

Abstract
In preclinical studies, neural stem cell (NSC)-based delivery of oncolytic virus has shown great promise in the treatment of malignant glioma. Ensuring the success of this therapy will require critical evaluation of the spatial distribution of virus after NSC transplantation. In this study, the patient derived GBM43 human glioma line was established in the brain of athymic nude mice, followed by administration NSCs loaded with conditionally replicating oncolytic adenovirus (NSC-CRAd-S-pk7). We determined the tumor coverage potential of oncolytic adenovirus by examining NSC distribution using magnetic resonance (MR) imaging and by three-dimensional reconstruction from ex vivo tissue specimens. We demonstrate that unmodified NSCs and NSC-CRAd-S-pk7 exhibit a similar distribution pattern with most prominent localization occurring at the tumor margins. We were further able to visualize the accumulation of these cells at tumor sites via T2-weighted MR imaging as well as the spread of viral particles using immunofluorescence. Our analyses reveal that a single administration of oncolytic virus-loaded NSCs allows for up to 31% coverage of intracranial tumors. Such results provide valuable insights into the therapeutic potential of this novel viral delivery platform.
Introduction
The most common primary malignant brain tumor affecting adult patients is glioblastoma (GBM), a WHO grade IV astrocytoma. This disease carries a very poor prognosis with patients exhibiting a median survival of about 14.6 months 1 and a 5-year survival rate of only 9.8% under a care regimen of surgical debulking, radiotherapy, and adjuvant chemotherapy with temozolomide 2. This poor prognosis reflects the inability of these therapies to adequately target evasive glioblastoma cells. Local recurrence is common secondary to (a) small, invasive tumor foci that may be spatially non-contiguous with the main tumor burden and (b) the presence of chemo- and radio-resistant populations of glioma cells that exist within the tumor 3. Thus, novel therapeutics are needed that can both target infiltrative disease while overcoming mechanisms of resistance in these cancer cells.
Neural stem cells (NSCs) have recently been used as delivery vehicles due to their inherent tumor-tropism, migrating towards a tumor mass as well as smaller micro-tumor foci found at or well beyond the tumor’s infiltrating edge 4, 5. Preclinical studies have demonstrated the effectiveness of NSCs for prodrug carboxylesterase-mediated conversion of CPT-11 (irinotecan) to SN-38 6–8, gene therapy 5, biomolecule/antibody expression 9, 10, and oncolytic virus delivery 11–15 in the context of a spectrum of brain tumor types including malignant glioma, medulloblastoma, and secondary brain metastases. In fact, this concept has already made its way into the clinical arena: the HB1.F3.CD NSC line for cytosine deaminase-mediated conversion of 5-Flucytosine to 5-Flurouracil is currently undergoing clinical trial testing in recurrent high-grade glioma patients (completed safety study NCT01172964; phase I dose escalation study in progress NCT02015819).
Enhancing oncolytic adenovirus delivery is an especially intriguing therapeutic avenue because these viruses have been shown to target the tumorigenic glioma cancer stem cell population as well 12, 16, 17. In fact, conditionally replicating adenoviruses that have been modified to possess survivin promoter-driven E1A gene expression and a polylysine addition to the viral fiber (CRAd-S-pk7) can effectively prolong survival in mice bearing intracranial glioma xenografts 18. NSC-mediated delivery of CRAd-S-pk7 has been shown to even further improve survival compared to free oncolytic virus in several murine preclinical models of human glioma, suggesting this platform’s potential use in glioma patients 11–15. However, there are a few areas of investigation that may be important in moving this therapy forward. First, it is important to determine the distribution pattern of NSCs within a tumor mass as these cells will mostly be targeting residual glioma cells at the tumor margins after surgical resection. Second, it is essential to quantify the therapeutic tumor coverage of oncolytic virus loaded NSCs to rule out inhibition of tropism as compared to unloaded NSCs. A previous study by Lin et al. 2007 examined the three dimensional distribution of HB1.F3.C1 NSCs in an intracranial glioma mouse model 19. In this study, confocal microscopy of serial tissue sections allowed for three-dimensional modeling of NSC and glioma cell spatial distribution providing insight into the potential tumor coverage of unloaded NSCs in the context of pro-drug therapy 19. However, to date, no method has been applied to analyze the tumor coverage potential of NSCs carrying oncolytic virus.
Our aims in this study were to determine the distribution patterns of CRAd-S-pk7-loaded NSCs and quantify the tumor coverage potential of these cells (Figure 1). In order to do this, both quantitative and qualitative methods were employed, including calculating the distance of individual NSCs to the tumor border, visualizing intratumoral accumulation of NSCs via MR imaging, and reconstructing three-dimensional images from ex vivo tissue specimens. We first demonstrated that unmodified NSCs and NSC-CRAd-S-pk7 both share a similar distribution pattern with localization mainly at the tumor borders. We were further able to visualize the accumulation of these cells at the tumor site via T2-weighted MR imaging as well as the spread of viral particles via immunofluorescence. Finally, our analysis revealed that a single administration of NSC-CRAd-S-pk7 allowed for up to 31% coverage of intracranial tumors.
Figure 1.

Neural stem cells can increase the therapeutic distribution of oncolytic virus. (A) Replication competent oncolytic viruses must rely on diffusion and pressure gradients to achieve distribution within a tumor. Neural stem cells have the potential to distribute within malignant gliomas (see text), suggesting their function as therapeutic carrier cells. (B) In this report, we examine the distribution of CRAd-S-pk7 oncolytic virus-loaded and ferumoxytol-labeled NSCs (NSC-CRAd-S-pk7) in an intracranial xenograft mouse model of GBM to determine the therapeutic coverage potential of this platform.
Materials and Methods
Cell culture
GBM43 glioma cells, derived from human GBM primary tissue, were obtained from Dr. David James (Northwestern University, Chicago, IL, USA) and maintained according to a protocol approved by the institutional review board at Northwestern University. Cells were maintained as serially passaged subcutaneous xenografts in athymic mice 20. Prior to intracranial injection, GBM43 cells were thawed, washed, and resuspended in serum free Dulbecco’s Modified Eagle’s media (DMEM)(Invitrogen, Carlsbad, CA, USA) at a concentration of 5 × 104 cells/2 μL for stereotactic intracranial injections.
This study used the established clonal, v-myc immortalized human NSC line HB1.F3.CD 21, a line that is established as a Master Cell Bank at City of Hope. Briefly, HB1.F3.CD cells were thawed and cultured in DMEM supplemented with 10% heat-activated fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and 2 mM L–glutamine (Invitrogen, Carlsbad, CA, USA).
Labeling of NSCs with ferumoxytol
HB1.F3.CD cells were labeled in culture with ferumoxytol according to a protocol previously reported 22, 23. Herparin-protamine sulfate-ferumoxytol (HPF) complex was prepared by mixing 2 U/mL heparin and 40 μg/mL protamine sulfate (American Pharmaceuticals Partners, LLC, Lake Zurich, IL, USA) with 100 μg/mL ferumoxytol (AMAG Pharmaceuticals, Inc., Schaumburg, IL, USA). After 24 h in culture with HPF, labeled HB1.F3.CD cells were washed with phosphate-buffered saline (PBS), trypsinized, and either prepared for oncolytic virus loading (NSC-CRAd-S-pk7 groups) or for intracranial injections directly (NSC groups).
Oncolytic viral vector and loading into NSCs
CRAd-S-pk7 is a replication-competent adenoviral vector that contains the wild-type adenovirus replication protein, E1A, under the control of the human survivin promoter, a gene that is known to be overexpressed in malignant glioma 24, 25. This viral vector was created via homologous recombination using a shuttle plasmid containing the survivin promoter upstream of the E1A gene and an adenoviral vector backbone modified to contain a poly-lysine (pk7) addition onto the C-terminus of the wild-type fiber protein 18.
To load HB1.F3.CD cells, cell suspensions were incubated with DMEM containing 10% FBS and 50 IU/cell of CRAd-Survivin-pk7 viruses. 50 IU/cell is a concentration that has been determined to produce the highest adenovirus replication and infectious progeny while minimizing toxicity towards NSCs 11. HB1.F3.CD cells were incubated with the virus for 2 hours, and during this time, the tube containing the cells was lightly mixed every 15 min to achieve maximum loading capacity. After this 2-hour incubation, HB1.F3.CD cells were washed twice with DMEM and once with PBS in preparation for intracranial implantation.
In vivo intracranial tumor implantation and NSC administration
For intracranial glioma xenograft establishment, 5 × 104 GBM43 cells in 2 μL of serum free DMEM were implanted into the right frontal lobe of athymic nude mice using a stereotactic instrument. Cells were loaded into a 25 μL Hamilton syringe with a 30-gauge needle and injected over 2 minutes at 2 mm lateral and 0.5 mm anterior to bregma at a depth of 3.3 mm from the surface of the skull. On day 6 following GBM43 engraftment, 5 × 105 NSC or NSC-CRAd-S-pk7 cells in 2 μL PBS were injected either adjacent to the tumor site (caudolateral to tumor) or into the contralateral frontal lobe. All animal studies were conducted under approved City of Hope (no. 04011) and Children’s Hospital of Los Angeles (no. 285) institutional animal care and use committee protocols. Mice from each group were euthanized on day 4 or day 7 after NSC injection, and brain tissue was collected for histological processing and analysis.
MR imaging
Mice underwent MRI scanning before NSC injection and on days 1, 4 and 7 after NSC injection to track the distribution of NSCs in the brain. Mice were inserted in the prone position into a small animal MRI scanner (PharmaScan 300, Bruker BioSpin Division, Billerica, MA, USA) 7T magnet using the 19 mm inner diameter transmit receive coil. ParaVision 4.0 scanner software was set to use Rapid Acquisition with Relaxation Enhancement (RARE) spin echo sequence for fast T2-weighted imaging (TE 50, TR 3 000, RARE Factor 8) with a 256 × 256 in-plane matrix and 2.56 cm field of view. MRI images were reconstructed at native resolution. For each mouse, 22 axial images with 0.4 mm thick slices and 0.02 mm gap between slices were acquired. This produced 0.1 mm × 0.1 mm per pixel in-plane resolution with an effective slice thickness of 0.42 mm. DICOM data was stored in the small animal imaging PACS server and processed using the Mayo ANALYZE (AnalyzeDirect, Inc., Overland Park, KS, USA) and OsiriX (Open-Source Software for Navigating in Multidimensions) image analysis software packages. Mice were anesthetized with isofluorane throughout the entire imaging procedure.
Histological staining and immunofluorescence
Brain tissue was harvested and fixed in 4% paraformaldehyde for 72–96 hours. Tissues were paraffin embedded and sectioned at 10 μm in thickness, and hematoxylin and eosin (H&E) and Prussian blue staining was done every 10th section to visualize the tumor and to detect ferumoxytol-labeled NSCs. Prussian blue staining was performed using the Accustain iron stain kit (Sigma-Aldrich, St. Louis, MO, USA). Images were captured with a 10× lens using Nikon-Eclipse 2E200U. Viral hexon protein staining was done using an anti-hexon FITC-conjugated antibody (Millipore, Billerica, MA, USA). In brief, sections were blocked using 1% BSA + 0.3% Triton-X for 1 h and then incubated with the antibody diluted in 1% BSA overnight at 4 °C. Sections were then washed in PBS and mounted using ProLong® Gold Antifade Mountant with DAPI (Life Technologies, Waltham, MA, USA). Imaging was performed at the University of Chicago Integrated Light Microscopy Facility. Images were captured with a Zeiss Axiovert 200m inverted epifluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY, USA) with a Hamamatsu Orca ER CCD camera (Hamamatsu Photonics, Skokie, IL, USA) for fluorescence imaging run by SlideBook 5.5 software (Intelligent Imaging Innovations, Denver, CO, USA).
Quantitative distribution analysis and three-dimensional reconstruction of tumor volume and NSC location
The Automatic Cellular Imaging System (ACIS from ChromaVision Medical Systems, Inc., San Juan Capistrano, CA, USA) was used to generate high-resolution images of Prussian blue stained histological sections (10 μm thick). For analysis of distribution, mouse tumor outlines were generated in ImageJ (http://rsb.info.nih.gov/ij/) by first segmenting the mouse tumor using the Simple Interactive Object Extraction (SIOX) segmentation tool. Next, the tumor edge was identified using the Image Edge plug-in (Deriche, 1.00 alpha). NSCs were segmented in ImageJ using the color threshold tool (Hue: 147–187, Saturation: 0–255, Brightness: 90–190). Regions within some glial cells contained small portions of color that fell within the color threshold selection. To remove these non-NSC selections, ImageJ’s Remove Outlier tool (Radius=2, threshold=50) was applied. Lastly, to account for color thresholding that selected multiple parts of the same stem cell, the stem cell selections were dilated to combine disparate selections within a couple pixel distances apart. After segmentation of the tumor border and NSCs, the minimum distance of each individual NSC to the tumor border was calculated in Matlab R2013a (MathWorks, Natick, MA, USA) using the function bwdist. Image scale was approximately 1 μm/pixel. A visual representation of the work-flow can be found in Supplemental Figure 1.
Three-dimensional reconstruction was performed using Reconstruct software 26. For each tumor, 10 serial brain sections separated by 200 μm were imported into Reconstruct and aligned manually based on anatomical landmarks visualized in H&E stained sections. The tumors were outlined by eye based on cell density in H&E stained sections, and NSCs were visualized based on blue color in Prussian blue stained sections. Tumor and NSC areas in each section were recorded. To produce three-dimensional images, structures of interest were segmented based on color (e.g. Prussian blue label for NSCs) and cell density (e.g. H&E staining of tumor areas). Tumor volume was quantified according to the Cavalieri principle by extrapolating the numbers of tumor-identified pixels over the 200 μm separating individual slides. Tumor coverage was estimated by drawing a 100 μm radius from the edge of Prussian blue-marked areas and determining the numbers of tumor area-containing pixels within this area.
Statistical Analysis
All statistical analyses were performed using Graphpad Prism 4 (GraphPad Software Inc., San Diego CA). To compare NSC vs. NSC-CRAd-S-pk7 distribution within the tumor vicinity (Figure 2C), Kolmogorov–Smirnov test was applied with a sample size of n=18 per group. To compare NSC vs. NSC-CRAd-S-pk7 tumor coverage (Figure 3B), paired t-test was used with a sample size of n=9 per group. Numerical data was reported as mean ± standard error of the mean. All reported p values were two-sided and considered to be statistically significant only if p<0.05.
Figure 2.

Quantitative analysis and visualization of NSC and NSC-CRAd-S-pk7 distribution within the tumor vicinity. A) Schematic of experimental design: athymic nude mice were administered GBM 43 cells into the right hemisphere with administration of stem cells either adjacent to or in the contralateral cortical hemisphere to the established GBM43 tumor. B) Histogram of NSCs or NSC-CRAd-S-pk7 distribution after either adjacent or contralateral injection. Cells were found to be distributed mostly at or adjacent to the tumor border (negative values = within tumor; positive values = outside tumor). C) Comparison of NSC and NSC-CRAd-S-pk7 distribution at day 4 after adjacent administration revealed no difference (n = 18 per group; K-S test; p=0.97). D) Reconstruction of tumor area and stem cell location from brain tissue sections collected on day 4. The majority of stem cells were distributed around the periphery of tumors. Scale bars = 200 μm.
Figure 3.
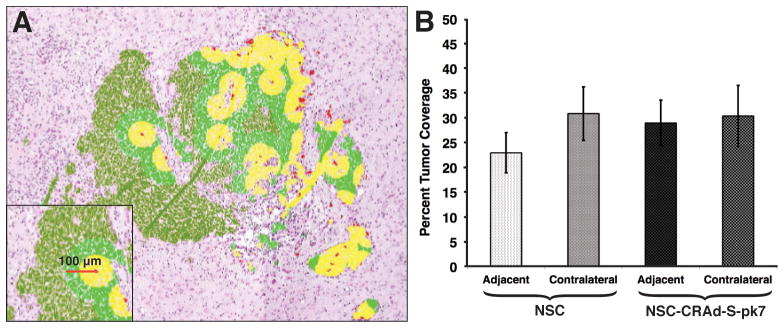
Determination of tumor coverage by NSC and NSC-CRAd-S-pk7. A) Visual representation of tumor coverage analysis. NSCs were identified by histology based on Prussian blue staining. A therapeutic diameter of 100 μm was used in the calculation of tumor coverage. B) Percent tumor coverage was determined for both NSC and NSC-CRAd-S-pk7 after either adjacent or contralateral administration. No difference was observed between adjacent NSC vs NSC-CRAd-S-pk7 groups (paired t-test; p = 0.37) or for contralateral NSC vs NSC-CRAd-S-pk7 groups (paired t-test; p = 0.96) (n = 9 for all groups)
Results
CRAd-S-pk7 loading does not alter the distribution potential of NSCs in glioma tumor xenografts
In order to determine if the distribution pattern of NSCs was altered after oncolytic virus loading, we compared the location of NSCs and NSC-CRAd-S-pk7 in reference to the border of a tumor. Athymic nude mice bearing established GBM43 tumors in the right fontal cerebral hemisphere were administered either NSCs or NSC CRAd-S-pk7 adjacent to (i.e. caudolateral to the tumor) or contralateral to the tumor (experimental design depicted in Figure 2A). Both cell lines were modified with ferumoxytol, an ultra small iron oxide nanoparticle that allows for cell tracking in vivo by MR imaging and ex vivo with Prussian blue staining 22. At day 4 and 7 after NSC administration, brain tissue was harvested, and NSCs and NSC-CRAd-S-pk7 identified histologically via Prussian blue staining. In all groups, NSCs distributed themselves mainly at or just outside the tumor border, with a smaller portion of cells penetrating into the interior of the tumor (Figure 2B). NSCs and NSC-CRAd-S-pk7 distribution at day 4 were comparable with no significant difference (n = 18 sections per group; K-S test: p=0.97) (Figure 2C). Interestingly, stem cell accumulation at the tumor site did not depend on its size (Supplemental Figure 2), suggesting another factor may be involved in determining migration efficiency.
To better visualize distribution patterns in these groups, we performed three-dimensional reconstructions using Reconstruct Software. Brain tissue sections were collected from mice sacrificed at either day 4 or day 7 after NSC or NSC-CRAd-S-pk7 administration and stained with Prussian blue to label the stem cells. As with the quantitative data, these reconstructed images demonstrated NSC distribution mostly around the tumor periphery whether oncolytic virus had been loaded or not (Figure 2D and Supplemental Figure 3).
Quantification of therapeutic tumor coverage by NSCs
To better understand the extent of therapeutic benefit that can be achieved with oncolytic virus-loaded NSCs, it is important to determine the spatial extent of tumor volume coverage. Data used to make the three-dimensional reconstruction images were analyzed in order to determine the tumor coverage potential of NSCs and NSC-CRAd-S-pk7. Each Prussian blue-stained NSC was assigned a therapeutic radius of 100 μm, and using this radius, we determined the numbers of tumor area-containing pixels within a given NSC’s area-of-effect (visualized in Figure 3A). Brains harvested on day 4 were used in the analysis to examine the extent of tumor coverage. Four different conditions were analyzed: NSCs administered adjacent to or contralateral to an established tumor as well as NSC-CRAd-S-pk7 administered adjacent to or contralateral to an established GBM43 intracranial xenograft. Our results demonstrate that a single administration of oncolytic virus-loaded NSCs was enough to achieve between 22.95 – 30.87 % coverage of the tumor (Figure 3B). There was no significant difference between NSC and NSC-CRAd-S-pk7 groups. Interestingly, the tumor coverage potential did not differ whether stem cells had been administered adjacent or contralateral to the GBM43 tumors, demonstrating the great migratory behavior of these cells.
MR imaging allows for visualization of ferumoxytol-labeled NSC-CRAd-S-pk7 cells at the tumor site
To follow and optimize the use of oncolytic virus-loaded NSCs in a clinical setting, it is important to develop a non-invasive tracking method to monitor these cells in vivo. Ferumoxytol labeling of NSCs allows for T2-weighted MRI tracking, a method that has already undergone preclinical testing and FDA approval for HB1.F3.CD cell tracking in glioma patients 22. NSC-CRAd-S-pk7 cells loaded with ferumoxytol were administered either adjacent to established GBM43 xenografts or in the contralateral cortical hemisphere. T2-weighted MRI images of mice were then acquired at either day 4 or day 7 after stem cell administration to assess for accumulation at the tumor site. Hypointense areas were found in the tumor vicinity on day 4 after adjacent administration of NSC-CRAd-S-pk7 cells when compared with pre-injection images (Figure 4A and Supplemental Figure 4). Similar changes were observed in the contralateral injection group by day 7 (Figure 4D). In order to confirm that these hypointense areas were indeed caused by ferumoxytol-loaded NSCs, brain tissue was harvested and subjected to histological Prussian blue staining. Labeled NSCs were found in locations that corresponded to the hypointense regions seen on MRI (Figures 4B and D).
Figure 4.
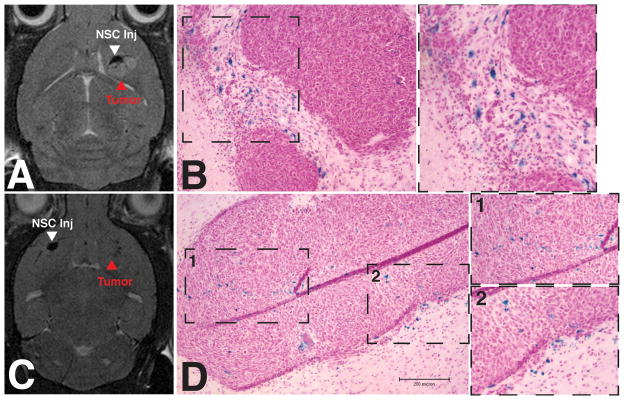
MRI visualization of NSC-CRAd-S-pk7 migration. NSC-CRAd-S-pk7 were administered either adjacent to (A,B) or in the contralateral cortical hemisphere (C,D) opposite established GBM43 tumors. T2-weighted MRI scans and brain tissue sections were acquired at day 4. Prussian blue staining of tissue sections from these same animals were done to confirm the presence of ferumoxytol-labeled stem cells.
NSC-CRAd-S-pk7 cells allow for specific delivery of oncolytic virus to intracranial tumors
To ensure that oncolytic virus was indeed being delivered to tumor cells, athymic nude mice bearing established GBM43 tumors with adjacent administration of NSC-CRAd-S-pk7 were sacrificed at day 7 after injection to visualize viral distributions. Immunofluorescence using an antibody against viral hexon protein allowed for the detection of viral particles. We observed extensive distribution of viral particles throughout the tumor bed at a distance from the administration site with no observable staining in the surrounding normal brain parenchyma (Figure 5A). Furthermore, areas containing high densities of viral particles also showed numerous atypical cells representing tumor cell destruction (Figure 5B).
Figure 5.
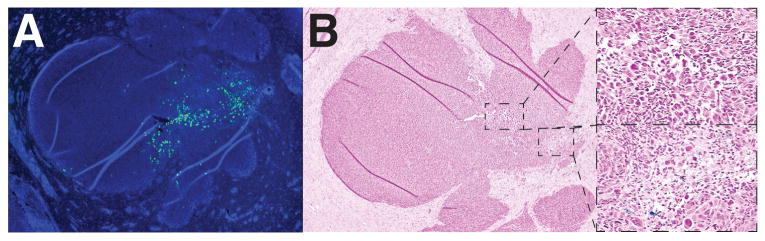
NSC-CRAd-S-pk7 successfully deliver oncolytic virus to the tumor area. A) Immunofluorescence staining of viral hexon protein with anti-hexon FITC-conjugated antibody demonstrated viral particle distribution only within the tumor area by day 7. B) Corresponding Prussian blue stained tissue sections demonstrated few ferumoxytol-labeled cells, with regions containing atypical cells corresponding to hexon-positive areas.
Discussion
NSC-mediated delivery of oncolytic virus has been a very promising strategy for inducing glioma cell destruction in several in vivo studies 11, 12, 15. Our results complement these previous reports by examining the critical questions as to 1) whether viral loading into NSCs alters their distributional pattern after administration and 2) the extent of NSC-CRAd-S-pk7 therapeutic coverage. While other studies have demonstrated by qualitative methods that NSCs loaded with oncolytic virus preserve their migratory potential 11, 12, the data we present examine this in a much more detailed way. We demonstrate the majority of NSCs (whether loaded with oncolytic virus or not) distribute themselves at the border of the tumor or just outside it. This peripheral distribution was further confirmed when we visualized NSC distribution using reconstruction of images giving a three-dimensional view of the tumor area. The distribution pattern observed suggests that this platform may be best used in conjunction with other glioblastoma therapies that target the bulk of a tumor such as surgical debulking and radiotherapy. In the current phase I clinical trial using NSCs for prodrug conversion (NCT02015819), NSCs serve as an adjuvant to gross surgical resection by homing to residual infiltrating tumor cells. Also of note, while NSC distribution was mostly peripheral, viral particles were dispersed within the interior of the glioma tumor. This suggests that viral release and infectivity of surrounding cancer cells allow for deeper tumor penetration and broader extension into the diffusely invading tumor periphery.
Other studies have also quantified the tumor coverage potential of NSCs in the context of pro-drug conversion. Lin et al. 2007 demonstrated that, with administration of 2 × 105 NSCs and with a final glioma tumor volume of 849 881 – 1 735 366 μm3, about 75–94% of the tumor could be covered assuming a killing radius of 23 μm 19. Although these tumor coverage values were greater than the values we report, the authors used a tumor cell line that is less aggressive compared to GBM43, the stem cell line used in the study had different modifications (CM-DiI), and a different detection method (confocal microscopy) was used for quantifying NSC distribution and tumor volume. While our results show between 23–31% coverage of the tumor, this is only after a single dose of 5 × 105 NSC-CRAd-S-pk7 cells. With multiple doses or initial administration of a greater quantity of oncolytic virus-loaded NSCs, it may be possible to achieve more extensive coverage. It also is important to note that preclinical experiments that used a similar experimental design to the one in our study have led to significant survival improvements in mice bearing intracranial glioma xenografts 11–15. Furthermore, our results pertain to coverage of a dense mass of tumor cells, while in humans these NSCs will be delivered after extensive tumor resection. As such, we might expect a more extensive coverage of scattered islands of tumor cells that have a greater surface area-to-volume ratio.
Our results also demonstrate that loading NSC-CRAd-S-pk7 with ferumoxytol could be a clinically useful way to monitor their distribution in vivo. Gutova et al. 2013 demonstrated that ferumoxytol allowed for T2-weighted MRI monitoring of the same HB1.F3.CD cell line used in our study 22. Our results expand on this by demonstrating that this iron oxide nanoparticle can be loaded in conjunction with CRAd-S-pk7 without impairing migratory function. Through T2-weighted MR imaging, we were able to demonstrate that ferumoxytol-loaded NSC-CRAd-S-pk7 cells could accumulate within the tumor site with hypointense signal observed as early as 4 days after NSC administration. This dual loading feature also speaks to the flexibility of NSCs to serve as delivery vehicles of a wide variety of compounds where both tumor-destruction capabilities and non-invasive imaging feedback are possible.
While NSC-mediated delivery of therapeutics is a promising avenue, there are still several ways in which such platforms may be optimized to enable improved tumor penetration and distribution. First, their delivery potential may be improved if oncolytic virus replication is delayed. This could be accomplished with exogenously stimulated promoters that control viral replication. Thus, improvement in the control over viral proliferation may delay toxicity to these carrier cells, providing additional time to achieve more extensive distribution. Enhancing the engraftment efficiency of NSCs should also improve tumor coverage. Administration of stem cells within gel scaffolds composed of extracellular matric proteins like laminin, fibronectin, and thrombin have improved stem cell survival, migration, and distribution in other disease models 27, 28 and may serve a role in stem cell therapy for GBM. Another method to improve stem cell distribution is to up-regulating receptors/signaling pathways in NSCs of that are critical for tumor-tropic migration. A number of factors have been found to be involved in NSC migration towards cancer cells including HIF-1 α, hepatocyte growth factor/c-MET, and VEGF/VEGF receptor 29, 30. By overexpressing these critical molecules in NSCs, more extensive distribution may be achieved.
In conclusion, our results demonstrate that NSCs loaded with replication competent oncolytic virus can achieve significant coverage of an intracranial glioma after only a single administration of these cells. Furthermore, not only is the migratory capacity of these cells maintained, their final spatial distribution around the tumor periphery indicates a clear role in targeting infiltrating glioma cells in the surrounding normal tissue.
Supplementary Material
Supplemental Figure 1. Schematic of analysis of NSC location determination and NSC distributional pattern within the tumor area.
Supplemental Figure 2. Scatter-plot of tumor-localized stem cells vs tumor volume.
Supplemental Figure 3. Reconstruction of tumor area and stem cell location from brain tissue sections collected on day 4 & 7.
Supplemental Figure 4. Example T2-weighted MRI images before and after NSC-CRAd-S-pk7 administration. Stem cells were administered either adjacent to or contralateral to an established GBM43 tumor (right side). Hypointense regions within the tumor area were detected in mice by day 4, demonstrating ferumoxytol-labeled NSC migration (dotted white boxes represent tumor area).
Acknowledgments
The authors thank Ms. Lingjiao Zhang, M.S. for her expertise in the statistical analysis of our studies.
Footnotes
Conflict of Interest: A.J.A and K.S.A are share-holders, directors, and officers of TheraBiologics, Inc. a clinical stage biopharmaceutical company focused on the development of stem cell-mediated cancer therapy. R.A.M is a director of TheraBiologics, Inc.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Nicholas MK, Lukas RV, Chmura S, Yamini B, Lesniak M, Pytel P. Molecular heterogeneity in glioblastoma: therapeutic opportunities and challenges. Sem Oncol. 2011;38(2):243–53. doi: 10.1053/j.seminoncol.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97(23):12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benedetti Gene therapy of experimental brain tumors using neural progenitor cells. Nature Med. 2000;6:447–450. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- 6.Gutova M, Shackleford GM, Khankaldyyan V, Herrmann KA, Shi XH, Mittelholtz K, et al. Neural stem cell-mediated CE/CPT-11 enzyme/prodrug therapy in transgenic mouse model of intracerebellar medulloblastoma. Gene Ther. 2013;20(2):143–50. doi: 10.1038/gt.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metz MZ, Gutova M, Lacey SF, Abramyants Y, Vo T, Gilchrist M, et al. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl Med. 2013;2(12):983–92. doi: 10.5966/sctm.2012-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seol HJ, Jin J, Seong DH, Joo KM, Kang W, Yang H, et al. Genetically engineered human neural stem cells with rabbit carboxyl esterase can target brain metastasis from breast cancer. Cancer Lett. 2011;311(2):152–9. doi: 10.1016/j.canlet.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Frank RT, Edmiston M, Kendall SE, Najbauer J, Cheung CW, Kassa T, et al. Neural stem cells as a novel platform for tumor-specific delivery of therapeutic antibodies. PLoS One. 2009;4(12):e8314. doi: 10.1371/journal.pone.0008314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehtesham M, Kabos P, Gutierrez MAR, Chung NHC, Griffith TS, Black KL, et al. Induction of Glioblastoma Apoptosis Using Neural Stem Cell-mediated Delivery of Tumor Necrosis Factor-related Apoptosis-inducing Ligand. Cancer Res. 2002;62(24):7170–7174. [PubMed] [Google Scholar]
- 11.Ahmed AU, Thaci B, Alexiades NG, Han Y, Qian S, Liu F, et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011;19(9):1714–26. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed AU, Thaci B, Tobias AL, Auffinger B, Zhang L, Cheng Y, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst. 2013;105(13):968–77. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyler MA, Ulasov IV, Sonabend AM, Nandi S, Han Y, Marler S, et al. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009;16(2):262–78. doi: 10.1038/gt.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thaci B, Ahmed AU, Ulasov IV, Tobias AL, Han Y, Aboody KS, et al. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012;19(6):431–42. doi: 10.1038/cgt.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed AU, Tyler MA, Thaci B, Alexiades NG, Han Y, Ulasov IV, et al. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm. 2011;8(5):1559–72. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alonso MM, Jiang H, Gomez-Manzano C, Fueyo J. Targeting brain tumor stem cells with oncolytic adenoviruses. Methods Mol Biol. 2012;797:111–25. doi: 10.1007/978-1-61779-340-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai R, Rabkin SD, Yip S, Sgubin D, Zaupa CM, Hirose Y, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulasov IV, Zhu ZB, Tyler MA, Han Y, Rivera AA, Khramtsov A, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther. 2007;18(7):589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- 19.Lin D, Najbauer J, Salvaterra PM, Mamelak AN, Barish ME, Garcia E, et al. Novel method for visualizing and modeling the spatial distribution of neural stem cells within intracranial glioma. Neuroimage. 2007;37(Suppl 1):S18–26. doi: 10.1016/j.neuroimage.2007.03.076. [DOI] [PubMed] [Google Scholar]
- 20.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-Oncol. 2005;7(2):164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, et al. Neural Stem Cell–Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Sci Transl Med. 2013;5(184):184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutova M, Frank JA, D’Apuzzo M, Khankaldyyan V, Gilchrist MM, Annala AJ, et al. Magnetic resonance imaging tracking of ferumoxytol-labeled human neural stem cells: studies leading to clinical use. Stem Cells Transl Med. 2013;2(10):766–75. doi: 10.5966/sctm.2013-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thu MS, Najbauer J, Kendall SE, Harutyunyan I, Sangalang N, Gutova M, et al. Iron labeling and pre-clinical MRI visualization of therapeutic human neural stem cells in a murine glioma model. PLoS One. 2009;4(9):e7218. doi: 10.1371/journal.pone.0007218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakravarti A, Noll E, Black PM, Finkelstein DF, Finkelstein DM, Dyson NJ, et al. Quantitatively Determined Survivin Expression Levels Are of Prognostic Value in Human Gliomas. J Clin Oncol. 2002;20(4):1063–1068. doi: 10.1200/JCO.2002.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 25.Xie D, Zeng YX, Wang HJ, Wen JM, Tao Y, Sham JS, et al. Expression of cytoplasmic and nuclear Survivin in primary and secondary human glioblastoma. Br J Cancer. 2006;94(1):108–14. doi: 10.1038/sj.bjc.6602904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218(1):52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- 27.Bensai_d W, Triffitt JT, Blanchat C, Oudina K, Sedel L, Petite H. A biodegradable fibrin scaffold for mesenchymal stem cell transplantation. Biomaterials. 2003;24(14):2497–2502. doi: 10.1016/s0142-9612(02)00618-x. [DOI] [PubMed] [Google Scholar]
- 28.Tate CC, Shear DA, Tate MC, Archer DR, Stein DG, LaPlaca MC. Laminin and fibronectin scaffolds enhance neural stem cell transplantation into the injured brain. J Tissue Eng Regen M. 2009;3(3):208–217. doi: 10.1002/term.154. [DOI] [PubMed] [Google Scholar]
- 29.Gutova M, Najbauer J, Frank RT, Kendall SE, Gevorgyan A, Metz MZ, et al. Urokinase Plasminogen Activator and Urokinase Plasminogen Activator Receptor Mediate Human Stem Cell Tropism to Malignant Solid Tumors. Stem cells. 2008;26(6):1406–1413. doi: 10.1634/stemcells.2008-0141. [DOI] [PubMed] [Google Scholar]
- 30.An JH, Lee SY, Jeon JY, Cho KG, Kim SU, Lee MA. Identification of Gliotropic Factors That Induce Human Stem Cell Migration to Malignant Tumor. J Proteome Res. 2009;8(6):2873–2881. doi: 10.1021/pr900020q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Schematic of analysis of NSC location determination and NSC distributional pattern within the tumor area.
Supplemental Figure 2. Scatter-plot of tumor-localized stem cells vs tumor volume.
Supplemental Figure 3. Reconstruction of tumor area and stem cell location from brain tissue sections collected on day 4 & 7.
Supplemental Figure 4. Example T2-weighted MRI images before and after NSC-CRAd-S-pk7 administration. Stem cells were administered either adjacent to or contralateral to an established GBM43 tumor (right side). Hypointense regions within the tumor area were detected in mice by day 4, demonstrating ferumoxytol-labeled NSC migration (dotted white boxes represent tumor area).