

Abstract
During brain development the duration of miniature inhibitory postsynaptic currents (mIPSCs) mediated by GABAA receptors (GABAARs) progressively reduces, to accommodate the temporal demands required for precise network activity. Conventionally, this synaptic plasticity results from GABAAR subunit reorganisation. In particular, in certain developing neurones synaptic α2-GABAARs are replaced by α1-GABAARs. However, in thalamocortical neurones of the mouse ventrobasal (VB) thalamus, the major alteration to mIPSC kinetics occurs on postnatal (P) day 10, some days prior to the GABAAR isoform change. Here, whole-cell voltage-clamp recordings from VB neurones of mouse thalamic slices revealed that early in postnatal development (P7–P8), the mIPSC duration is prolonged by local neurosteroids acting in a paracrine or autocrine manner to enhance GABAAR function. However, by P10, this neurosteroid ‘tone’ rapidly dissipates, thereby producing brief mIPSCs. This plasticity results from a lack of steroid substrate as pre-treatment of mature thalamic slices (P20–24) with the GABAAR-inactive precursor 5α-dihydroprogesterone (5α-DHP) resulted in markedly prolonged mIPSCs and a greatly enhanced tonic conductance, mediated by synaptic and extrasynaptic GABAARs, respectively. In summary, endogenous neurosteroids profoundly influence GABAergic neurotransmission in developing VB neurones and govern a transition from slow to fast phasic synaptic events. Furthermore, the retained capacity for steroidogenesis in the mature thalamus raises the prospect that certain physiological or pathophysiological conditions may trigger neurosteroid neosynthesis, thereby providing a local mechanism for fine-tuning neuronal excitability.
Key points.
During neuronal development synaptic events mediated by GABAA receptors are progressively reduced in their duration, allowing for rapid and precise network function.
Here we focused on ventrobasal thalamocortical neurones, which contribute to behaviourally relevant oscillations between thalamus and cortex.
We demonstrate that the developmental decrease in the duration of inhibitory phasic events results predominantly from a precisely timed loss of locally produced neurosteroids, which act as positive allosteric modulators of the GABAA receptor.
The mature thalamus retains the ability to synthesise neurosteroids, thus preserving the capacity to enhance both phasic and tonic inhibition, mediated by synaptic and extrasynaptic GABAA receptors, respectively, in physiological and pathophysiological scenarios associated with perturbed neurosteroid levels.
Our data establish a potent, endogenous mechanism to locally regulate the GABAA receptor function and thereby influence thalamocortical activity.
Introduction
During postnatal brain development, demand for temporal precision within neural network activity increases and in accordance, synaptic events mediated by GABAARs become progressively reduced in their duration (Brickley et al. 1996; Okada et al. 2000; Vicini et al. 2001; Jüttner et al. 2001; Goldstein et al. 2002; Bosman et al. 2005; Takahashi et al. 2005; Fritschy&Panzanelli, 2014; Deidda et al. 2014). We reported that miniature inhibitory postsynaptic currents (mIPSCs) of thalamocortical neurones of the ventrobasal (VB) thalamus undergo considerable changes during development. In particular, their duration is greatly reduced over a critical, but relatively short, period of 24–48 h that occurs between postnatal days (P) 9–10 and P10–11 (Peden et al. 2008). Conventionally, such changes have been ascribed to alterations in the subunit composition of the synaptic GABAARs. These inhibitory receptors are composed of five subunits, selected from a repertoire of 19 proteins (Olsen&Sieghart, 2008). Most neuronal GABAARs contain two α subunits, two β subunits and either a γ or a δ subunit, permitting ∼20–30 distinct naturally occurring receptor subtypes, which differ in their pharmacology, channel biophysics, anatomical location and developmental expression (Olsen&Sieghart, 2008; D'Hulst et al. 2009; Fritschy&Panzanelli, 2014; Rudolph&Möhler, 2014).
The developmental expression of the α1 subunit has often been associated with a more rapid mIPSC decay time (Okada et al. 2000; Peden et al. 2008). In apparent agreement, our immunohistochemical and electrophysiological studies using both α1 ‘knock-out’ (α10/0) and α1 ‘knock-in’ (α1H101R) mice revealed synaptic GABAARs of P20 VB neurones to express the α1β2γ2 isoform, whereas those of younger animals were primarily composed of α2-GABAARs (Peden et al. 2008). However, the rapid transition of mIPSC kinetics between P 9 and 11 occurs at a stage when the synaptic receptors still predominantly contain the α2 subunit and is evident prior to the increased incorporation of the α1 subunit. These findings are complemented by the observation that identical changes in mIPSC decay kinetics occur in VB neurones derived from α10/0 mice (Peden et al. 2008). Thus, mechanisms additional to GABAAR subunit composition appear to influence the development of synaptic current kinetics during this early postnatal period. In this regard, modulation of GABAAR function by endogenous ligands, such as neurosteroids, represents a viable alternative mechanism.
Certain naturally occurring steroids, synthesised both in the periphery and within the central nervous system (CNS), are potent positive allosteric modulators of GABAAR function (Belelli&Lambert, 2005; Carver&Reddy, 2013; Zorumski et al. 2013). Their brain levels are dynamically regulated in a number of physiological scenarios (e.g. development, stress, puberty, pregnancy and during the ovarian cycle; Purdy et al. 1991; Paul&Purdy, 1992; Grobin&Morrow, 2001; Maguire&Mody, 2009; Shen et al. 2010; Zorumski et al. 2013; Gunn et al. 2014). Additionally, perturbations in their levels occur in several pathophysiological conditions (e.g. panic attacks, major depression, postpartum depression, premenstrual tension and schizophrenia; Purdy et al. 1991; Eser et al. 2006; Smith et al. 2006; Uzunova et al. 2006; Longone et al. 2008; Zorumski et al. 2013, Gunn et al. 2014). Intriguingly, neurosteroidogenic enzymes are expressed in certain neurones (Agis-Balboa et al. 2006; Tsutsui, 2008; Do Rego et al. 2009; Castelli et al. 2013), suggesting that these local neuromodulators have the potential to rapidly influence neuronal excitability in a paracrine or an autocrine fashion. Here, we have recorded from VB neurones across the postnatal development period of P7–24 to investigate the influence of endogenous neurosteroids on the profile of inhibitory neurotransmission. By exploiting the steroid-sequestering actions of the cyclic oligosaccharide γ-cyclodextrin (γ-CD; Adam et al. 2002; Shu et al. 2004), together with inhibition of neurosteroid synthesis by finasteride, we demonstrate that during a critical postnatal period, GABAergic inhibitory neurotransmission in the thalamus is greatly influenced by local neurosteroids acting in a paracrine, or autocrine manner.
Methods
Ethical approval
All studies were conducted in accordance with Schedule 1 of the UK Government Animals (Scientific Procedures) Act, 1986, following approval by the University of Dundee Ethical Review Committee under the auspices of a Home Office Project Licence 60/4005 to D. Belelli.
Breeding of mice
The α4 subunit ‘knockout’ (α40/0) mice were generated on a single C57BL6 background at the University of Pittsburgh as previously described (Chandra et al. 2006). Heterozygous breeding pairs were housed at the University of Dundee and brain slices were prepared from first or second generation of wild-type (WT), or α40/0 offspring.
Thalamic slice preparation
Thalamic slices were prepared as previously described (Belelli et al. 2005) from P7–24 WT, or α40/0 mice of either sex. Mice were killed by cervical dislocation, the brain dissected and rapidly placed in ice-cold oxygenated (95% O2/5%CO2) artificial cerebrospinal fluid (aCSF) containing (in mm): 234 sucrose, 2.95 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 10 MgSO4, 10 glucose, (pH 7.4; 335–340 mosmol l−1). Whilst maintained in oxygenated aCSF, the brain was sectioned in the horizontal plane using a Vibratome series 1000 PLUS Sectioning System (Intracell, Royston, Hertfordshire, UK). Tissue slices were cut at 300–350 μm thickness for P20–P24 mice and 400 μm for younger animals. The slices were immediately transferred onto a nylon mesh platform housed within a chamber containing circulating oxygenated extracellular solution (in mm: 126 NaCl, 26 NaHCO3, 2.95 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 10 glucose (306–309 mosmol l−1)) and maintained at room temperature for a minimum of 1 h prior to recording.
Whole-cell voltage-clamp recordings
Patch pipettes were pulled from thick-wall borosilicate glass (Garner Glass Co., Claremont, CA, USA) using a Narashige PC-10 electrode puller (Narashige, Japan). Open tip resistances of 3–6 MΩ were obtained when pipettes were filled with intracellular solution (ICS) containing (in mm): 140 CsCl, 10 Hepes, 10 EGTA, 2 MgCl2, 1 CaCl2, 2 Mg-ATP and 5 QX-314 (pH 7.2–7.3, 290–300 mosmol l−1). Neurones of the ventrobasal (VB) complex were visually identified using an Olympus BX51 microscope (Olympus), equipped with differential interference contrast/infrared optics and a CCD camera. Whole-cell voltage-clamp recordings were acquired at a holding potential of −60 mV using an Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA, USA) and filtered at 2 kHz. Isolation of mIPSCs was achieved using the solutions outlined above and by supplementing the extracellular solution (ECS) with 2 mm kynurenic acid and 0.5 μm tetrodotoxin (TTX) to inhibit ionotropic glutamate receptors and action potential-dependent neurotransmitter release, respectively. Data acquisition and digitisation (10 kHz) were performed using a NIDAQ mx interface (National Instruments, Austin, TX, USA) and stored to a computer hard-drive using WinEDR software (Strathclyde University, UK). Series resistance was compensated for by up to 80% and recordings were considered invalid if the series resistance changed by more than 20%, or if it exceeded 15 MΩ.
Drug and solution application
Bicuculline methobromide and TTX were prepared as concentrated stock solutions in distilled water. The neurosteroids 5α-pregnan-3α-ol-20-one (5α3α) and 5α-dihydroprogesterone (5α-DHP), the 5α-reductase inhibitor finasteride and the 3α-hydroxysteroid dehydrogenase (3α-HSD) inhibitor indomethacin were prepared as concentrated stock solutions (1000X) in DMSO. Drug stock solutions were diluted to the final required concentration in ECS (giving a final DMSO concentration of 0.1%). Kynurenic acid was dissolved directly into the ECS. Similarly, α- and γ-cyclodextrin (α-CD, γ-CD) were dissolved directly into the extracellular and intracellular solution. Two protocols were implemented for cyclodextrin studies. In the first protocol, named ‘cyclodextrin pre-incubation’, thalamic slices were pre-incubated in the holding chamber at room temperature with α-, or γ-CD (1 mm, >1 h). Subsequently, during the recording the tissue was continuously perfused with ECS (35°C) containing the particular cyclodextrin (1 mm), coupled with inclusion of this cyclic oligosaccharide (0.5 mm) in the recording pipette. In the second protocol, cyclodextrin was only included in the recording pipette (0.5 mm). For studies involving drug pre-incubation of thalamic brain slices prior to recording, the compound (indomethacin, finasteride or 5α-DHP) was dissolved into the holding chamber ECS (at room temperature), from a 1000X DMSO stock for the stated period. The final DMSO concentration (0.1%) had no effect on the mIPSCs, or the tonic current. Once transferred to the recording chamber, slices were maintained in ECS with the compound. The stated incubation times relate to the total time the brain slice was exposed to the compound before recording. In most cases where α- or γ-CD was applied solely to the intracellular compartment, mIPSCs were only included for analysis if they were recorded at least 6 min after acquiring the whole-cell configuration. For experiments designed to assess the temporal profile of intracellular α-, or γ-CD effects, mIPSCs were included for analysis as soon as the whole-cell capacitance transients were accurately cancelled and compensated for by the patch-clamp amplifier circuitry. Typically, these adjustments were achieved within 1 min of attaining the ‘whole-cell’ recording mode. Thalamic slices were superfused using a gravity-based perfusion system set to a flow rate of 3–5 ml min–1 and recycled to a 50 ml oxygenated reservoir using a peristaltic pump (Minipuls 3, Gilson, Luton, UK). Drugs dissolved in ECS were applied to the slice from separate reservoirs. For all experiments, the temperature in the recording chamber was maintained at 35°C.
mIPSC analysis
Offline analysis of digitised data was performed using WinEDR/WinWCP software (Strathclyde University, UK). Identification of mIPSCs was conducted by an algorithmic detection protocol. To eliminate events affected by dendritic filtering, detected mIPSCs were subjected to a filtering protocol whereby events were discarded if their 10–90% rise time occurred outwith the limits of a Gaussian distribution, or were >1 ms. Following visual inspection of individual mIPSCs and deletion of spurious events, parameters from typically 50 or more mIPSCs were analysed with respect to their peak amplitude, 10–90% rise time, and time taken to decay from peak by 90% (T90). Subsequently, mIPSCs recorded from a single neurone were averaged by alignment of the mid-point of their rising phase and the resulting averaged mIPSC was fitted with either a mono-exponential (y(t) = Ae(−t/τ)), or bi-exponential (y(t) = A1e(−t/τ1) + A2e(−t/τ2)) decay function where y(t) is the current amplitude at any given time t, A is the current amplitude and τ is the decay time constant. To assess whether the decay phase of the averaged mIPSC was best fitted by a mono- or bi-exponential, the standard deviation of the residuals was measured and an F test applied. For the vast majority of mIPSCs analysed, the decay was best described by a bi-exponential function. Accordingly, a mean weighted decay constant (τW) was calculated to depict the relative contribution of each decay component. Values for τW were derived from the following equation:
where τ1 and τ2 are the decay time constants for the first and second exponential functions, and P1 and P2 are the proportions of current amplitude described by each component, i.e.
Frequency analysis of mIPSCs was conducted for each recording whereby events were detected by running an offline event detection protocol (WinEDR) based on the rate of rise time threshold. For control experiments, and those involving drug pre-incubation, the number of events was determined for two bins located towards the beginning and end of a 5 min recording period. The duration of each bin varied with developmental age such that recordings from young animals (P7–8, in which mIPSCs occurred relatively infrequently) required a bin size ∼1 min, whereas ∼20 s bins were utilised for P20–24 animals. The accuracy of the event detection protocol was verified by visually inspecting the current recordings to ensure that all events were included and that any detected spurious noise was removed. A mean inter-event interval was then generated and converted to a frequency value.
The resident tonic current and the steroid-induced current
The resident tonic current was calculated by determining the outward current produced by bath-applied bicuculline methobromide (30 μm). Similarly, the effect of 5α-DHP pre-incubation on the tonic conductance was assessed by comparing the bicuculline methobromide-evoked outward current for control recordings (i.e. no pre-incubation) vs. recordings from VB neurones following drug pre-incubation. For experiments where 5α3α was applied acutely, the enhancement of the tonic current was calculated by determining the difference in holding current before and after neurosteroid application. In each case, the mean DC current of a 30–60 s current section was determined from sequential 25.6–102.4 ms epochs, depending on the mIPSC frequency of the cell. Epochs containing mIPSCs, or sections of recording which exhibited an unstable baseline, were not included in the determination of mean holding current.
Statistical analysis
All data are expressed as the arithmetic mean value ± standard error of the mean (SEM). To calculate statistical significance of mean data, Student's t tests (paired, or unpaired, Microsoft Excel) and ANOVA (followed post hoc by Tukey's HSD, SigmaStat, Systat Software Inc., San Jose, CA, USA) were used as appropriate. To compare the cumulative probability distributions of individual mIPSC properties at different developmental stages, the Kolmogorov–Smirnoff (KS) test was used (SPSS software, Chicago, IL, USA).
Results
Developmental changes to the properties of VB mIPSCs
We previously reported that the duration of VB mIPSCs is greatly reduced over a critical, but relatively short, period of 24–48 h, between P8/9 and P10/11 (Peden et al. 2008). In confirmation, here, age had a significant effect on all mIPSC parameters tested (P < 0.001, one-way ANOVA). In particular, the mIPSCs of P7 VB neurones exhibited a relatively prolonged decay (τW = 8.5 ± 0.4 ms; n = 15), which was greatly reduced in duration by P10 (τW = 4.7 ± 0.3 ms; n = 7) and further decreased by P20–24 (τW = 3.2 ± 0.1 ms; n = 28, Fig. 1A–D, Table 1). Additionally, in comparison to P7, the mIPSCs recorded from P10 and P20–24 VB neurones occurred much more frequently (Fig. 1A and D; Table 1) and by P20–24, both the mIPSC peak amplitude and rise time were reduced relative to earlier time points (Table 1).
Figure 1. Postnatal development influences phasic inhibition of VB neurones.
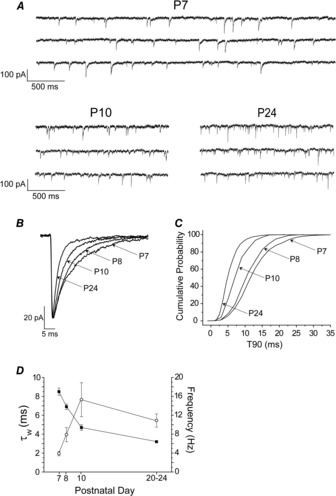
A, representative current traces recorded from VB neurones obtained from P7, P10 and P24 WT mice. B, representative, superimposed, averaged mIPSCs normalised to P7 peak amplitude, showing a progressive decrease in decay time with age. C, combined cumulative probability curves for the time taken for individual mIPSCs to decay from peak amplitude to 10% of peak (T90) for P7 (1160 events, n = 15 cells), P8 (559 events, n = 9 cells), P10 (526 events, n = 7 cells) and P20–24 (3642 events, n = 28 cells). Note the progressive leftward shift in T90 distribution indicating a developmental decrease in the mIPSC decay time (P < 0.001; KS test). D, graph illustrating the progressive decrease of the mIPSC τW (filled squares) and the concomitant increase of mIPSC frequency (open circles) during development (P < 0.001 in each case, one-way ANOVA). Each symbol represents the mean ± SEM.
Table 1.
The properties of mIPSCs recorded from WT VB neurones at different stages of development
| P7 (n = 15 cells) | P8 (n = 9 cells) | P10 (n = 7 cells) | P20–24 (n = 28 cells) | |
|---|---|---|---|---|
| Peak amplitude (pA) | −82 ± 4 | −92 ± 6 | −93 ± 9 | −66 ± 3∗ |
| Rise time (ms) | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1∗∗∗ |
| τW (ms) | 8.5 ± 0.4 | 6.9 ± 0.3∗∗ | 4.7 ± 0.3∗∗∗ | 3.2 ± 0.1∗∗∗ |
| Frequency (Hz) | 3.9 ± 0.5 | 7.9 ± 1.5 | 15.3 ± 3.6∗∗ | 10.9 ± 1.4∗∗ |
Note that all the mIPSC parameters determined here are significantly different across the developmental ages investigated (P < 0.001, one-way ANOVA). Asterisks indicate level of significance vs. those of P7 neurones as determined by Tukey's post hoc test.
P < 0.05
P < 0.01
P < 0.001.
Inhibitory synaptic transmission of immature VB thalamocortical neurones is influenced by locally synthesised neurosteroids
To investigate whether local neurosteroid synthesis contributes to the relatively prolonged mIPSCs of immature VB neurones, P7 thalamic slices were pre-incubated with the 5α-reductase inhibitor finasteride (50 μm; for 4 h). This treatment had no effect on the mIPSC peak amplitude or rise time (P = 0.99 and 0.75, respectively, one-way ANOVA, Table 2), but significantly reduced the mIPSC τW of P7 VB neurones (control τW = 8.5 ± 0.4 ms; n = 15; finasteride τW = 6.0 ± 0.3 ms, n = 8; P < 0.001, one-way ANOVA, Fig. 2A and D, Table 2).
Table 2.
A summary of the effects of finasteride (50 μm), γ-CD (1 mm pre-incubated/0.5 mm ICS only) and α-CD (1 mm pre-incubated/0.5 mm ICS only), upon the properties of the mIPSCs of VB neurones derived from 7-day-old mice
| Control (n = 15 cells) | 50 μm finasteride (n = 8 cells) | 1 mm γ-CD (n = 9 cells) | 1 mm α-CD (n = 7 cells) | 0.5 mm γ-CD ICS only (n = 8 cells) | 0.5 mm α-CD ICS only (n = 8 cells) | |
|---|---|---|---|---|---|---|
| Peak amplitude (pA) | −82 ± 4 | −80 ± 7 | −90 ± 5 | −93 ± 3 | −74 ± 8 | −83 ± 5 |
| Rise time (ms) | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 |
| τW (ms) | 8.5 ± 0.4 | 6.0 ± 0.3∗∗∗ | 5.8 ± 0.3∗∗∗ | 8.8 ± 0.7 | 5.7 ± 0.2∗∗∗ | 8.2 ± 0.3 |
| Frequency (Hz) | 3.9 ± 0.5 | 10.2 ± 3.4∗ | 7.4 ± 1.6 | 5.1 ± 1.2 | 3.8 ± 1.2 | 3.8 ± 1.2 |
Note that τW and frequency were significantly (P < 0.001 and P < 0.05, respectively) affected by drug treatment whereas peak amplitude and rise time were not (one-way ANOVA). Asterisks indicate level of significance vs. control as determined by Tukey's post hoc test.
P < 0.05
P < 0.001 vs. control.
Abbreviations: CD, cyclodextrin; ICS, intracellular solution.
Figure 2. Early in development endogenous neurosteroids prolong GABAAR-mediated mIPSCs of VB neurones.
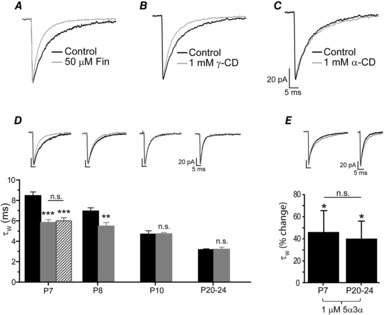
A–C, superimposed and averaged mIPSCs normalised to peak amplitude, recorded from P7 VB neurones for control and following finasteride treatment (A), γ-CD pre-incubation (B), or α-CD pre-incubation (C). D, bar graph comparing the effect of γ-CD pre-incubation (grey bars) on the mean mIPSC τW vs. control (black bars) across postnatal development. Note that for comparison, the effect of finasteride (50 μm) treatment is included for P7 VB neurones (hatched bar). The data were obtained from 7–28 neurones and are shown as the mean ± SEM. Representative corresponding averaged and superimposed mIPSCs are given above each developmental time point for control (black trace) and γ-CD (grey trace). ∗∗P < 0.01, ∗∗∗P < 0.001 vs. control, one-way ANOVA (Tukey's post hoc test for P7 and unpaired t test for P8). E, bar graph illustrating the effects of acute 5α3α (1 μm) on the mIPSC τW recorded from P7 (n = 3) and P20–24 (n = 5) VB neurones (∗P < 0.05 for each). Compared to P20–24, the percentage change in τW following 5α3α application at P7 was not statistically significantly different (two-way mixed ANOVA). The inset shows averaged superimposed mIPSCs before and after application of 5α3α (1 μm) for representative P7 and P20 neurones. Note for panels A–E the current calibration bar refers to the averaged control mIPSC. Abbreviations: n.s., not significant; CD, cyclodextrin; Fin, finasteride; 5α3α, 5α-pregnan-3α-ol-20-one.
To further investigate whether early in development the duration of phasic inhibitory events is influenced by neurosteroids we utilised γ-CD, which sequesters lipophilic molecules, including steroids (Davis&Brewster, 2004; Chisari et al. 2010). Previous studies demonstrated the neurosteroid-scavenging properties of γ-CD (composed of 8 cyclic sugars, Shu et al. 2004, 2007; Akk et al. 2005). For P7 VB neurones, γ-CD treatment (>1 h pre-incubation, see Methods) resulted in mIPSCs with a significantly reduced τW (γ-CD = 5.8 ± 0.3 ms, n = 9; control = 8.5 ± 0.4 ms; n = 15, P < 0.001, one-way ANOVA, Fig. 2B and D, Table 2), indeed, a value similar to that of finasteride-treated neurones (6.0 ± 0.3 ms, P = 0.99, one-way ANOVA, n = 8). The averaged mIPSC decay was best described by two exponentials (τfast and τslow). Further analysis revealed that γ-CD had no effect on τfast (P7 control τfast = 3.6 ± 0.3 ms, n = 15, vs. P7 γ-CD τfast = 3.2 ± 0.2 ms, n = 9, P = 0.36, Student's unpaired t test), but selectively influenced τslow (P7 control τslow = 13.2 ± 0.8 ms, n = 15, vs. P7 γ-CD τslow = 9.8 ± 0.5 ms, n = 9, P < 0.05, one-way ANOVA). Indeed, in the presence of γ-CD the value of P7 τslow is now not significantly different from that of untreated P10 neurones (P10 control τslow = 8.98 ± 2.1 ms, n = 7, P = 0.65, one-way ANOVA). Similarly, for P7 neurones, finasteride treatment selectively reduced the τslow component (P7 control τslow = 13.2 ± 0.8 ms, n = 15, vs. P7 finasteride τslow = 7.3 ± 0.4 ms, n = 6, P < 0.01, one-way ANOVA), such that it was now not significantly different from that of untreated P10 neurones (P = 0.41, one-way ANOVA). These observations are consistent with endogenous neurosteroid altering the kinetics of entry and exit from desensitised states of the synaptic GABAARs (Jones&Westbrook, 1995; Zhu&Vicini, 1997).
Importantly, the sensitivity of mIPSCs to γ-CD treatment declined as development progressed, such that it had no effect on the duration of mIPSCs recorded from P10, or P20–24 VB neurones (P10 control τW = 4.7 ± 0.3 ms, n = 7, vs. P10 γ-CD τW = 4.7 ± 0.1 ms, n = 6, P = 0.96, Student's unpaired t test; P20–24 control τW = 3.2 ± 0.1 ms, n = 28, vs. P20–24 γ-CD τW = 3.2 ± 0.2 ms, n = 9, P = 0.85, Student's unpaired t test; Fig. 2D). As a control we utilised the structurally similar α-CD, as it is ineffective in sequestering pregnane steroids (Shu et al. 2004), as a consequence of having a smaller pore diameter (6 cyclic sugars) than that of γ-CD (Davis&Brewster, 2004). The equivalent treatment with α-CD had no effect on the mIPSC τW of P7 neurones (Fig. 2C, Table 2). It is conceivable that the synaptic GABAARs become less steroid sensitive during this critical developmental period. As a precedent, during parturition rat synaptic GABAARs of oxytocin-releasing neurones become neurosteroid insensitive (Koksma et al. 2003). However, here the prolongation of mIPSC decay observed upon acute application of 5α3α (1 μm) was not significantly different between recordings obtained from P7 and P20–24 neurones (Fig. 2E, n = 3 and 5, respectively; P = 0.67, two-way mixed ANOVA). Although interpretation of this comparison is complicated by the presence of a neurosteroid tone at P7 and that the mIPSCs are mediated by different synaptic GABAARs (α2- and α1-GABAARs for P7 and P20–24, respectively; Peden et al. 2008) these data confirm that synaptic GABAARs are neurosteroid sensitive at both P7 and P20–24. Therefore, collectively our data suggest that the developmental decrease of the VB mIPSC duration is accounted for by a programmed loss of endogenous neurosteroid.
Immunohistochemical and in situ hybridisation studies suggest that certain neurones can synthesise GABA-active neurosteroids, raising the prospect that they might act in an autocrine fashion to fine-tune neuronal inhibition (Agis-Balboa et al. 2006; Tsutsui, 2008; Castelli et al. 2013). In support, a study utilising an antibody raised against GABAAR-active neurosteroids suggested that thalamic VB neurones contain the steroid 5α3α (Saalmann et al. 2007). To explore whether P7 VB neurones are a potential source of these GABAAR neuromodulators, we investigated the impact of including the membrane-impermeant γ-CD (0.5 mm) solely in the recording pipette. This treatment (recordings made >6 min after achieving the whole-cell recording configuration) significantly reduced the mIPSC τW (5.7 ± 0.2 ms, P < 0.001, one-way ANOVA, n = 8) in comparison to control mIPSCs (τW = 8.5 ± 0.4 ms, n = 15, Fig. 3A and C, Table 2). In this respect, intracellular γ-CD was equi-effective with those recordings made with either the γ-CD pre-incubation protocol, or following finasteride pre-treatment (P = 0.99, one-way ANOVA, Fig. 3C, Table 2). In contrast, addition of α-CD (0.5 mm) solely to the ICS, had no effect on the mIPSC τW (τW = 8.2 ± 0.3 ms, n = 8, P = 0.99 vs. control, one-way ANOVA, Fig. 3B and C, Table 2). When delivered from a recording electrode, relatively large molecules such as cyclodextrin require time (dependent upon molecular weight, pipette resistance, series resistance and cell geometry/volume) to equilibrate with the intracellular milieu (Marty&Neher, 1995). In agreement, upon achieving the whole-cell recording configuration, intracellular γ-CD (0.5 mm) caused a time-dependent decrease of the mIPSC τW, which approached equilibrium after 6–11 min (Fig. 3D). By contrast, both standard ICS and ICS containing α-CD (0.5 mm) were inert in this respect (Fig. 3D).
Figure 3. Intracellular γ-cyclodextrin reduces the duration of P7 VB mIPSCs.
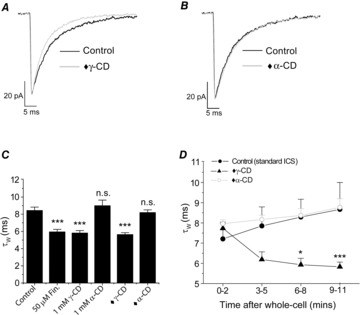
A and B, superimposed and averaged P7 mIPSCs, normalised to peak amplitude, for control and following intracellular γ-CD (A, ♦γ-CD), or intracellular α-CD (B, ♦α-CD) treatment. The current calibration bar refers to the averaged control mIPSC. C, bar graph (mean ± SEM) comparing the effects of finasteride pre-incubation, γ/α-CD pre-incubation (1 mm extracellular + 0.5 mm intracellular) and intracellular ♦γ/α-CD (0.5 mm) application alone on the mIPSC τW (n = 7–15 neurones, ∗∗∗P < 0.001 vs. control, one-way ANOVA with Tukey's post hoc test). D, graph illustrating the impact of standard ICS, ICS + ♦α-CD and ICS + ♦γ-CD on the mIPSC τW with time after acquisition of the whole-cell recording configuration. Each data point is composed of data from 4–8 neurones. ∗P < 0.05, ∗∗∗P < 0.001, mixed ANOVA with Tukey's post hoc test. Abbreviations: CD, cyclodextrin; Fin, finasteride; ICS, intracellular solution; n.s., not significant; ♦, intracellular 0.5 mm γ- or α-cyclodextrin.
Neurosteroid synthesis in thalamic slices
Phasic inhibition
Our findings suggest that young (P7) thalamic tissue produces neurosteroids that influence GABAergic synaptic signalling. However, by P10 there is an abrupt suppression of neurosteroid synthesis, resulting in a clear reduction of the mIPSC decay time. To determine whether more mature thalamus retains the ability to synthesise neuroactive steroids we investigated the impact on GABAergic inhibition of incubating P20–24 thalamic slices with 5α-DHP, the immediate precursor of 5α3α. Limited (30–60 min) exposure to 5α-DHP (3 μm) had no effect on the mIPSC τW of P20–24 VB neurones (P = 0.98 vs. control, one-way ANOVA, Fig. 4A and B). However, a more extended incubation (>120 min) with this steroid caused a significant (P < 0.001, one-way ANOVA) prolongation of P20–24 mIPSCs (5α-DHP τW = 8.1 ± 0.5 ms, n = 11 vs. control τW = 3.2 ± 0.1 ms, n = 28; Fig. 4A and B). Whereas γ-CD had no significant effect on mIPSCs of P20–24 VB neurones (Fig. 2D, control τW = 3.2 ± 0.1 ms, n = 28 vs. γ-CD τW = 3.3 ± 0.2 ms, n = 9, P = 0.85, Student's unpaired t test), intracellular γ-CD (0.5 mm) clearly reduced the effect of 5α-DHP pre-treatment (3 μm; >120 min) on the mIPSC duration of such neurones (τW = 5.3 ± 0.2 ms, n = 5, P < 0.001, one-way ANOVA, Fig. 4B). These results suggest that 5α-DHP is converted to a GABAAR-active steroid by thalamic tissue. Therefore, we used indomethacin (see Methods) to inhibit the 3α-HSD-mediated conversion of 5α-DHP to the GABA-active 5α3α (Griffin&Mellon, 2001). The effect of 5α-DHP (3 μm) to prolong P20–24 mIPSCs was substantially suppressed by indomethacin (30 μm) incubation, (τW = 4.2 ± 0.3 ms, n = 8; P < 0.001, one-way ANOVA; Fig. 4B). Therefore, in P20–24 thalamus, 3α-HSD is functional, i.e. retaining the capacity to synthesise the GABAAR-active steroid 5α3α.
Figure 4. The neurosteroid precursor 5α-dihydroxyprogesterone prolongs the decay of mIPSCs recorded from P20 and P7 VB neurones.
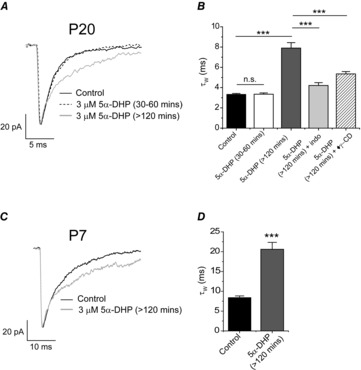
A, superimposed and averaged mIPSCs, normalised to peak amplitude, recorded from P20 VB neurones under control conditions (black trace), following pre-incubation of 5α-DHP (3 μm) for 30–60 min (dashed black trace) and >120 min (grey trace). B, bar graph (mean ± SEM) illustrating the mean P20–P24 mIPSC τW for control and the influence of various 5α-DHP treatments. Note that treatment with either indomethacin (30 μm; pre-incubation for 1 h and subsequent co-incubation with 3 μm 5α-DHP for >120 min) or intracellular γ-CD (0.5 mm ♦γ-CD) significantly reduced the prolongation of the mIPSC decay produced by 5α-DHP (3 μm pre-incubated >120 min) alone. Data obtained from 5–15 neurones (∗∗∗P < 0.001, one-way ANOVA with Tukey's post hoc test). C, superimposed and averaged mIPSCs, normalised to peak amplitude, recorded from P7 VB neurones under control conditions (black trace) and following pre-incubation with 5α-DHP (3 μm) for >120 min (grey trace). D, bar graph illustrating the mean τW for control (n = 15 neurones) and following pre-incubation of P7 thalamic slices with 5α-DHP (>120 min, 3 μm; n = 6 neurones; ∗∗∗P < 0.001 vs. control, Student's unpaired t test). Note for A and C the calibration bar for current refers to the averaged control mIPSC. Abbreviations: n.s., not significant; ICS, intracellular solution; γ-CD, γ-cyclodextrin; indo, indomethacin; 5α-DHP, 5α-dihydroprogesterone; ♦, intracellular.
We next investigated whether the endogenous neurosteroid production at P7 can be further augmented. Following 5α-DHP incubation (3 μm, >120 min), mIPSCs recorded from P7 VB neurones were substantially prolonged (5α-DHP τW = 21.4 ± 1.6 ms, n = 6, vs. control τW = 8.5 ± 0.4 ms, P < 0.001, Student's unpaired test, n = 15, Fig. 4C and D), indicating that neurosteroid production and the associated GABAAR modulation is not maximal at this developmental time point.
Tonic inhibition
Mature mouse VB neurones additionally possess a tonic conductance mediated by extrasynaptic GABAARs composed primarily of α4β2δ receptors (Porcello et al. 2003; Belelli et al. 2005; Chandra et al. 2006; Peden et al. 2008). Application of bicuculline (30 μm) to P20–24 VB neurones revealed a standing tonic current (−96 ± 9 pA; n = 27, Fig. 5B). For such VB neurones, acutely applied 5α3α induced an inward current (100 nm = −32 ± 4 pA, n = 11; 1 μm = −55 ± 20 pA, n = 6, Fig. 5A and B). We have previously shown the VB tonic current to be greatly reduced by deletion of the δ, or the α4 subunit (Herd et al. 2009, 2013; see also Chandra et al. 2006; Fig. 5B). Furthermore, the steroid-induced inward current evident for WT neurones (5α3α, 100 nm) was absent for α40/0 neurones (−4 ± 3 pA, n = 5, Fig. 5B), revealing this effect of the neurosteroid to be mediated by extrasynaptic α4-GABAARs.
Figure 5. Neurosteroid enhancement of the tonic current mediated by extrasynaptic α4βδ GABAARs in P20–24 VB neurones.
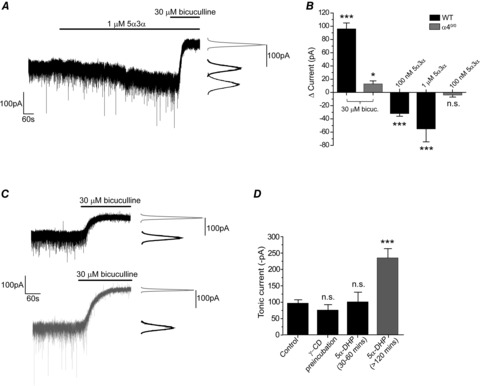
A, a representative current trace recorded from a P21 VB neurone. Acute application of 5α3α (1 μm) evoked an inward current. Bicuculline (30 μm), in addition to blocking the current produced by 5α3α, revealed the resident tonic current. The corresponding all-points histograms are shown to the right of the trace. B, bar graph illustrating the impact of the α4-subunit deletion on the mean tonic current (mean ± SEM) of P20–24 VB neurones as determined by the change in holding current following bicuculline (30 μm) treatment. Compared to WT (n = 27), the α40/0 VB neurones (n = 12) exhibited a markedly decreased tonic conductance. For WT neurones, acutely applied 5α3α (100 nm and 1 μm) enhanced the tonic current (i.e. produced an inward current). By contrast, for VB neurones derived from α40/0 mice 100 nm 5α3α was inert in this regard. ∗P < 0.05 vs. control, ∗∗∗P < 0.001 vs. control, Student's paired t test. C, representative current traces recorded from WT P20–24 VB neurones under control conditions (top trace) and following pre-incubation with 5α-DHP (3 μm; >120 min; lower trace). The corresponding all-points histograms are shown to the right of each trace. D, bar graph depicting the magnitude of the tonic current (mean ± SEM) recorded from P20–24 VB neurones for control and following the (i) γ-CD pre-incubation, (ii) 5α-DHP pre-incubation for 30–60 min, and (iii) 5α-DHP pre-incubation for >120 min. Data obtained from 4–27 neurones. ∗∗∗P < 0.001 vs. control, one-way ANOVA, Tukey's post hoc test. Abbreviations: n.s., not significant; ICS, intracellular solution; γ-CD, γ-cyclodextrin; indo, indomethacin; 5α-DHP, 5α-dihydroprogesterone; 5α3α, 5α-pregnan-3α-ol-20-one. Note the data depicted in B pertaining to the resident tonic current (i.e. revealed by bicuculline) in WT and α40/0 neurones is reproduced from Herd et al. 2013.
Our finding that P20–24 mIPSCs are not sensitive to γ-CD treatment suggests that neurosteroid levels are relatively low at this developmental stage. Nevertheless, recombinant GABAARs incorporating the δ-subunit are particularly sensitive to neurosteroid modulation (Belelli et al. 2002; Brown et al. 2002; Wohlfarth et al. 2002). With regard to untreated P20–24 VB neurones, following γ-CD pre-incubation we observed no significant change in the standing tonic current revealed by bicuculline (control = −96 ± 9 pA, n = 27, vs. γ-CD pre-incubation = −75 ± 15 pA, n = 4, P = 0.86, one-way ANOVA, Fig. 5D). However, following pre-incubation with 5α-DHP (>120 min), the magnitude of the tonic current revealed by bicuculline was significantly greater (control = −96 ± 9 pA, n = 27, vs. 5α-DHP = −234 ± 27 pA, n = 6, P < 0.001, one-way ANOVA, Fig. 5C and D). By contrast, no such effect was observed following a relatively short pre-incubation period (30–60 min) with 3 μm 5α-DHP (−100 ± 28 pA, n = 5, P = 0.99 vs. control, one-way ANOVA, Fig. 5D). Therefore, although neurosteroid production is limited at P20–24, presumably due to a lack of substrate, it may be reinitiated by providing a precursor to the GABA-active steroid, which enhances both synaptic and tonic inhibition.
Neurosteroids and GABA release
As described above, the frequency of mIPSCs recorded from P10 and P20–24 VB neurones was much greater than that of P7 neurones (Fig. 1D; Table 1). Whilst exploring the postsynaptic effects of finasteride, we noted that incubation with this 5α-reductase inhibitor additionally greatly increased the mIPSC frequency of P7 VB neurones (control = 3.9 ± 0.5 Hz; n = 15; finasteride = 10.2 ± 3.4 Hz, n = 8; P < 0.05, one-way ANOVA, Fig. 6A and B; Table 2). One interpretation of these data posits that for P7 nucleus reticularis (nRT)–VB synapses the local neurosteroid tone is sufficient to presynaptically suppress the vesicular release of GABA. Therefore, it is conceivable that the increased mIPSC frequency, evident for P10–24 neurones, reflects a loss of this neurosteroid tone. In agreement, prolonged incubation of P20–24 thalamic slices with 5α-DHP (3 μm), in addition to enhancing phasic and tonic inhibition, markedly reduced the VB mIPSC frequency (control = 10.9 ± 1.4 Hz, n = 28, vs. 3 μm 5α-DHP = 3.9 ± 0.8 Hz, n = 11, P < 0.01, one-way ANOVA, Fig. 6A and B). The effect on release appears to be mediated by 5α3α as co-incubation with 5α-DHP and indomethacin had no effect on frequency (9.9 ± 1.9 Hz, n = 8, P = 0.91, one-way ANOVA, Fig. 6B). Furthermore, in contrast to P20–24 neurones, for P7 VB neurones, 3 μm 5α-DHP alone (prolonged incubation) had no effect on mIPSC frequency, suggesting that for this developmental stage the effect of the native steroid to influence GABA release is maximal (5α-DHP = 2.8 ± 0.5 Hz, n = 5, vs. control = 3.9 ± 0.5 Hz, n = 15, P = 0.23, one-way ANOVA, Fig. 6B).
Figure 6. Neurosteroids influence the mIPSC frequency of VB neurones.
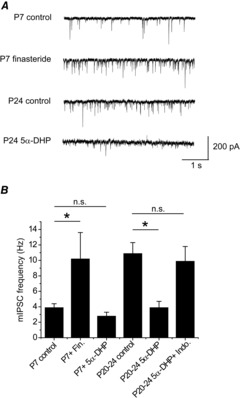
A, representative traces depicting the frequency of mIPSCs for an untreated P7 neurone, a P7 neurone pre-treated with 50 μm finasteride, an untreated P24 neurone and a P24 neurone pre-treated with 3 μm 5α-DHP. B, summary bar graph of mIPSC τW for various manipulations of neurosteroid synthesis at P7 and P20–24. Data from 5–28 neurones. ∗P < 0.05, one-way ANOVA with Tukey's post hoc test. Abbreviations: 5α-DHP, 5α-dihydroprogesterone; Indo, indomethacin; Fin, finasteride; n.s., not significant.
Discussion
During postnatal development, fast excitatory and inhibitory synapses are subject to considerable molecular reorganisation resulting in brief synaptic currents, permitting increased temporal precision and fidelity. For GABAergic inhibitory neurotransmission, alterations to GABAAR subunit composition are primarily implicated in mediating these important kinetic changes (Takahashi et al. 2005). In thalamus, the duration of GABAergic inhibition plays an important role in thalamocortical oscillations (Huguenard&McCormick, 2007; Rovó et al. 2014). Here, we demonstrate for thalamic relay neurones that the substantial alteration of mIPSC kinetics, which occurs during a developmentally critical (P10) window, results not from changes in GABAAR expression, but from decreased local neurosteroid production. Although this GABA-modulatory tone dissipates with maturation, the neurosteroid synthesising enzymes remain functional, permitting the adult thalamus to potentially respond to physiological/pathophysiological stimuli with renewed steroidogenesis and thereby ‘fine-tune’ neuronal inhibition.
In thalamus a developmentally regulated endogenous neurosteroid tone influences phasic inhibition
Studies of a variety of CNS neurones report that an α2/3- to α1-GABAAR subunit switch probably underlies the developmental decrease in the duration of IPSCs (Takahashi et al. 2005; Fritschy&Panzanelli, 2014). In support, in thalamic neurones α1-subunit over-expression reduced the mIPSC decay time (Okada et al. 2000). Furthermore, for developing thalamic VB neurones synaptic α2-GABAARs are replaced by α1-GABAARs (Peden et al. 2008). However, in these neurones the major change to mIPSC kinetics occurs during a critical, brief (P9–10) time window, which precedes the GABAAR subunit switch (Peden et al. 2008). Therefore, what underpins this rapid change of synaptic α2-GABAAR function? Using finasteride to inhibit synthesis, or γ-CD to sequester neurosteroids, we demonstrate that during postnatal development, thalamic synaptic inhibition is influenced by an endogenous neurosteroid tone that rapidly dissipates by P10, consequently reducing mIPSC duration. Previous studies demonstrated γ-CD, but not α-CD, to complex with 5α3α (Shu et al. 2004). In agreement, only γ-CD reduced the mIPSC decay of P7 VB neurones. Furthermore, γ-CD had no effect on >P10 mIPSCs, consistent with previous observations revealing it to be inert on recombinant GABAARs (Shu et al. 2004).
The effect of γ-CD may reflect sequestration of alternative endogenous GABAAR modulators. A study of mouse nRT neurones reported the presence of an endogenous benzodiazepine-site ‘agonist’, sufficient to prolong spontaneous IPSCs (Christian et al. 2013). Irrespective of whether γ-CD complexes such ‘endozepines’, VB inhibitory synapses are not subject to this form of modulation (Christian et al. 2013). In neocortex, endocannabinoids influence synaptic inhibition by a direct interaction with GABAARs (Golovko et al. 2014). However, crucially, the impact of γ-CD treatment on P7 VB neurones was mimicked by inhibiting neurosteroid synthesis with finasteride. Furthermore, although γ-CD had no effect on the mIPSCs of P20–24 VB neurones, it reduced their duration when prolonged by prior 5α-DHP treatment (i.e. by sequestering newly synthesised 5α3α; see below). Collectively, these findings implicate local GABAAR-active neurosteroids in the developmental change to mIPSC duration. Note that after P10, the mIPSC duration is further reduced in a γ-CD-insensitive manner. This subsequent change exhibits a temporal profile consistent with the replacement of synaptic α2-GABAARs with α1-GABAARs (Peden et al. 2008). The neurosteroid-mediated change in mIPSCs could result from an alteration of the sensitivity of synaptic GABAARs to this endogenous modulator, governed by various factors including post-translational receptor modification (Koksma et al. 2003; Belelli&Lambert, 2005). However, P7 and P20–24 VB mIPSCs were similarly sensitive to exogenous 5α3α. Given the presence of an endogenous neurosteroid tone at P7, and that the different subtypes of synaptic GABAARs mediate these mIPSCs at P7 cf. P20–24, caution is warranted in making a meaningful quantitative comparison of the effects of exogenous neurosteroid at the two developmental stages. Nevertheless these data, together with those obtained with the 5α3α precursor DHP, establish that the synaptic GABAARs at both developmental stages are neurosteroid sensitive, therefore suggesting that this perturbation of mIPSC kinetics primarily results from a dynamic change in the level of local neurosteroid.
Our recent findings suggest that the developmental changes to IPSC kinetics will be complex. VB neurones express extrasynaptic GABAARs that mediate an inhibitory tonic conductance (Belelli et al. 2005; Cope et al. 2005; Jia et al. 2005). Utilising paired recordings of nRT and VB (P15–23) neurones, we demonstrated that GABA, released in response to physiological frequencies of nRT excitation, not only activates synaptic (α1β2γ2) receptors, but additionally engages extrasynaptic (α4β2δ) GABAARs, by GABA ‘spill-over’, to produce a slow IPSC component (Herd et al. 2013; see also Rovó et al. 2014). Intriguingly, at P8–9, when the IPSC duration is prolonged by resident neurosteroid, the expression of extrasynaptic α4-GABAARs is modest (Peden et al. 2008) and therefore these receptors presumably have only a limited impact on IPSC duration. However, with further development (>P10) the loss of the neurosteroid influence on the IPSC duration is temporally coincident with increased α4-GABAAR expression (Peden et al. 2008), presumably leading to an increased slow component of the IPSC mediated by GABA ‘spill-over’.
P20–24 VB neurones synthesise GABAAR-active neurosteroids that enhance phasic and tonic inhibition
The abrupt cessation during development of a neurosteroid influence on synaptic GABAARs may result from changes to enzyme expression, or loss of steroid substrate(s). To explore whether more mature thalamus can synthesise GABAAR-active neurosteroids we investigated the 5α3α precursor, 5α-DHP. Acute or short application of 5α-DHP had no effect on the mIPSCs of P20–24 VB neurones, confirming that this steroid does not directly interact with synaptic GABAARs. However, a >2 h application produced a clear prolongation of the mIPSC decay and significantly increased the tonic conductance. The requirement for a prolonged incubation with DHP is not known, but may reflect time required for penetration of the steroid into the slice preparation (Gedell et al. 2004), equilibration of the steroid with intracellular membrane compartments (Li et al. 2007) and conversion to the GABAAR-active 5α3α. Given the coordinated effects of synaptic and extrasynaptic GABAAR activation on IPSC duration (Herd et al. 2013), enhancement of neurosteroidogenesis during physiological/pathophysiological scenarios, or in response to psychoactive drugs (see below), should greatly influence thalamic inhibition and consequently behaviourally relevant thalamocortical network oscillations (Huguenard&McCormick, 2007). However, note the neurosteroid effects on GABA release also requires consideration in this regard (see below).
Although 5α-DHP may engage genomic mechanisms to alter gene expression (Schumacher et al. 2014), both the prolongation of GABA-mediated synaptic currents and the increased tonic current were substantially prevented by the 3α-HSD inhibitor indomethacin (30 μm), or by intracellular γ-CD. Therefore, the effects of 5α-DHP on phasic and tonic inhibition result from the local conversion of this precursor into 5α3α by 3α-HSD. Consequently, the decreased neurosteroid tone observed later in development probably results from a lack of substrate, suggesting that one, or more steps ‘upstream’ from 5α-DHP synthesis are inactive. Similarly, in neonatal spinal dorsal horn neurones, a developmentally regulated neurosteroid presence prolongs the mIPSC decay, an effect that similarly dissipates upon maturity (Inquimbert et al. 2008).
Developmental changes in quantal GABA release are influenced by neurosteroids
Temporally coincident with the postsynaptic changes to phasic inhibition, there is a large increase by P10 of mIPSC frequency, which remains elevated for P20–24 neurones. For P7 neurones, finasteride greatly increased mIPSC frequency, such that it approached that of older neurones. Furthermore, increasing 5α3α synthesis with 5α-DHP clearly reduced the elevated mIPSC frequency of P20–24, but not of P7 neurones. This 5α-DHP effect on P20–24 neurones was prevented by co-incubation with indomethacin, implicating the involvement of 5α3α. These results suggest that early in development (P7), the neurosteroid tone is sufficient to suppress spontaneous vesicular GABA release from nRT terminals, an effect mimicked by exogenous 5α-DHP when added to more mature neurones. These findings are difficult to reconcile with the relatively depolarised chloride reversal potential reported for nRT neurones, where GABAAR activation results in a net Cl− efflux and consequently depolarisation (Ulrich&Huguenard, 1997; Sun et al. 2012), i.e. neurosteroid enhancement of presynaptic GABAARs should increase, not decrease, vesicular release. However, other GABAAR modulators including zolpidem (1 μm), pentobarbital (100 μm) and etomidate (3 μm), at concentrations that produced a clear prolongation of P18–24 VB mIPSCs, had no effect on their frequency (Belelli et al. 2005; authors' unpublished observations), suggesting that this neurosteroid effect may not be mediated by presynaptic GABAARs, but involve other targets, e.g. voltage-gated T-type calcium channels (Jevtovic-Todorovic et al. 2009).
Putative autocrine and paracrine actions of neurosteroids
Studies of steroidogenic enzyme expression indicate that neurosteroid synthesis occurs in both glia and neurones (Do Rego et al. 2009), suggesting neurosteroids act as either paracrine or autocrine messengers to influence neuronal GABAARs (Belelli&Lambert, 2005; Lambert et al. 2009). However, a subsequent investigation revealed that expression of 5α-reductase and 3α-HSD enzymes is restricted primarily to particular neuronal populations in mouse brain (Agis-Balboa et al. 2006). Studies using an antibody raised against the neurosteroid suggest that the distribution of the 5α3α steroid in adult rat brain exhibits a similar neuronal localisation, with staining restricted predominantly to intracellular and membrane compartments of principal neurones (Saalmann et al. 2007; Tokuda et al. 2011). Adult rodent nRT neurones exhibited staining for the 5α-reductase enzyme and for the neurosteroid in nRT and VB neurones (Agis-Balboa et al. 2006; Saalmann et al. 2007; Castelli et al. 2013). Clearly, it would be of interest to perform such immunohistochemical investigations earlier in development.
Therefore, in certain neurones the neurosteroids may act in an autocrine fashion to fine-tune neuronal inhibition. In this scenario, the neurosteroid accesses the GABAAR by lateral diffusion in the plasma membrane, without the necessity for being secreted in a paracrine manner into the extracellular milieu from surrounding neurones or glial cells (Agis-Balboa et al. 2006; Chisari et al. 2010), a scenario consistent with the demonstration of a neurosteroid transmembrane binding site on the GABAAR and the lipophilic nature of the neurosteroid (Chisari et al. 2010). Although low nanomolar aqueous concentrations of 5α3α enhance GABAAR function in vitro, given their lipophilicity, much greater concentrations (μm) may accumulate in the membrane, raising the prospect of a relatively low affinity steroid binding site on the GABAAR (Chisari et al. 2010), i.e. consistent with the observation that γ-CD efficiently sequesters the neurosteroid of VB neurones. Note that our studies with finasteride and γ-CD do not address the neurosteroid source. Indeed, our observation that the developmental changes to GABA release also appear to be governed by local neurosteroids suggests that their ambient concentrations are sufficient to be biologically active, and/or that nRT neurones may synthesise neurosteroids (Agis-Balboa et al. 2006: Saalmann et al. 2007). Further studies are required to clarify the relative contribution of autocrine and paracrine neurosteroids to inhibition in developing thalamus.
Do neurosteroids influence GABAAR-mediated inhibition in the adult brain?
The neurosteroid tone dissipates by P10, prompting the question as to whether they play a role in the adult. Indeed, it will be of interest to establish whether a thalamic neurosteroid tone re-emerges at >P24. Certainly, as described above, the synthetic enzymes and the neurosteroid are present in particular neurones of adult rodents (Agis-Balboa et al. 2006; Saalmann et al. 2007; Castelli et al. 2013; Gunn et al. 2014). Furthermore, when provided with substrate, the mature thalamus can synthesise GABAAR-active neurosteroids. Irrespective of the source, in adults neurosteroid levels are not static, but change in a variety of physiological and pathophysiological scenarios, including ageing, stress, pregnancy, the oestrous cycle, postpartum depression, premenstrual tension, inflammatory pain and certain forms of epilepsy (Belelli&Lambert, 2005; Schumacher et al. 2014). Furthermore, certain antidepressants and various psychoactive drugs also perturb their levels (Belelli&Lambert, 2005; Lambert et al. 2009). Note that peripheral endocrine glands such as the adrenal cortex and ovaries provide an important additional source of brain GABAAR-active neurosteroids as they can readily cross the blood–brain barrier (Belelli&Lambert, 2005; Schumacher et al. 2014). How these endocrine messengers synergise with local autocrine and paracrine steroids to influence neuronal inhibition remains to be clarified. In adults, what initiates local neurosteroid production in the brain? A recent report demonstrated activation of NMDA receptors to increase the staining for 5α-reduced neurosteroids in CA1 hippocampal neurones (Tokuda et al. 2011). By enhancing inhibition, such a mechanism may provide a balance in situations associated with excessive glutamate-induced excitation (Chisari et al. 2010; Tokuda et al. 2011).
Neurosteroid influence on thalamic neuronal excitability during development
What are the likely effects of neurosteroids on the excitability of mouse thalamic relay neurones? Clearly this will depend on the developmental age studied. Developed (P18–27) VB neurones express both synaptic α1β2γ2 GABAARs and extrasynaptic α4β2δ GABAARs, and their activation produces a hyperpolarisation of the resting membrane potential (Belelli et al. 2005; Cope et al. 2005; Peden et al. 2008; Herd et al. 2009; authors' unpublished observations). Although here we have not directly investigated the effects of the neurosteroid upon relay neurone excitability, our previous studies on etomidate may be instructive. Low concentrations of this general anaesthetic, in common with 5α3α, enhance both synaptic and extrasynaptic GABAAR function, consequently increasing the tonic conductance, thereby causing membrane hyperpolarisation and prolonging the neurally evoked IPSP (Herd et al. 2014). Functionally, the increased tonic conductance suppresses tonic spiking and the associated hyperpolarisation may favour burst firing due to relief of T-type calcium channel inactivation (Cope et al. 2005; Herd et al. 2014). Additionally, prolongation of the IPSP caused a delay in the timing of post-inhibitory rebound burst firing. As neurosteroid synthesis also resulted in an enhanced tonic conductance and prolonged synaptic inhibition, presumably it would exert a similar influence to etomidate upon thalamocortical excitability. However, caution is warranted in making this assumption, as in contrast to etomidate (Belelli et al. 2005), the neurosteroid additionally acted upon nRT neurones to substantially reduce the frequency of mIPSCs. Nevertheless, given that the architecture of thalamocortical network oscillations is considered to be partly dependent on the duration of synaptic inhibition (Kim et al. 1997; Huguenard&McCormick, 2007), a neurosteroid-induced IPSP prolongation may simplistically be predicted to lengthen the period of rebound-dependent oscillations (e.g. sleep spindles). However, in contrast, more modest increases in IPSC decay time have been shown to paradoxically reduce rebound latency and network synchrony (Sohal et al. 2006), suggesting that any spatio-temporal variation of endogenous neurosteroidogenesis may differentially impact upon network operations. Adding further complexity, an enhanced tonic conductance increases temporal jitter in rebound burst timing and may reduce network synchrony (Sohal et al. 2006; Bright et al. 2007). Future computational modelling studies are thus required to dissect the relative impact of a dynamic steroid-induced modulation of tonic and phasic inhibition on thalamic network functions.
Even less predictable are the effects of the neurosteroid tone upon developing thalamocortical activity. Immature (P8–9) VB neurones express synaptic α2βγ2 not α1β2γ2 GABAARs and the tonic current is only ∼25% of that exhibited by more mature (P23–27) neurones (Peden et al. 2008). Adding complexity, at P7 we illustrate here that the endogenous neurosteroid tone is sufficient to act presynaptically to substantially reduce the quantal release of GABA onto VB neurones. Hence, at this developmental stage the neurosteroid appears to act both pre- and postsynaptically to reduce and facilitate the actions of GABA, respectively. Furthermore, determining whether GABA is excitatory or inhibitory will be fundamental to elucidating the action of neurosteroids on developing thalamus. GABA in (P9–12) rat VB neurones, in common with older mouse VB neurones, is reported to be hyperpolarising (Ulrich&Huguenard, 1997; Herd et al. 2009; authors' unpublished observations). However, critically, whether GABA causes a depolarisation or hyperpolarisation in P7 VB neurones to date has not been investigated. An additional consideration concerns the intracellular concentrations of Cl− within different neuronal compartments (cell body, dendrites, axon terminals), which may not be uniform, reflecting differential expression of cation–chloride co-transporters, e.g. KCC2 and NKCC1 (Trigo et al. 2008). Clearly a specific study on young thalamocortical neuronal activity is now warranted to elucidate the physiological relevance of the changing neurosteroid tone during development.
Conclusion
We reveal an important developmental role for neurosteroids, underpinning the dramatic changes to synaptic inhibition that occur early in thalamocortical neurone development. This precisely timed neurosteroid loss appears to be temporally harmonised with the emergence of an increased contribution from extrasynaptic GABAARs, allowing a transition from immature to mature inhibition. Interestingly, this attenuation of neurosteroid tone at P9–10 coincides precisely with the onset of coordinated, targeted movements and mobility in mice (Dehorter et al. 2011) and in many neurones with the dramatic change in the effects of GABA from depolarising to hyperpolarising (e.g. Tyzio et al. 2007). Although the neurosteroid influence dissipates by P10, we demonstrate that upon provision of substrate, these thalamic neurones retain the ability to synthesise GABAAR-active neurosteroids. A future challenge will be elucidating whether under certain physiological or pathophysiological circumstances local neurosteroids play a role in thalamocortical inhibition of mature circuits and, if so, determining the molecular mechanism(s) that triggers the synthesis of these potent neuromodulators.
Glossary
- α10/0
GABAA receptor α1 subunit ‘knockout’
- α40/0
GABAA receptor α4 subunit ‘knockout’
- 5α3α
5α-pregnan-3α-ol-20-one
- α-CD
α-cyclodextrin
- γ-CD
γ-cyclodextrin
- 5α-DHP
5α-dihydroprogesterone, or 5α-pregnane-3,20-dione
- 3α-HSD
3α-hydroxysteroid dehydrogenase
- CNS
central nervous system
- ECS
extracellular solution
- GABAAR
γ-aminobutyric acid type A receptor
- ICS
intracellular solution
- IPSC
inhibitory postsynaptic current
- KS
Kolmogorov–Smirnov statistical test
- mIPSC
miniature inhibitory postsynaptic current
- nRT
nucleus reticularis of the thalamus
- P
postnatal day
- τw
weighted decay time constant
- T90
time taken for the peak amplitude of an mIPSC to decrease by 90%
- TTX
tetrodotoxin
- VB
ventrobasal complex of the thalamus
- WT
wild-type
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
All experiments were performed and analysed by A.R.B. and M.B.H. A.R.B., D.B., M.B.H. and J.J.L. designed the experiments. All authors contributed to the interpretation of data, the drafting of the article or revising it critically for important intellectual content. We hereby confirm that all authors approved the final version of the manuscript and that all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The studies were supported by an A. J. Clark Studentship from the British Pharmacological Society (A.R.B.), Epilepsy Research UK (M.B.H.), Tenovus Tayside and the Anonymous Trust.
Author's present address
A. R. Brown: Pfizer Neusentis, The Portway, Granta Park, Cambridge CB21 6GS, UK.
References
- Adam JM, Bennett DJ, Bom A, Clark JK, Feilden H, Hutchinson EJ, Palin R, Prosser A, Rees DC, Rosair GM, Stevenson D, Tarver GJ. Zhang MQ. Cyclodextrin-derived host molecules as reversal agents for the neuromuscular blocker rocuronium bromide: synthesis and structure-activity relationships. J Med Chem. 2002;45:1806–1816. doi: 10.1021/jm011107f. [DOI] [PubMed] [Google Scholar]
- Agis-Balboa RC, Pinna G, Zhubi A, Maloku E, Veldic M, Costa E. Guidotti A. Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc Natl Acad Sci U S A. 2006;103:14602–14607. doi: 10.1073/pnas.0606544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF. Mennerick S. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Casula A, Ling A. Lambert JJ. The influence of subunit composition on the interaction of neurosteroids with GABAA receptors. Neuropharmacology. 2002;43:651–661. doi: 10.1016/s0028-3908(02)00172-7. [DOI] [PubMed] [Google Scholar]
- Belelli D. Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA. Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurones: a molecular target for hypnotics. J Neurosci. 2005;25:11513–11520. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S. Brussaard AB. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol. 2005;94:338–346. doi: 10.1152/jn.00084.2005. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG. Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI. Brickley SG. Synaptic release generates a tonic GABAA receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ. Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM. Reddy DS. Neurosteroid interactions with synaptic and extrasynaptic GABAA receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology (Berl) 2013;230:151–188. doi: 10.1007/s00213-013-3276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli PM, Casti A, Casu A, Frau R, Bortalato M, Spiga S. Ennas MG. Regional distribution of 5α-reductase type 2 in the adult rat brain: An immunohistochemical analysis. Psychoneuroendocrinology. 2013;38:281–293. doi: 10.1016/j.psyneuen.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Jia F, Liang J, Peng Z, Suryanarayanan A, Werner DF, Spigelman I, Houser CR, Olsen RW, Harrison NL. Homanics GE. GABAA receptor α4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci U S A. 2006;103:15230–15235. doi: 10.1073/pnas.0604304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari M, Eisenman LN, Covey DF, Mennerick S. Zorumski CF. The sticky issue of neurosteroids and GABAA receptors. Trends Neurosci. 2010;33:299–306. doi: 10.1016/j.tins.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Herbert AG, Holt RL, Peng K, Sherwood KD, Pangratz-Fuehrer S, Rudolph U. Huguenard JR. Endogenous positive allosteric modulation of GABAA receptors by diazepam binding inhibitor. Neuron. 2013;78:1063–1074. doi: 10.1016/j.neuron.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW. Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurones. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ME. Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–1035. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- Deidda G, Bozarth IF. Cancedda C. Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front Cell Neurosci. 2014;8:1–23. doi: 10.3389/fncel.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Michel FJ, Marissal T, Rotrou Y, Matrot B, Lopez C, Humphries MD. Hammond C. Onset of pup locomotion coincides with loss of NR2C/D-mediated cortico-striatal EPSCs and dampening of striatal network immature activity. Front Cell Neurosci. 2011;5:24. doi: 10.3389/fncel.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hulst C, Atack JR. Kooy RF. The complexity of the GABAA receptor shapes unique pharmacological profiles. Drug Discov Today. 2009;14:866–875. doi: 10.1016/j.drudis.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Do Rego JL, Seong JY, Burel D, Leprince J, Luu-The V, Tsutsui K, Tonon MC, Pelletier G. Vaudry H. Neurosteroid biosynthesis: enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front Neuroendocrinol. 2009;30:259–301. doi: 10.1016/j.yfrne.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Eser D, Schüle C, Baghai TC, Romeo E, Uzunov DP. Rupprecht R. Neuroactive steroids and affective disorders. Pharmacol Biochem Behav. 2006;84:656–666. doi: 10.1016/j.pbb.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Fritschy JM. Panzanelli P. GABAA receptors and plasticity of inhibitory neurotransmission in the central nervous system. Eur J Neurosci. 2014;39:1845–1865. doi: 10.1111/ejn.12534. [DOI] [PubMed] [Google Scholar]
- Gedell JA, Turnquist PA, Maciver MB. Pearce RA. Determination of partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br J Anaesth. 2004;93:810–817. doi: 10.1093/bja/aeh272. [DOI] [PubMed] [Google Scholar]
- Goldstein PA, Elsen FP, Ying SW, Ferguson C, Homanics GE. Harrison NL. Prolongation of hippocampal miniature inhibitory postsynaptic currents in mice lacking the GABAA receptor α1 subunit. J Neurophysiol. 2002;88:3208–3217. doi: 10.1152/jn.00885.2001. [DOI] [PubMed] [Google Scholar]
- Golovko T, Min R, Lozovaya N, Falconer C, Yatsenko N, Tsintsadze T, Tsintsadze V, Ledent C, Harvey RJ, Belelli D, Lambert JJ, Rozov A. Burnashev N. Control of inhibition by the direct action of cannabinoids on GABAA receptors. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu045. &(in press; DOI: 10.1093/cercor/bhu045) [DOI] [PubMed] [Google Scholar]
- Griffin LD. Mellon SH. Biosynthesis of the neurosteroid 3α-hydroxy-4-pregnen-20-one (3αHP), a specific inhibitor of FSH release. Endocrinology. 2001;142:4617–4622. doi: 10.1210/endo.142.11.8477. [DOI] [PubMed] [Google Scholar]
- Grobin AC. Morrow AL. 3α-hydroxy-5α-pregnan-20-one levels and GABAA receptor-mediated 36Cl− flux across development in rat cerebral cortex. Brain Res Dev Brain Res. 2001;131:31–39. doi: 10.1016/s0165-3806(01)00242-5. [DOI] [PubMed] [Google Scholar]
- Gunn BG, Cunningham L, Mitchell S, Swinny JD, Lambert JJ. Belelli D. GABAA receptor acting neurosteroids: a role in the development and regulation of the stress response. Front Neuroendocrinol. 2014 doi: 10.1016/j.yfrne.2014.06.001. &(in press; DOI: 10.1016/j.yfrne.2014.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd MB, Brown AR, Lambert JJ. Belelli D. Extrasynaptic GABAA receptors couple presynaptic activity to postsynaptic inhibition in the somatosensory thalamus. J Neurosci. 2013;33:14850–14868. doi: 10.1523/JNEUROSCI.1174-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd MB, Foister N, Chandra D, Peden DR, Homanics GE, Brown VJ, Balfour DJK, Lambert JJ. Belelli D. Inhibition of thalamic excitability by 4,5,6,7-tetrahydroisoxazolo[4,5-c]pyridine-3-ol: a selective role for δ-GABAA receptors. Eur J Neurosci. 2009;29:1177–1187. doi: 10.1111/j.1460-9568.2009.06680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd MB, Lambert JJ. Belelli D. The general anaesthetic etomidate inhibits the excitability of mouse thalamocortical neurons by modulating multiple modes of GABAA receptor-mediated inhibition. Eur J Neurosci. 2014;40:2487–2501. doi: 10.1111/ejn.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Inquimbert P, Rodeau JL. Schlichter R. Regional differences in the decay kinetics of GABAA receptor-mediated miniature IPSCs in the dorsal horn of the rat spinal cord are determined by mitochondrial transport of cholesterol. J Neurosci. 2008;28:3427–3437. doi: 10.1523/JNEUROSCI.5076-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Covey DF. Todorovic SM. Are neuroactive steroids promising therapeutic agents in the management of acute and chronic pain. Psychoneuroendocrinology. 2009;34:S178–185. doi: 10.1016/j.psyneuen.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL. Goldstein PA. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol. 2005;94:4491–4501. doi: 10.1152/jn.00421.2005. [DOI] [PubMed] [Google Scholar]
- Jones MV. Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Jüttner R, Meier J. Grantyn R. Slow IPSC kinetics, low levels of α1subunit expression and paired-pulse depression are distinct properties of neonatal inhibitory GABAergic synaptic connections in the mouse superior colliculus. Eur J Neurosci. 2001;13:2088–2098. doi: 10.1046/j.0953-816x.2001.01587.x. [DOI] [PubMed] [Google Scholar]
- Kim U, Sanchez-Vives MV. McCormick DA. Functional dynamics of GABAergic inhibition in the thalamus. Science. 1997;278:130–134. doi: 10.1126/science.278.5335.130. [DOI] [PubMed] [Google Scholar]
- Koksma JJ, Van Kesteren RE, Rosahl TW, Zwart R, Smit AB, Lüddens H. Brussaard AB. Oxytocin regulates neurosteroid modulation of GABAA receptors in supraoptic nucleus around parturition. J Neurosci. 2003;23:788–797. doi: 10.1523/JNEUROSCI.23-03-00788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JJ, Cooper MA, Simmons RD, Weir CJ. Belelli D. Neurosteroids: endogenous allosteric modulators of GABAA receptors. Psychoneuroendocrinology. 2009;34:S48–58. doi: 10.1016/j.psyneuen.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Li P, Shu HJ, Mennerick S, Zorumski CF, Covey DF, Steinbach JH. Akk G. Neurosteroid migration to intracellular compartments reduces steroid concentration in the membrane and diminishes GABAA receptor potentiation. J Physiol. 2007;584:789–800. doi: 10.1113/jphysiol.2007.142794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longone P, Rupprecht R, Manieri GA, Bernardi G, Romeo E. Pasini A. The complex roles of neurosteroids in depression and anxiety disorders. Neurochem Int. 2008;52:596–601. doi: 10.1016/j.neuint.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Maguire J. Mody I. Steroid hormone fluctuations and GABAAR plasticity. Psychoneuroendocrinology. 2009;34:S84–90. doi: 10.1016/j.psyneuen.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A. Neher E. Tight-seal whole-cell recording. In: Neher E, Sakmann B, editors; Single-Channel Recording. 2nd edn. New York: Plenum Press; 1995. pp. 42–45. [Google Scholar]
- Okada M, Onodera K, Van Renterghem C, Sieghart W. Takahashi T. Functional correlation of GABAA receptor α subunits expression with the properties of IPSCs in the developing thalamus. J Neurosci. 2000;20:2202–2208. doi: 10.1523/JNEUROSCI.20-06-02202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW. Sieghart W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acid A receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM. Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Peden DR, Petitjean CM, Herd MB, Durakoglugil MS, Rosahl TW, Wafford K, Homanics GE, Belelli D, Fritschy JM. Lambert JJ. Developmental maturation of synaptic and extrasynaptic GABAA receptors in mouse thalamic ventrobasal neurones. J Physiol. 2008;586:965–987. doi: 10.1113/jphysiol.2007.145375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Huntsman MM, Mihalek RM, Homanics GE. Huguenard JR. Intact synaptic GABAergic inhibition and altered neurosteroid modulation of thalamic relay neurones in mice lacking δ subunit. J Neurophysiol. 2003;89:1378–1386. doi: 10.1152/jn.00899.2002. [DOI] [PubMed] [Google Scholar]
- Purdy RH, Morrow AL, Moore PH., Jr Paul SM. Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci U S A. 1991;88:4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovó Z, Mátyás F, Barthó P, Slézia A, Lecci S, Pellegrini C, Astori S, Dávid C, Hangya B, Lüthi A. Acsády L. Phasic, nonsynaptic GABA-A receptor-mediated inhibition entrains thalamocortical oscillations. J Neurosci. 2014;34:7137–7147. doi: 10.1523/JNEUROSCI.4386-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U. Möhler H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol. 2014;54:483–507. doi: 10.1146/annurev-pharmtox-011613-135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalmann YB, Kirkcaldie MT, Waldron S. Calford MB. Cellular distribution of the GABAA receptor-modulating 3α-hydroxy, 5α-reduced pregnane steroids in the adult rat brain. J Neuroendocrinol. 2007;19:272–284. doi: 10.1111/j.1365-2826.2006.01527.x. [DOI] [PubMed] [Google Scholar]
- Schumacher M, Mattern C, Ghoumari A, Oudinet JP, Liere P, Labombarda F, Sitruk-Ware R, De Nicola AF. Guennoun R. Revisiting the roles of progesterone and allopregnanolone in the nervous system: resurgence of the progesterone receptors. Prog Neurobiol. 2014;113:6–39. doi: 10.1016/j.pneurobio.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Shen H, Sabaliauskas N, Sherpa A, Fenton AA, Stelzer A, Aoki C. Smith SS. A critical role for α4βδGABAA receptors in shaping learning deficits at puberty in mice. Science. 2010;327:1515–1518. doi: 10.1126/science.1184245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF. Mennerick S. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci. 2004;24:6667–6675. doi: 10.1523/JNEUROSCI.1399-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HJ, Zeng CM, Wang C, Covey DF, Zorumski CF. Mennerick S. Cyclodextrins sequester neuroactive steroids and differentiate mechanisms that rate limit steroid actions. Br J Pharmacol. 2007;150:164–175. doi: 10.1038/sj.bjp.0706973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS, Ruderman Y, Frye C, Homanics G. Yuan M. Steroid withdrawal in the mouse results in anxiogenic effects of 3α,5β-THP: a possible model of premenstrual dysphoric disorder. Psychopharmacology. 2006;186:323–333. doi: 10.1007/s00213-005-0168-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Pangratz-Fuehrer S, Rudolph U. Huguenard JR. Intrinsic and synaptic dynamics interact to generate emergent patterns of rhythmic bursting in thalamocortical neurons. J Neurosci. 2006;26:4247–4255. doi: 10.1523/JNEUROSCI.3812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Wu CS, Renger JJ, Uebele VN, Lu HC. Beierlein M. GABAergic synaptic transmission triggers action potentials in thalamic reticular nucleus neurons. J Neurosci. 2012;32:7782–7790. doi: 10.1523/JNEUROSCI.0839-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T. Postsynaptic receptor mechanisms underlying developmental speeding of synaptic transmission. Neurosci Res. 2005;53:229–240. doi: 10.1016/j.neures.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Trigo FF, Marty A. Stell B. Axonal GABAA receptors. Eur J Neurosci. 2008;28:841–848. doi: 10.1111/j.1460-9568.2008.06404.x. [DOI] [PubMed] [Google Scholar]
- Tokuda K, Izumi Y. Zorumski CF. Ethanol enhances neurosteroidogenesis in hippocampal pyramidal neurons by paradoxical NMDA receptor activation. J Neurosci. 2011;31:9905–9909. doi: 10.1523/JNEUROSCI.1660-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui K. Neurosteroids in the Purkinje cell: biosynthesis, mode of action and functional significance. Mol Neurobiol. 2008;37:116–125. doi: 10.1007/s12035-008-8024-1. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y. Khazipov R. Timing of the developmental switch in GABAA mediated signalling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia. 2007;48:96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- Ulrich D. Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova V, Sampson L. Uzunov DP. Relevance of endogenous 3α-reduced neurosteroids to depression and antidepressant action. Psychopharmacology (Berl) 2006;186:351–361. doi: 10.1007/s00213-005-0201-6. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL. Homanics GE. GABAA receptor α1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;21:3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT. Macdonald RL. Enhanced neurosteroid potentiation of ternary GABAA receptors containing the δ subunit. J Neurosci. 2002;22:1541–1549. doi: 10.1523/JNEUROSCI.22-05-01541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W. Vicini S. Neurosteroid prolongs GABAA channel deactivation by altering kinetics of desensitized states. J Neurosci. 1997;17:4022–4031. doi: 10.1523/JNEUROSCI.17-11-04022.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, Paul SM, Izumi Y, Covey DF. Mennerick S. Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37:109–122. doi: 10.1016/j.neubiorev.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]