, Student's paired t test) or Vmin (–47.2 ± 2.5 vs. –50.3 ± 1.8 mV ms–1, n = 6, P = 0.4, Student's paired t test) over the same time period in whole-cell mode.
Figure 2. hCSF lowers the threshold for action potential firing in CA1 pyramidal neurons.
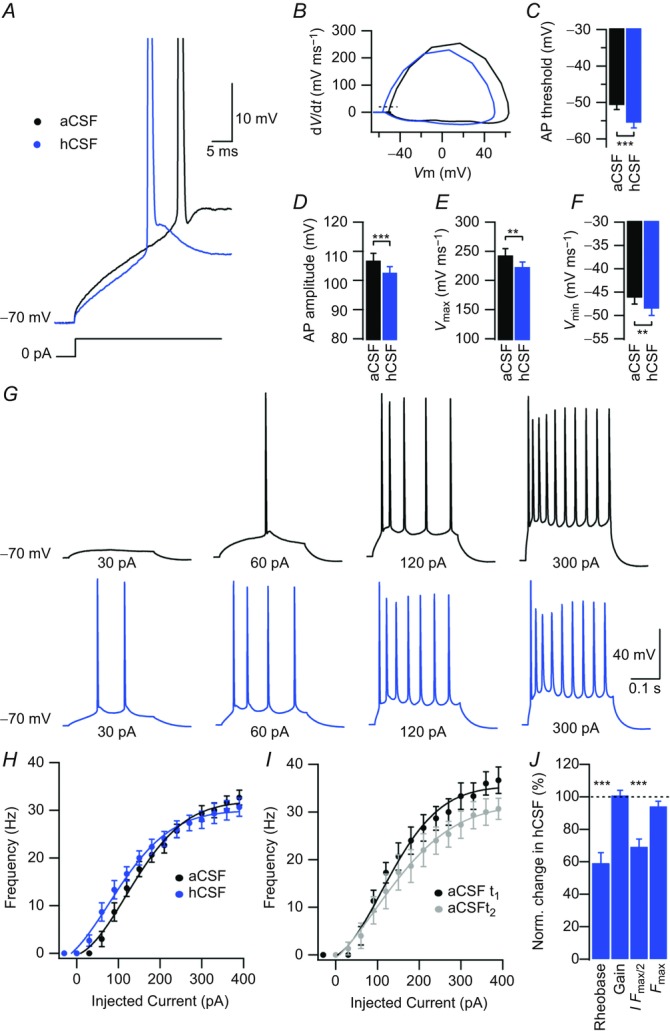
A, example sweeps of APs in aCSF and hCSF evoked by a 300 ms depolarizing current pulse from a Vm of –70 mV. Sweeps are expanded in time and show first APs evoked by depolarizing current injection. B, phase plane plot of the APs shown in A, with aCSF in black and hCSF in blue. Dashed line shows threshold as defined by a dV/dt of 20 mV ms–1. C, summary bar graph showing the AP threshold in aCSF and hCSF. D, summary bar graph showing AP amplitude in aCSF and hCSF. E, summary bar graph showing maximal AP upstroke velocity in aCSF and hCSF. F, summary bar graph showing maximal AP downstroke velocity in aCSF and hCSF. G, example sweeps showing AP output in aCSF (black) and hCSF (blue) in response to depolarizing current pulses of increasing magnitude as indicated. H, frequency–current relationship in aCSF (black) and hCSF (blue). I, control experiment showing rundown of frequency–current relationship over time; t1 (black) corresponds to time point of aCSF recording (H, black) and t2 (grey) to time point of hCSF recording (H, blue). J, summary bar graph showing normalized change in rheobase, gain, I Fmax/2 and Fmax in hCSF. Data were extracted from frequency–current experiments summarized in H. Error bars represent SEM. ∗∗P < 0.01; ∗∗∗P < 0.001.
Frequency–current (F–I) curves were constructed for each experiment in aCSF and hCSF, where hCSF seemed to consistently produce a left shift (Fig. 2H). We also noted a rundown of firing frequency over time in whole-cell mode (n = 6, Fig. 2I). F–I curves showed that hCSF decreased the injected current required to evoke a single AP (rheobase) (59.2 ± 6.4%, n = 13, P = 0.0002, Student's paired t test, Fig. 2J), as well as the current required to reach half maximal firing frequency (I Fmax/2) (69.2 ± 4.8%, n = 13, P = 0.00013, Student's paired t test, Fig. 2J), without affecting slope (gain).
The excitability-increasing effect of hCSF on CA1 pyramidal neurons is mediated by G-protein coupled receptors
To investigate whether G-protein-dependent mechanisms were involved in the effects of hCSF on CA1 pyramidal neurons, we used the non-hydrolysable GTP analogue GTPγS to render G-protein signalling constitutively active. The net result of intracellular application of GTPγS (0.3 mm) was a decreased excitability at baseline, as indicated by a more hyperpolarized Vrest (–63.7 ± 1.1 vs. –58.9 ± 0.7 mV, df = 16, P = 0.002, Student's unpaired t test, Fig. 3C aCSF, cf. Fig. 1C aCSF) and higher I Fmax/2 (203.4 ± 14.5 vs. 136.7 ± 8.1 pA, df = 20, P = 0.0003, Student's unpaired t test, Fig. 3G aCSF, cf. Fig. 2H aCSF). Accordingly, very few CA1 pyramidal neurons displayed spontaneous firing at Vrest. CA1 pyramidal neurons also showed a lower Rin in the presence of GTPγS (87.4 ± 7.3 vs. 108.1 ± 5.8 MΩ, n = 16, P = 0.04, Student's paired t test, Fig. 3D aCSF, cf. Fig. 1J aCSF).
Figure 3. Intracellularly applied GTPγS occludes the excitability-increasing effect of hCSF on CA1 pyramidal neurons.
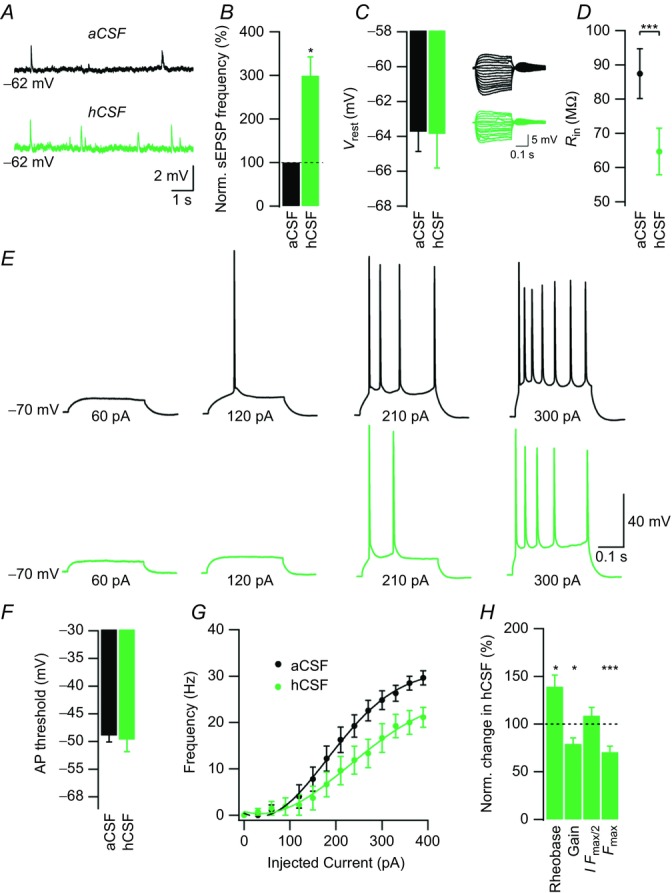
A, example sweeps of current-clamp recordings in aCSF and hCSF at Vrest. B, summary bar graph showing normalized effect of hCSF on sEPSP frequency. C, summary bar graph showing Vrest in aCSF and hCSF. D, example sweeps of voltage responses to a series of current pulses (cf. Fig. 1H) in aCSF (black sweeps) and hCSF (green sweeps) from a Vm of –70 mV. Summary graph showing Rin in aCSF and hCSF. E, example sweeps showing AP output in aCSF (black) and hCSF (green) in response to depolarizing current pulses of increasing magnitude as indicated. F, summary bar graph showing the AP threshold in aCSF and hCSF. G, frequency–current relationship in aCSF (black) and hCSF (green). H, summary bar graph showing normalized change in rheobase, gain, I Fmax/2 and Fmax in hCSF. Data were extracted from frequency–current experiments summarized in G. Error bars represent SEM. ∗P < 0.05; ∗∗∗P < 0.001.
In the presence of intracellularly applied GTPγS, hCSF increased the frequency of sEPSPs (313.9 ± 66.1%, n = 9, P = 0.036, Student's paired t test, Fig. 3A and B) but had no effect on the Vrest of CA1 pyramidal neurons (–63.7 ± 1.1 vs. –63.9 ± 2 mV, n = 9, P = 0.91, Student's paired t test, Fig. 3A and C), indicating that the same effects of hCSF on presynaptic neurons were present in these experiments. hCSF decreased Rin of CA1 pyramidal neurons to a similar extent in the presence of GTPγS (87.4 ± 7.3 vs. 64.7 ± 6.8 MΩ, n = 9, P = 0.0001, Student's paired t test, Fig. 3D), suggesting that the net change in Rin was largely independent of G-protein. Somatic current injections showed that the slope of the F–I curve (gain) was decreased when hCSF was applied in the presence of GTPγS (79.3 ± 6.3%, n = 9, P = 0.017, Student's paired t test, Fig. 3G and H), indicating a decreased excitability of CA1 neurons. hCSF had no effect on the AP threshold (–49 ± 1.1 vs. –49.7 ± 2.2 mV, n = 9, P = 0.56, Student's paired t test, Fig. 3F) but increased rheobase in the presence of intracellular GTPγS (139.1 ± 12.3%, n = 9, P = 0.01, Student's paired t test, Fig. 3H). Finally, we found a significant decrease in maximal firing frequency (Fmax) in hCSF (70.5 ± 6.3%, n = 9, P = 0.001, Student's paired t test, Fig. 3G and H).
hCSF differentially affects excitatory and inhibitory spontaneous synaptic transmission onto CA1 pyramidal neurons
We next studied the effect of hCSF on glutamatergic synaptic transmission by recording AMPA receptor-mediated mEPSCs from CA1 pyramidal neurons in the presence of 0.5 μm TTX. hCSF had no effect on mEPSC frequency (100 ± 18%, n = 4, P = 0.99, Student's paired t test) or amplitude (91 ± 7%, n = 4, P = 0.23, Student's paired t test). Instead, we found that hCSF markedly increased the frequency of AP-dependent sEPSCs to 447 ± 96% (n = 7, P = 0.01, Student's paired t test, Fig. 4A and B), with no effect on sEPSC amplitude (104 ± 11%, n = 7, P = 0.74, Student's paired t test, Fig. 4A and C).
Figure 4. hCSF increases the frequency of spontaneous EPSCs, but not IPSCs, in CA1 pyramidal neurons.

A, example sweeps of sEPSCs in aCSF and hCSF. B and C, summary graphs showing normalized sEPSC frequency (B) and amplitude (C) when switching from aCSF to hCSF. D, example sweeps of sIPSCs in aCSF and hCSF. E and F, summary graphs showing normalized sIPSC frequency (E) and amplitude (F) when switching from aCSF to hCSF. G, one experiment illustrating the differential effect of hCSF on sEPSCs (recorded at –70 mV) and sIPSCs (recorded at 0 mV). H, bar graph summarizing the effect of hCSF on sEPSC frequency, sIPSC frequency and the ratio of sEPSC/sIPSC frequency (E/I ratio) in six experiments such as the one illustrated in G. Error bars represent SEM. ∗P < 0.05.
To determine the effect of hCSF on spontaneous GABAAergic transmission, we recorded sIPSCs. Interestingly, we found no increase in either sIPSC frequency (90.2 ± 6.6%, n = 7, P = 0.25, Student's paired t test, Fig. 4D and E) or sIPSC amplitude (80.7 ± 6.4%, n = 7, P = 0.034, Student's paired t test, Fig. 4D and F) in hCSF, indicating a differential effect on excitatory and inhibitory synaptic transmission. We further corroborated these findings by recording both sEPSCs and sIPSCs from single CA1 pyramidal neurons (Fig. 4G), where wash-in of hCSF produced a strong shift in the excitation/inhibition ratio (n = 6, P = 0.02, Student's paired t test, Fig. 4G and H).
hCSF potentiates evoked excitatory synaptic transmission and decreases PPR at CA3–CA1 synapses
To examine the effect of hCSF on evoked excitatory synaptic transmission, we performed extracellular field recordings in P10–20 rats. Wash-in of hCSF caused a rapid and large increase of the fEPSP in CA1 stratum radiatum to 271 ± 36% (n = 15, P = 0.0003, Student's paired t test, Fig. 5B) and a small increase of the fibre volley to 108 ± 3% (n = 15, P = 0.03, Student's paired t test, Fig. 5B). The effect of hCSF was reversible upon washout for both fEPSP (99.2 ± 1.3% vs. 108.9 ± 5.4%, n = 13, P = 0.15, Student's paired t test) and fibre volley (100.2 ± 0.5% vs. 97.7 ± 2.1%, n = 13, P = 0.24, Student's paired t test). To exclude that the effect of hCSF on excitatory synaptic transmission was restricted to developing synapses we repeated the experiments in slices from young adult rats (P40–78). The increase in fEPSP (fEPSP slope 256 ± 14%, n = 7, P = 0.0003, Student's paired t test) was of similar magnitude in slices from these rats.
Figure 5. hCSF potentiates evoked excitatory synaptic transmission at CA3–CA1 synapses.

A, one experiment illustrating the effect of hCSF on the fEPSP slope (upper graph) and fibre volley (lower graph). Example sweeps taken at times indicated are shown on top: aCSF (black), hCSF (blue) and washout (grey). B, summary graph showing the effect of hCSF on the fEPSP slope (upper graph) and fibre volley (lower graph). C, summary graph showing the paired-pulse ratio (PPR) in aCSF and hCSF. Example sweeps from paired-pulse recordings in aCSF (black) and hCSF (blue). D, summary graph of the effect of dialysed hCSF (D. hCSF, green) on the fEPSP slope and fibre volley. E, summary graph of PPR in aCSF (black), control hCSF (blue), D. hCSF (green) and after washout (grey). Error bars represent SEM. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
As hCSF did not increase mEPSC amplitude or frequency (cf. above), the most likely explanation for the large increase of the evoked fEPSP, considering only a very small contribution from the increased fibre volley, would be an AP-dependent increase of release probability at CA3–CA1 synapses. As the paired-pulse ratio (PPR) can indicate a change in release probability, we also tested PPR before and after the application of hCSF. The PPR was reduced substantially from 2.15 ± 0.13 to 1.61 ± 0.14 (n = 15, P = 0.0009, Student's paired t test, Fig. 5C), consistent with an increased release probability. However, as can be seen in example sweeps from paired-pulse recordings (Fig. 5C), the second fEPSP in hCSF in particular was associated with prominent population spike activity. The large increase in spiking activity is expected as we use physiological (i.e. low) concentrations of Ca2+ and Mg2+, and then further increase the excitability by introducing hCSF. Spike activity during the fEPSP can result in an underestimation of the measured fEPSP slope, and thus an overestimation of a PPR decrease. To minimize the risk of underestimation of the fEPSP slope, we used a very early and short (first 0.8 ms) period of the fEPSP to measure the slope. Nevertheless, we also analysed the PPR data using an even shorter interval (0.6 ms). With this shorter interval the PPR was reduced from 2.04 ± 0.11 to 1.61 ± 0.13 (n = 15, P = 0.0003, Student's paired t test), supporting the notion that potentiation of the fEPSP by hCSF is associated with a true decrease of the PPR, and consistent with an increased release probability at CA3–CA1 synapses.
To test the specificity of the hCSF effect, we added 1% human serum to aCSF to match the normal protein content of hCSF (Burtis et al. 2006). aCSF supplemented with 1% serum had, however, no effect on the fEPSP (103 ± 5.4%, n = 4, P = 0.65, Student's paired t test) or fibre volley (100.9 ± 1.4% vs. 101.1 ± 0.8%, n = 4, P = 0.88), indicating that the substances responsible for the effects of hCSF may be largely derived from the CNS. In an attempt to isolate an active fraction of hCSF substances according to molecular weight, we performed membrane dialysis to remove substances ≤ 8 kDa from the hCSF (see Methods). We tested this dialysed hCSF against control hCSF from the same original pool and found that it significantly reduced the fEPSP magnitude (from 358 ± 51% in control hCSF to 216.5 ± 28% in dialysed hCSF, n = 4, P = 0.01, Student's paired t test, Fig. 5D). The decrease in fEPSP magnitude was accompanied by an increase in the PPR (from 1.32 ± 0.12 in control hCSF to 1.83 ± 0.18 in dialysed hCSF, n = 4, P = 0.008, Student's paired t test, Fig. 5E), together suggesting that low molecular weight neuromodulators (e.g. amino acids, peptides, amines) account for the effects of hCSF on synaptic transmission. One such low molecular weight substance that might contribute to the effect of hCSF is glutamate. We tested this idea by supplementing an aCSF with 1 μm glutamate, a concentration that is in the upper range of what has been measured in healthy hCSF (Alfredsson et al. 1988; Fiszman et al. 2010; Buvanendran et al. 2012; Coccaro et al. 2013). The addition of glutamate did not produce effects similar to hCSF in CA1 pyramidal neurons. Rather, we found that 1 μM glutamate decreased spontaneous firing (36.3 ± 9.8%, n = 6, P = 0.025, Student's paired t test).
Discussion
Here we show that human CSF strongly increases the excitability of pyramidal neurons in vitro. In CA1 pyramidal neurons, hCSF depolarizes Vrest and lowers the AP threshold to boost spontaneous firing, and decreases Rin. Clamping G-protein activity in CA1 pyramidal neurons with GTPγS completely occluded effects on excitability, suggesting that neuromodulators in hCSF act via G-protein coupled receptors to increase spontaneous excitatory activity. Both evoked and spontaneous glutamatergic synaptic transmission are markedly increased in hCSF, whereas spontaneous GABAAergic synaptic transmission is not. As for the augmentation of evoked glutamatergic synaptic transmission, our results suggest that hCSF increases the release probability at CA3–CA1 synapses.
We found that hCSF lowered Rin, depolarized Vrest and decreased the firing threshold of CA1 pyramidal neurons, indicating a net increase of inward current. As clamping G-protein activity in CA1 pyramidal neurons completely occluded effects of hCSF on Vrest and AP threshold, without noticeably affecting the net change in Rin, the G-protein-dependent mechanisms responsible for the excitability-increasing effects contributed little to the overall decrease in Rin. This suggests instead that other tonic conductances (e.g. potassium, chloride, mixed cationic conductances) are enhanced by hCSF to reduce Rin in a G-protein-independent manner. The lowered firing threshold of CA1 pyramidal neurons in hCSF probably resulted from neuromodulation of ion channels in the axon initial segment. For example, dopaminergic modulation of voltage-gated calcium channels in the axon initial segment has been reported as a mechanism to alter AP threshold without noticeably affecting Rin (Bender et al. 2012). Direct modulation of voltage-gated sodium channels is another mechanism that could alter the threshold (Cantrell & Catterall, 2001). We observed a strongly increased spontaneous glutamatergic synaptic transmission in hCSF. This increase was, however, not accompanied by an increase in spontaneous GABAAergic transmission, indicating clear specificity of the hCSF effects. Although the effects of hCSF on interneurons require closer examination, our results suggest that the basal relationship between excitation and inhibition (E/I ratio) in vivo is, at least in part, determined by neuromodulators in the CSF.
Pyramidal neurons in vivo are bombarded with synaptic activity and operate in a high-conductance state (Destexhe et al. 2003). In contrast, pyramidal neurons studied in in vitro brain slices show much sparser synaptic activity, commonly ascribed to the loss of neuronal inputs from tissue slicing. Our results show that pyramidal neurons in the brain slice can exhibit in vivo-like properties if exposed to hCSF. In the presence of hCSF, CA1 pyramidal neurons show a much lower Rin, more depolarized Vrest, greater Vm fluctuations and a strongly increased excitatory synaptic activity, all properties associated with in vivo pyramidal neurons in the high-conductance state (Destexhe et al. 2003). The synaptic bombardment of pyramidal neurons in vivo is predicted to alter their input–output function, resulting in a decreased gain and an increased responsiveness to low amplitude inputs (Destexhe et al. 2003; Fellous et al. 2003). We observed no effect of hCSF on gain (Fig. 2H and J), but a substantial leftward shift of the input–output function. It is, however, possible that an effect on gain could have been masked, in part, by rundown of firing frequency over time in whole-cell mode (Fig. 2I). Moreover, although hCSF increased the frequency of spontaneous excitatory synaptic activity several hundred per cent in our experiments, the resulting frequency still appears lower than that typically observed in vivo, presumably because of a lower number of functional presynaptic inputs in the slice. Thus, the increased synaptic activity caused by hCSF does not probably, in itself, account for the change in input–output function that we observe. Rather, the leftward shift in input–output function resulted from effects of hCSF on intrinsic properties of CA1 neurons, which in turn can help explain the increased spontaneous excitatory synaptic activity and AP firing observed in vivo.
We repeatedly measured concentrations of electrolytes and glucose in hCSF samples over the course of this study. Although Na+, K+, Cl− and Mg2+ occur at concentrations normally matched in the standard aCSF, we recognize that Ca2+ is present at only 1 mm in hCSF (see Table 1), as opposed to the 2 mm used by most studies in aCSF. Reducing the extracellular concentration of Ca2+ from 2 to 1 mm will act to lower synaptic release probability while simultaneously increasing intrinsic excitability. This will probably enhance spontaneous firing in neurons, but at the same time increase presynaptic filtering of arriving APs. As we carefully matched our aCSF to measured pH, osmolality and electrolyte/glucose concentrations in hCSF, and because the increased excitability was entirely occluded by GTPγS, the effects of hCSF on pyramidal neurons are unlikely to have resulted from differences in these measured variables between the aCSF and hCSF. The differential effect of hCSF on excitatory and inhibitory neurons further supports this conclusion. Rather, our results indicate that endogenous neuromodulators in the hCSF act via G-protein-dependent mechanisms to increase excitability. A wide range of such neuromodulators has been found (Jackson, 1980; Post et al. 1988; Veening & Barendregt, 2010), including amino acids, amines and peptides. We do not yet know the identity of the substances responsible for the neuromodulatory effects of hCSF, but several neuropeptides have been shown to depolarize and increase spontaneous firing in hippocampal and neocortical pyramidal neurons, including orexin, cholecystokinin, neurotensin and vasoactive intestinal peptide (Audinat et al. 1989; Haas & Gahwiler, 1992; Wang et al. 2011; Yan et al. 2012).
The CSF has previously been proposed to convey neuromodulatory messages to neurons in different brain areas (Nicholson, 1999; Veening & Barendregt, 2010). Although a number of interesting anatomical findings have added plausibility to this concept (Cupedo & de Weerd, 1980; Jackson, 1980; Buma et al. 1989; Calle et al. 2005), direct physiological evidence has thus far been missing. Our results suggest that ambient neuromodulators in CSF are present at sufficient concentration as to profoundly increase the excitability of pyramidal neurons, and thus add physiological plausibility to the theory of CSF acting as a conduit for non-synaptic transmission. In addition to effects of neuromodulators at ambient levels in CSF, there is the possibility that specific neuromodulators could be released into the CSF at high concentration in a more temporally restricted manner. This form of release has been suggested as a mechanism by which the CSF could act to induce different brain states (Veening & Barendregt, 2010), such as sleep or active wake. Thus, the CSF may include an ambient activity-promoting fraction of neuromodulators, as suggested by the present study, as well as a more dynamic component whose release may be spatiotemporally regulated to induce specific changes in signalling properties of recipient neurons.
Our findings reveal a previously unrecognized function of the CSF in promoting spontaneous excitatory neuronal activity, and thus help explain differences in activity observed between pyramidal neurons in vivo and in vitro. It will be important for future work to examine the effects of the CSF in other cell types and brain areas, as well as on oscillatory activity and plasticity.
Glossary
- aCSF
artificial cerebrospinal fluid
- AP
action potential
- CSF
cerebrospinal fluid
- CV
coefficient of variation
- fEPSP
field excitatory postsynaptic potential
- GTPγS
guanosine 5′-[γ-thio]triphosphate
- hCSF
human cerebrospinal fluid
- mEPSC
miniature excitatory postsynaptic current
- NPH
normal pressure hydrocephalus
- PPR
paired-pulse ratio
- Rin
input resistance
- sEPSC
spontaneous excitatory postsynaptic current
- sEPSP
spontaneous excitatory postsynaptic potential
- sIPSC
spontaneous inhibitory postsynaptic current
- Vm
membrane potential
- Vrest
resting membrane potential
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
A.B. performed the experiments and analysed the data. U.A. and H.Z. provided theoretical and practical considerations regarding hCSF. I.R. and P.W. assisted in data analysis. J.D. initiated experimentation on hCSF. All authors took part in experimental design and interpretation of data. A.B. and E.H. wrote the article. All authors have approved and read the final version of this manuscript, and only authors qualified for authorship are recognized.
Funding
This work was supported by the Swedish Research Council (K2013-12600, K2010-63P-21562-01-4, K2011-61X-20401-05-6), Swedish State Support for Clinical Research (ALFGBG-136991, ALFGBG-144341), Swedish Brain Foundation (FO2011-003), Swedish Alzheimer Association (13-03-076) and the Wolfson Foundation.
References
- Alfredsson G, Wiesel FA. Tylec A. Relationships between glutamate and monoamine metabolites in cerebrospinal fluid and serum in healthy volunteers. Biol Psychiatry. 1988;23:689–697. doi: 10.1016/0006-3223(88)90052-2. [DOI] [PubMed] [Google Scholar]
- Audinat E, Hermel JM. Crepel F. Neurotensin-induced excitation of neurons of the rat's frontal cortex studied intracellularly in vitro. Exp Brain Res. 1989;78:358–368. doi: 10.1007/BF00228907. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Ford CP. Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2012;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman MW. Palay SL. The fine structure of ependyma in the brain of the rat. J Cell Biol. 1963;19:415–439. doi: 10.1083/jcb.19.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman MW. Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buma P, Veening J. Nieuwenhuys R. Ultrastructural characterization of adrenocorticotrope hormone (ACTH) immunoreactive fibres in the mesencephalic central grey substance of the rat. Eur J Neurosci. 1989;1:659–672. doi: 10.1111/j.1460-9568.1989.tb00372.x. [DOI] [PubMed] [Google Scholar]
- Burtis CA, Ashwood ER. Bruns DE. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. St Louis, MO: Elsevier Saunders; 2006. [Google Scholar]
- Buvanendran A, Kroin JS, Della Valle CJ, Moric M. Tuman KJ. Cerebrospinal fluid neurotransmitter changes during the perioperative period in patients undergoing total knee replacement: a randomized trial. Anesth Analg. 2012;114:434–441. doi: 10.1213/ANE.0b013e31823dc5fb. [DOI] [PubMed] [Google Scholar]
- Calle M, Claassen IE, Veening JG, Kozicz T, Roubos EW. Barendregt HP. Opioid peptides, CRF, and urocortin in cerebrospinal fluid-contacting neurons in Xenopus laevis. Ann NY Acad Sci. 2005;1040:249–252. doi: 10.1196/annals.1327.035. [DOI] [PubMed] [Google Scholar]
- Cantrell AR. Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Coccaro EF, Lee R. Vezina P. Cerebrospinal fluid glutamate concentration correlates with impulsive aggression in human subjects. J Psychiatr Res. 2013;47:1247–1253. doi: 10.1016/j.jpsychires.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo RN. de Weerd H. Serotonergic intraventricular axons in the habenular region. Phagocytosis after induced degeneration. Anat Embryol (Berl) 1980;158:213–226. doi: 10.1007/BF00315907. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Rudolph M. Pare D. The high-conductance state of neocortical neurons in vivo. Nat Rev Neurosci. 2003;4:739–751. doi: 10.1038/nrn1198. [DOI] [PubMed] [Google Scholar]
- Fellous JM, Rudolph M, Destexhe A. Sejnowski TJ. Synaptic background noise controls the input/output characteristics of single cells in an in vitro model of in vivo activity. Neuroscience. 2003;122:811–829. doi: 10.1016/j.neuroscience.2003.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszman ML, Ricart KC, Latini A, Rodriguez G. Sica RE. In vitro neurotoxic properties and excitatory aminoacids concentration in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Relationship with the degree of certainty of disease diagnoses. Acta Neurol Scand. 2010;121:120–126. doi: 10.1111/j.1600-0404.2009.01200.x. [DOI] [PubMed] [Google Scholar]
- Goldstein DA. Massry SG. Divalent ions in blood and cerebrospinal fluid: effect of hypercalcemia, hyperphosphatemia, renal failure and parathyroid hormone. Adv Exp Med Biol. 1980;128:289–297. doi: 10.1007/978-1-4615-9167-2_34. [DOI] [PubMed] [Google Scholar]
- Haas HL. Gahwiler BH. Vasoactive intestinal polypeptide modulates neuronal excitability in hippocampal slices of the rat. Neuroscience. 1992;47:273–277. doi: 10.1016/0306-4522(92)90243-u. [DOI] [PubMed] [Google Scholar]
- Jackson IMD( Significance and function of neuropeptides in cerebrospinal fluid. In: Wood JH, editor. Neurobiology of Cerebrospinal Fluid. New York: Plenum Press; 1980. pp. 625–644. [Google Scholar]
- Joborn C, Hetta J, Niklasson F, Rastad J, Wide L, Agren H, Akerstrom G. Ljunghall S. Cerebrospinal fluid calcium, parathyroid hormone, and monoamine and purine metabolites and the blood–brain barrier function in primary hyperparathyroidism. Psychoneuroendocrinology. 1991;16:311–322. doi: 10.1016/0306-4530(91)90017-n. [DOI] [PubMed] [Google Scholar]
- Nicholson C. Signals that go with the flow. Trends Neurosci. 1999;22:143–145. doi: 10.1016/s0166-2236(98)01388-5. [DOI] [PubMed] [Google Scholar]
- Parent A. Comparative anatomy of the serotoninergic systems. J Physiol (Paris) 1981;77:147–156. [PubMed] [Google Scholar]
- Post RM, Rubinow DR, Kling MA, Berrettini W. Gold PW. Neuroactive substances in cerebrospinal-fluid - normal and pathological regulatory mechanisms. Ann NY Acad Sci. 1988;531:15–28. doi: 10.1111/j.1749-6632.1988.tb31808.x. [DOI] [PubMed] [Google Scholar]
- Smith DE, Johanson CE. Keep RF. Peptide and peptide analog transport systems at the blood-CSF barrier. Adv Drug Deliv Rev. 2004;56:1765–1791. doi: 10.1016/j.addr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I. Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Wang S, Zhang AP, Kurada L, Matsui T. Lei S. Cholecystokinin facilitates neuronal excitability in the entorhinal cortex via activation of TRPC-like channels. J Neurophysiol. 2011;106:1515–1524. doi: 10.1152/jn.00025.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JG. Barendregt HP. The regulation of brain states by neuroactive substances distributed via the cerebrospinal fluid; a review. Cerebrospinal Fluid Res. 2010;7:1. doi: 10.1186/1743-8454-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh B, Manzano e Silva MJ, Frank CL, Vincze C, Czirok SJ, Szabo A, Lukats A. Szel A. The system of cerebrospinal fluid-contacting neurons. Its supposed role in the nonsynaptic signal transmission of the brain. Histol Histopathol. 2004;19:607–628. doi: 10.14670/HH-19.607. [DOI] [PubMed] [Google Scholar]
- Yan J, He C, Xia JX, Zhang D. Hu ZA. Orexin-A excites pyramidal neurons in layer 2/3 of the rat prefrontal cortex. Neurosci Lett. 2012;520:92–97. doi: 10.1016/j.neulet.2012.05.038. [DOI] [PubMed] [Google Scholar]