

Abstract
TLR-mediated recognition of microbial danger induces substantial changes in macrophage migration, adherence and phagocytosis. Recently, we described the LPS-regulated phosphorylation of many cytoskeleton-associated proteins by phosphoproteomics. The functional role of these cytoskeletal and motor proteins in innate immune cell responses is largely unexplored. Here, we first identified both long-tailed class I myosins Myo1e and Myo1f as important contributors to LPS-triggered macrophage spreading. Mouse bone marrow-derived macrophages and dendritic cells (DCs) deficient in Myo1e selectively secreted increased amounts of the chemokine CCL2. In addition, the cell surface expression of MHC class II (MHC-II) on both cell types was reduced in the absence of Myo1e. However, transcriptional changes in CCL2 and MHC-II were not observed in the absence of Myo1e, indicating that Myo1e regulates specific intracellular transport processes. The capacity of macrophages and DCs lacking Myo1e to stimulate antigen-specific CD4+ T-cell proliferation was impaired, consistent with the reduced MHC-II surface protein levels. Surprisingly, in Myo1e-deficient DCs, the proteolytic cleavage of endocytosed antigen was also increased. Together, our results provide evidence for a non-redundant function of the motor protein Myo1e in the regulation of TLR4-controlled, cytoskeleton-associated functional properties of macrophages and DCs, and in induction of a full MHC-II-restricted adaptive immune response.
Keywords: macrophages, Toll-like receptors, cytoskeleton, MHC class II, myosin
Introduction
The capacity to migrate in response to chemoattractants and to phagocytose dead cells and invading bacteria are two key functions of macrophages. Migration and phagocytosis require the remodeling of the cytoskeleton to generate the mechanical forces required for changes in cellular morphology. The dynamic polymerization and depolymerization of actin filaments is an essential process controlled by a large group of associated proteins. More than 150 proteins containing actin binding domains have already been identified [1] and thus might be important in microfilament organization. The rearrangement process is regulated by actin nucleation factors such as the Arp2/3 complex and formins, nucleation promoting factors like WASP and WAVE, and proteins responsible for filament depolymerization. Collectively, these proteins form an actin regulatory network, which can be rapidly modulated in response to extracellular stimuli [2, 3].
An important aspect of cell shape regulation is the mechanical linkage between the plasma membrane and the underlying actin filaments, which may be mediated by the class I myosins, members of the myosin superfamily of molecular motors. Class I myosins are single-headed molecular motors, composed of a single heavy chain containing the motor domain with an actin-binding site followed by a light chain-binding neck region and a C-terminal tail domain that includes membrane-binding and protein interaction domains. This specific structure allows a simultaneous interaction with actin filaments and membranes or other proteins necessary for active membrane deformation or intracellular vesicle trafficking. The mechanical forces required for these mechanisms are produced through hydrolysis of ATP [4]. In B lymphocytes, Myo1c and Myo1g are both highly expressed [5], localize to plasma membrane compartments, regulate actin reorganization and contribute to antigen presentation [6, 7]. In macrophages, Myo1g is recruited to the phagocytic cup during FcR-mediated phagocytosis [8]. Myo1f is highly expressed in granulocytes and regulates adhesion to fibronectin, likely through limiting the cell surface expression of the integrin CD18 by controlling granule exocytosis [9]. Myo1e, closely related to Myo1f, is a long-tailed myosin containing a C-terminal region with three tail homology (TH) domains that is more widely expressed than Myo1f and is involved in clathrin-mediated endocytosis [10, 11]. Myo1e also plays an important role in renal filtration as shown by Myo1e mutations in human patients with glomerular dysfunction [12] and the phenotype of podocyte-specific knockout mice [13, 14].
Sensing of infectious danger by macrophages through ligation of TLRs triggers rapid and robust cytoskeletal changes, including an integrin-mediated spreading response in cell culture that is dependent on actin polymerization [15], and p38 MAPK-dependent increases in phagocytosis and phagosome maturation [16]. Recently, we described the global changes in the macrophage phosphoproteome following stimulation of TLR4 using the LPS of Gram-negative bacteria, and observed a strong enrichment of cytoskeleton-associated proteins among the LPS-regulated phosphopeptides [17]. Among these more than 40 proteins with regulated phosphorylation are well-established actin polymerization factors like Arp3, but also several class I myosins and many cytoskeleton-associated proteins with yet unknown function in macrophage biology. The elucidation of the functional role of the TLR4-regulated cytoskeletal phosphoproteins requires the availability of suitable readout systems for phagocytosis, macrophage spreading and other cytoskeleton-associated cell function. To facilitate this endeavor, we have recently developed an automated image analysis algorithm to quantitatively measure macrophage spreading [15].
Here, we used siRNA knockdown of selected cytoskeletal proteins identified previously as strongly phosphorylated in response to LPS to determine the effects on spreading. We identified a contribution of Myo1e and Myo1f to the spreading response. Myo1e-deficient mice were used to study its role in the macrophage and dendritic cell (DC) response to TLR stimulation and in antigen presentation. Our data show a selective involvement in the control of CCL2 secretion and antigen presentation via regulation of MHC class II (MHC-II) surface expression.
Results
Cytoskeleton-associated proteins are enriched among the LPS-regulated phosphoproteome
In a recent phosphoproteome analysis of macrophages we observed a strong enrichment of cytoskeleton-associated proteins among the TLR4-regulated proteome [17]. Among the 41 LPS-regulated phosphoproteins annotated with the Gene Ontology term “cytoskeletal protein binding”, several actin polymerization factors (e.g. Actr3 (Arp3), Fhod1), capping proteins (Capzb), and motor proteins (Myo1e, Myo9b, Myo18a) showed strong and fast phosphorylation at one or multiple sites (Table 1). While some of these cytoskeleton-associated phosphoproteins have well-described functions in phagocytosis or motility of macrophages, others have not yet been associated with cytoskeleton-based macrophage responses.
Table 1.
LPS-induced phosphorylation of cytoskeleton-associated proteins.
| Gene Symbol | Fold change in phosphorylationa) Time after LPS | |
|---|---|---|
| 15 min | 4 h | |
| Actr3 (Arp3)b) | 88c) | 95 |
| Add1 | 6 | 5 |
| Capzb | 10 | 8 |
| Coro1b | 6 | 0 |
| Dbn1 | 11 | 2 |
| Epb4.1/1 | 5 | 5 |
| Epb4.1/2 | 2 | 1 |
| Eps8 | 6 | 5 |
| Fhod1 | 55 | 10 |
| Flna | 2 | 8 |
| Fmn1 | 19 | 0 |
| Frmd4b | 1 | 2 |
| Hdac6 | 3 | 1 |
| Inppl1 | 13 | 0 |
| Lcp1 | 10 | 9 |
| Lima1 | 17 | 10 |
| Lsp1 | 24 | 6 |
| Marcks | 6 | 5 |
| Mib2 | 2 | 1 |
| Mkl1 | 17 | 7 |
| Mkl2 | 15 | 8 |
| Msn | 1 | 4 |
| Mtss1 | 9 | 5 |
| Myh9 | 1 | 2 |
| Myo18a | 13 | 1 |
| Myo1e | 10 | 8 |
| Myo1f | 3 | 3 |
| Myo5a | 2 | 2 |
| Myo9b | 8 | 6 |
| Palld | 6 | 2 |
| Phactr4 | 8 | 2 |
| Plec1 | 1 | 2 |
| Ppp1r9b | 10 | 6 |
| Pxn | 3 | 5 |
| Snta1 | 2 | 1 |
| Svil | 2 | 1 |
| Tln1 | 2 | 2 |
| Tmod3 | 6 | 9 |
| Twf1 | 6 | 0 |
| Vasp | 8 | 4 |
| Wipf1 | 6 | 3 |
Shown are intensities of the most strongly regulated phosphorylation site of each detected protein in response to 15 min and 4 h LPS stimulation (Data from Weintz et al. 2010. [17]). Intensities were calculated by the ratio of stimulated to unstimulated condition.
Listed are only proteins with the GO annotation “actin binding” or “cytoskeletal protein binding” and at least a 1.5-fold increase in any phosphosite by LPS. Detailed information with respect to multiple phosphorylation sites is publicly accessible at www.phosida.com.
Numbers indicate the fold induction relative to unstimulated macrophages.
siRNA knockdown of cytoskeletal phosphoproteins regulates LPS-induced macrophage spreading
Triggering of TLR4 on macrophages by LPS induces a rapid spreading response (Fig. 1), which is dependent on actin polymerization [15]. We have recently described an automated image analysis algorithm to quantitatively measure the spreading response of macrophages [15]. Here, we used siRNA knockdown (KD) in primary bone marrow-derived macrophages [17, 18] to determine whether some of the cytoskeleton-associated phosphoproteins identified by us previously are involved in the TLR4-driven spreading response. Eight genes were selected for KD based on the strength and reproducibility of regulation by LPS. Efficiency of the KD on mRNA level was measured by qRT-PCR and was between 50% and 90% (Fig. 1B–I). Actr3 (encoding Arp3) is part of the essential actin polymerization factor Arp2/3, which is composed of seven subunits including Arp2 and ARPC1-5 [19]. Actr3 KD strongly reduced the basal and LPS-induced macrophage contact area (Fig. 1B). In contrast, KD of the formin-family actin polymerization factors Fmn1 and Fhod1, shown previously to be involved in stress fiber formation [20, 21], did not alter the TLR4-induced spreading (Fig. 1 C, D). Coronin1b has been described to antagonize Arp2/3-mediated actin polymerization [22, 23]. Knockdown of Coro1b led to a biphasic alteration in spreading, with increased contact size at the late 24 h time-point (Fig. 1E). KD of Mtss1, Lsp1 and Eps8 caused only minor changes in spreading (Fig. 1F–H). In contrast, knockdown of Myo1f caused a significant and robust reduction in basal and LPS-induced macrophage spreading (Fig. 1I).
Figure 1. siRNA KD of selected cytoskeletal-associated phosphoproteins.

(A) For analysis of LPS-induced spreading of C57BL/6 macrophages, the cells were stimulated or not in 8-well chamber slides for the indicated time points, followed by fixation, staining for CD11b, and automatic image analysis for quantitative determination of the contact area [15]. Representative fluorescence images from control and 24 h timepoint are shown. CD11b/APC surface staining illustrated in yellow and nuclei DAPI in light blue. Scale bar = 50 μm. (B–I) Spreading phenotype of siRNA-treated macrophages. Differentiated primary macrophages were electroporated with specific siRNA for selected cytoskeletal proteins 48 h prior LPS stimulation (100 ng/ml). Contact area quantification of (B) Actr3 (Arp3), (C) Fmn1, (D) Fhod1, (E) Coro1b, (F) Mtss1, (G) Lsp1, (H) Eps8 and (I) Myo1f siRNA-treated cells was measured by automatic segmentation software [15]. Shown are mean + SEM of at least 300 cells per condition from a single experiment. Media control (open symbols), LPS (closed symbols), non-silencing control RNA treated cells (circles), specific siRNA treated cells (red squares). Knockdown efficiency was determined by qRT-PCR shown as KD within the graphs. Statistical significance refers to nsRNA-treated, LPS-stimulated control cells compared to siRNA-treated, LPS-stimulated cells. * p <0.05, * * p <0.001, * * * p <0.0001, ns=not significant; Student’s t test for unpaired conditions.
The class I myosins Myo1e and Myo1f are required for efficient macrophage spreading
Myo1e and Myo1f belong to myosin class I, which includes myosins that are able to interact with actin filaments and lipid membranes [24]. Myo1e and Myo1f are so-called long-tailed class I myosins based on the presence of three tail homology regions (TH1, TH2 and a SH3 domain termed TH3, see Fig. 2A). Both Myo1e and Myo1f were strongly phosphorylated in the tail domain after triggering of TLR4, with several phosphosites located in the TH2 domain (required for binding to microtubules and microfilaments [25, 26]) and one threonine in the pleckstrin homology domain with the TH1 region (Fig. 2A).
Figure 2. Activation of Myo1e and Myo1f is required for TLR4-driven macrophage spreading.

(A) Schematic illustration of Myo1e and Myo1f with single protein domains. LPS-induced phosphorylation of threonine (T) or serine (S) residues detected in the phosphoproteome analysis are indicated [17]. The regulation of each phosphosite in response to 15 min and 4 h LPS stimulation is shown in the tables below. (B) Western blot control for Myo1e KO (left). Cell lysates taken at indicated time points after LPS stimulation. Grb2: loading control. Spreading behavior of primary Myo1e KO macrophages in response to LPS (right). Data are shown as mean + SEM of at least 300 cells per condition from one experiment representative of two independent experiments with comparable results. (C) Generation of “double-deficient” cells by Myo1f siRNA KD in Myo1e KO macrophages. Expression of mRNA levels and KD efficiency was determined by qRT-PCR. Fold changes were calculated in relation to the unstimulated WT condition and, in the case of Myo1f KD, in relation to the nsRNA-treated unstimulated control cells. Data are shown as mean + SD of duplicate samples from one experiment representative of two performed. (D) Impact of combined Myo1e deletion and Myo1f knockdown on spreading of macrophages. Representative immunofluorescence microscopy images (original magnification: 200-fold) are shown (left). Quantitation of spreading for Myo1e single- (red) and Myo1e and 1f double-deficient macrophages (green) (right). Shown are mean + SEM of at least 300 cells per condition from a single experiment representative of two independent experiments with similar results. Statistical significance refers to WT or nsRNA-treated LPS-stimulated control cells compared to KO or siRNA-treated LPS-stimulated cells. * p <0.05, * * p <0.001, * * * p <0.0001, ns=not significant; Student’s t test for unpaired conditions.
We used bone marrow-derived macrophages from Myo1e−/− mice [14] and observed a moderately reduced spreading response to LPS after 2h and 8h (Fig. 2B). Given the high homology of Myo1e and Myo1f at the protein level and the similarity of LPS-induced phosphorylation between both long-tailed myosins, we reasoned that they may be functionally redundant in macrophage spreading. Therefore, we performed siRNA knockdown of Myo1f in Myo1e−/− macrophages. qRT-PCR of Myo1e and Myo1f mRNA expression indicated that no compensatory increase of one isoform occurred when the other was genetically deleted or knocked down by siRNA. The transcription of both myosins was down-regulated in response to 24h LPS stimulation (Fig. 2C). While Myo1e-deficiency alone caused a moderate decrease in basal and early LPS-induced spreading, the additional knockdown of Myo1f led to significantly reduced contact area of macrophages at all time-points, indicating that both long-tailed class I myosins cooperate in macrophage spreading (Fig. 2D).
Myo1e selectively regulates the release of the chemokine CCL2 from macrophages and DCs
Triggering of TLR4 by LPS induces a rapid and massive induction of pro-inflammatory gene expression and secretion of cytokines and chemokines, which may induce a spreading response in leukocytes [27]. We therefore asked whether the reduced spreading observed in Myo1e-deficient macrophages was related to altered secretion of cytokines in response to LPS (Fig. 3A). The kinetics and levels of TNF, IL-10, IL-6 and IL-12p40 were unaltered in Myo1e−/− macrophages (except for slight but significant increase in IL-12p40 after 24h). However, the chemokine CCL2/MCP-1 was significantly elevated in the supernatants of LPS-stimulated Myo1e−/− macrophages compared to WT controls at the 8h and 24h time point. This effect was specific for CCL2, as it was not observed for CCL3 and CCL4, which were secreted in equal amounts or at transiently higher levels by WT macrophages (Fig. 3B). Increased secretion of CCL2 was also observed when Myo1e−/− bone marrow-derived dendritic cells were stimulated with LPS (Fig. 3C). Thus, Myo1e, which is expressed in macrophages and DC, regulated CCL2 production in both cell types comparably. We next asked whether increased CCL2 release was due to enhanced TLR signaling leading to stronger transcription in response to LPS. However, activating MAPK signaling as measured by phosphorylation of p38 and ERK1/2 MAPK was unaltered in Myo1e−/− macrophages (Fig. 3D). In addition, the mRNA expression of CCL2 was comparably induced to high levels in WT and Myo1e−/− macrophages (Fig. 3E). In addition, intracellular staining and FACS analysis showed that CCL2 protein levels were comparable after LPS stimulation in WT and Myo1e−/− macrophages (Fig. 3F). Therefore, Myo1e appears to regulate not the expression but rather the transport and secretion of CCL2 from macrophages and DC.
Figure 3. Selective chemokine secretion in LPS-activated Myo1e-deficient macrophages and DCs.

(A, B) Supernatants of LPS-stimulated macrophages were analyzed by ELISA for the presence of (A) cytokines and (B) chemokines. (C) CCL2 secretion of LPS-activated DCs was measured by ELISA. (D) Western blot for ERK1/2 and p38 phosphorylation (left). Cell lysates were taken at indicated time points (15 min to 24 h) after LPS stimulation. Grb2: loading control. Band intensities of pERK1 and pp38 were normalized against those of total ERK1 and p38 using Image J software. To normalize between experiments, the ratio of the WT 15 min timepoint was set to 100 % and used as reference for all other samples (right). Data are shown as mean and SD (n=3 samples) and are pooled from three independent experiments. (E) CCL2 mRNA expression was determined by qRT-PCR in WT and Myo1e KO macrophages. To normalize for differences between experiments in the absolute amounts of cytokines, the 24 h time point of the LPS-treated control cells in the ELISA experiments was set to 100% (shown as dashed line). Fold changes in CCL2 mRNA expression were first calculated by normalization to the expression of the house-keeping gene HPRT, subsequently the 24 h fold-change of LPS-treated WT cells was set to 100 %. (A, B, C, E) Data are shown as mean + SEM of pooled samples from at least 6 independent experiments, each with biological duplicates. (F) Intracellular staining for CCL2 in LPS-treated macophages. Brefeldin A was added together with LPS to block protein secretion. Cells were fixed, permeabilized and stained for flow cytometry. Data are shown as mean and SD of biological triplicates from one representative experiment of two performed (left) and histogram overlays of CCL2-PE stainings from unstimulated and 8 h LPS samples (right). * * p <0.001, * * * p <0.0001, ns=not significant; Student’s t test for unpaired conditions. Significant differences in CCL2 secretion were confirmed by the non-parametric Mann-Whitney Rank Sum test.
Myo1e is required for optimal MHC-II surface expression on macrophages
Integrins are important mediators of macrophage spreading in response to TLR stimulation [28]. Of interest, Myo1f-deficient neutrophils were shown to have higher cell surface levels of CD18, which has been linked to an increased adherence phenotype [9]. We therefore considered that impaired spreading in macrophages deficient in Myo1e may be related to altered cell surface expression of integrins. Analysis of macrophage surface protein levels of β1 integrin CD18, β2 integrin CD29 and the β3 integrin CD61 by flow cytometry did not reveal any difference under basal or LPS-stimulated conditions (data not shown). In addition, the integrin subunits CD11a, CD11b and CD11c were comparably expressed on WT and Myo1e−/− macrophages (data not shown).
We also measured the expression of the MHC-II molecule IAb, and observed a moderate but highly significant reduction of 20–25% on macrophages under resting conditions and 8h after LPS stimulation (Fig. 4A). In contrast, treatment with IFNγ induced substantial up-regulation of MHC-II in WT and Myo1e−/− macrophages, overcoming the phenotype of Myo1e-deficiency in resting macrophages (Fig. 4B). Expression levels of Iab1 encoding the MHC-II protein were not significantly different between WT and Myo1e−/− macrophages; in addition, the mRNA for the transcription factor CIITA, a master regulator of MHC-II expression, was expressed at equal levels in Myo1e−/− and WT macrophages (Fig. 4C). Therefore, the reduced expression of cell surface MHC-II in Myo1e−/− macrophages is more likely due to an intracellular transport defect rather than alterations in biosynthesis.
Figure 4. Reduced TLR4-driven MHC-II surface expression in Myo1e KO macrophages and DCs.

(A, B) WT and Myo1e KO macrophages were stimulated with (A) LPS or (B) IFN-γ (50 ng/ml) and MHC-II surface expression was determined by flow cytometry. A histogram overlay of WT cells is shown (left). Quantified and normalized MFI of MHC-II expression for different experiments is shown as mean + SEM of 23 (A) or 10 (B) samples, pooled from 13 (A) or 5 (B) experiments (right). (C) MHC-II and CIITa mRNA expression of LPS-treated macrophages was analyzed by qRT-PCR. Fold changes in MHC-II and CIITa mRNA expression were first calculated by normalization to the expression of the house-keeping gene Hprt, subsequently the maximum fold change of LPS-treated WT cells was set to 100% (shown as dashed line). Data shown as mean + SEM of biological duplicates pooled from five experiments. (D) MHC-II surface expression on LPS-activated WT and Myo1e KO DCs was determined by flow cytometry. A histogram overlay of WT cells is shown (left). Quantified and normalized MFI of MHC-II expression for different experiments. MFI were normalized for differences between experiments, the maximum MFI of treated WT cells in each experiment was set to 100%. Data are shown as mean + SEM of biological duplicates pooled from six experiments. * * p <0.001, * * * p <0.0001, ns=not significant; Student’s t test for unpaired conditions. Significant differences were confirmed by the non-parametric Mann-Whitney Rank Sum test.
DC express much higher levels of MHC-II protein on the cell surface. The basal levels were indistinguishable between WT and Myo1e−/− DC, but 24 h after LPS stimulation we observed a reproducible and significant reduction of 20% in the absence of Myo1e (Fig. 4D). Thus, Myo1e regulates MHC-II surface protein in macrophages and DC.
The reduced expression of MHC-II on Myo1e−/− macrophages and DC pointed to possible effects on antigen presentation and elicitation of T-cell responses. We therefore also investigated the expression of the co-stimulatory surface proteins CD80, CD86 and CD40 on macrophages by flow cytometry. Myo1e-deficiency did not alter the basal and LPS-induced surface levels of CD40 and CD86; in the case of CD80 a weak but significant reduction in the mean fluorescence intensity was found 24h after LPS stimulation in Myo1e−/− macrophages (data not shown).
Myo1e-deficient antigen-presenting cells are impaired in eliciting T-cell activation
Reduced MHC-II expression in the absence of Myo1e raised the question whether Myo1e-deficient antigen-presenting cells (APCs) are deficient in stimulating antigen-specific T-cell activation. To address this issue, we employed purified CD4+ T cells from OT-II transgenic mice which carry a TCR specific for an MHC-II-binding ovalbumin (OVA) peptide. Macrophages were fed ovalbumin overnight, washed and used for stimulation of OT-II CD4+ T cells whose antigen-specific proliferation was determined by 3H-thymidine incorporation assay. While high numbers of Myo1e−/− macrophages induced T-cell proliferation equally well as WT macrophages, a significant difference between the macrophage genotypes was found when limiting numbers of macrophages were used (Fig. 5A). As expected, OT-II stimulation by OVA-fed DC induced much stronger T-cell proliferation than that triggered by macrophages. At a low DC-OT-II ratio, differences between WT and Myo1e−/− DC were not significant, but when high numbers of DC were used a strong decrease in the 3H-thymidine incorporation elicited by Myo1e−/− DC was observed (Fig. 5B). This difference was also found when T-cell divisions were measured by the CFSE dilution assay (Fig. 5C), confirming that the proliferation of OT-II T cells is indeed reduced when the OVA-presenting APC lack Myo1e. Together, the reduced expression of MHC-II on the surface of Myo1e−/− APC correlates with an impaired capacity to elicit antigen-specific T-cell proliferation.
Figure 5. Impaired antigen-specific CD4+ T-cell activation by Myo1e KO APCs.
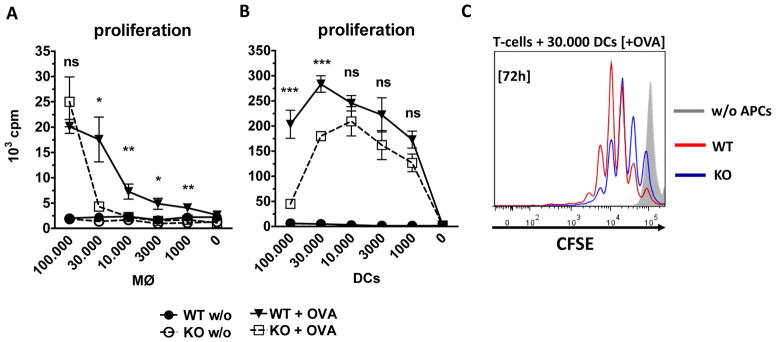
(A) In vitro OT-II assay to determine macrophage MHC-II-specific activation of CD4+ T cells. Macrophages were pre-incubated with 2 mg/ml OVA overnight and intensively washed in cRPMI before addition of MACS-isolated CD4+ T cells from the spleen and LN of OT-II mice. Macrophages numbers were titrated and added to 3 ×105 T cells. T-cell proliferation was measured by 3H-thymidine incorporation assay. (B) In vitro OT-II assay to determine DC MHC-II-specific activation of CD4+ T cells. (A, B) Data are shown as mean + SEM (n=6–9 samples) and are pooled from two to three experiments with biological triplicates each. (C) CFSE dilution assay to determine antigen-specific CD4+ T-cell division. T cells were labeled with CFSE before co-incubation with 3×104 DCs. After 72 h samples were analyzed by flow cytometry. Shown are representative histogram overlays from one experiment with two biological replicates. * p <0.05, * * p <0.001, * * * p <0.0001, ns=not significant; Student’s t test for unpaired conditions.
Proteolytic processing of exogenous antigen in Myo1e-deficient APC
Triggering of T cells by APC depends not only on high levels of MHC-II but also on the amount of antigen that is available for loading onto MHC molecules. Proteolytic cleavage of exogenous proteins in the endolysosome is required for generation of peptides to be loaded onto MHC-II. However, to achieve an optimal level of antigenic peptides, the degradative capacity of the APC has to be regulated [29]. We reasoned that Myo1e may also impact on the processing of exogenous antigen taken up by macrophages and DC, and therefore assessed the cleavage of endocytosed OVA using a FACS-based assay (Fig. 6). WT and Myo1e−/− macrophages displayed a comparable, fast increase in the signal intensity of DQ-OVA, indicating highly efficient cleavage (Fig. 6A). Only at the late 24h time point, Myo1e−/− macrophages exhibited a higher DQ-OVA signal than their WT counterparts. DC cleaved DQ-OVA more slowly than macrophages, and even after 24h had substantially lower signal. Interestingly, Myo1e−/− DC displayed significantly higher DQ-OVA signals than WT DC over the entire kinetic analysis, approaching the levels observed in WT macrophages (Fig. 6B). Thus, in the absence of Myo1e, the proteolysis of exogenous antigen in DC appears to proceed unchecked.
Figure 6. Contribution of Myo1e to antigen processing by APCs.

(A) Macrophages and (B) DCs were incubated with DQ-OVA (20 μg/ml) for the indicated times at 37°C or 4°C as control. Cells were fixed with 2% PFA/PBS to stop processing and subsequently analyzed by flow cytometry. Histogram overlays of WT and KO cells + DQ-OVA. WT (blue line); Myo1e KO (red line) are shown (left). For quantification and statistical analysis, the mean MFI values of WT macrophages after 24 h were set to 100 % and used to calibrate the data points from all conditions, including the DC samples (right). Data are shown as mean + SEM (n=6 samples) and are pooled from two independent experiments. * p <0.05, * * p <0.001, ns=not significant; Student’s t test for unpaired conditions.
Discussion
The starting point for the study described in this manuscript was our previous identification of multiple cytoskeleton-associated proteins as kinase targets in LPS-stimulated macrophages. By combining siRNA knockdown with automated quantitation of the macrophage spreading we aimed to start a functional investigation of the phosphoproteins regulated by TLR4 activation. The results obtained with a limited set of eight candidate genes provided a proof of concept validation of the actin polymerization factor subunit Arp3 as critical for macrophage spreading. The functional Arp2/3 complex is known to play a central role in macrophage chemotaxis and receptor mediated phagocytosis [30]. In our phosphoproteome studies of TLR4-activated macrophages a strong and consistent phosphorylation of the Arp3 subunit was identified, still unknown in its function. However, biochemical studies identified a crucial role for Arp3 in regulating the actin polymerization kinetic and linking the whole complex to the site of actin filaments [31]. Our results provide evidence for an essential role of Arp3 in formation of a functional Arp2/3 complex, required in TLR4-driven primary macrophage spreading. The utilized experimental strategy appears therefore to be well suited to determine the possible role of the other 40 cytoskeleton-associated phosphoproteins in macrophage spreading in future experiments. Here, the motor protein Myo1f was identified as new component of the TLR4-induced spreading response in macrophages.
Given the effect observed with Myo1f knockdown, we also turned to Myo1e which showed considerably stronger phosphorylation after LPS (Fig. 2). Myo1e and Myo1f collaborated for efficient LPS-induced spreading in macrophages, as demonstrated by the combination of Myo1f siRNA knockdown in Myo1e−/− macrophages. While Myo1f is described to be involved in integrin surface expression by regulation of granule exocytosis [9], Myo1e was observed to contribute to actin rearrangement itself [32]. In recent studies, Myo1e was identified to co-localize with Carmil, Fhod1, Arp3 and β3-intergins in early adhesion sites, suggested to transport the actin polymerization proteins to these structures and additionally to be involved in their stabilization [32]. Furthermore, proteomic studies identified Myo1e as a component of macrophage podosomes [33] and fibroblast integrin adhesome [34]. A contribution of Myo1e in LPS-induced integrin expression on macrophages was not observed in our experiments (data not shown). To our knowledge this is the first demonstration of a role for class I myosins in macrophage spreading. However, in B lymphocytes the class I myosins Myo1c and Myo1g have been shown to contribute to spreading induced by triggering CD44 or LFA-1 [6, 7].
In macrophages, both Myo1e (formerly known as Myo1c) and Myo1g are recruited to the phagocytic cup in Fc-receptor-triggered phagocytosis [8, 35]. Because of the involvement of Myo1e in receptor-mediated endocytosis [10, 11], we tested Myo1e-deficient macrophages in phagocytosis experiments using latex beads and heat-killed Escherichia (E.) coli bacteria, but observed no difference to WT control macrophages. In gentamicin protection assay the incubation with E. coli, Staphylococcus aureus or Salmonella revealed no difference in pathogen killing dependent on the macrophage genotype. (JW, unpublished data). Thus, although Myo1e is required for efficient LPS-induced spreading, it is not essential for phagocytosis or pathogen killing in macrophages.
We have not addressed here the cell biological aspects of Myo1e intracellular localization and interaction with other cytoskeletal proteins and the plasma membrane in macrophages. In fibroblasts, Myo1e co-localizes with clathrin- und dynamin-containing puncta at the plasma membrane [10], where it may coordinate actin assembly [11]. This localization of Myo1e is dependent on its SH3 domain, which is not present in the short tailed class I myosin isoforms. Recently published data from embryonic fibroblasts show that Myo1e moves to active lamellipodia and actin-rich early adhesions, again dependent on the SH3 domain, where it promotes adhesion stabilization [32]. In renal podocytes, Myo1e colocalizes with actin filaments at the site of newly established cell-cell contacts [36]. Together, these studies suggest a function of Myo1e in linking plasma membrane and actin filaments early in the process of cytoskeletal rearrangements following adhesion and cell-cell contact and identified the SH3 domain as a specific feature of the long-tailed myosins required for function. Whether Myo1e does localize to the same sites and interaction partners in macrophages during spreading and phagocytosis will be the subject of future work.
While most cytokines and chemokines analyzed here were secreted in equal amounts by LPS-stimulated WT and Myo1e-deficient macrophages, CCL2/MCP-1 was released in significantly higher concentrations by Myo1e−/− macrophages and DC. This selectively increased secretion of CCL2 is likely caused by altered transport and/or release from the cells, because the expression at the mRNA level and the intracellular content of CCL2 protein was not altered in the absence of Myo1e. A regulatory role of Myo1e in the vesicular trafficking of the chemokine CCL2 seems therefore likely. Interestingly, Myo1e is involved in granule exocytosis in frog oocytes [37]. Another example of a regulatory function for a long-tailed class I myosin is provided by the increased CD18 surface expression in Myo1f-deficient granulocytes [9]. Recent studies showed that CCL2 causes cytoskeletal rearrangements and morphological changes in human macrophages [38]. Whether the altered CCL2 secretion of Myo1e−/− macrophages is related to their impaired spreading response remains to be elucidated.
A surprising finding in Myo1e-deficient macrophages was the selective reduction in MHC-II surface expression levels, while co-stimulatory molecules were not altered. The reduction in MHC-II levels was moderate, but proved to be functionally relevant in experiments testing antigen-specific proliferation of OT-II cells in response to Myo1e-deficient macrophages or DC pulsed with OVA. Attempts to address the role of Myo1f in MHC-II expression by siRNA KD in WT and Myo1e−/− macrophages were not informative, because transfection of non-silencing control siRNA upregulated MHC-II levels (data not shown). Interestingly, Myo1e was recently assigned a possible function in regulating the transport of MHC-II to the plasma membrane in a systems biology investigation of pathways controlling antigen presentation [39]. The GTPase ARF7 was identified by genome-wide shRNA screening as involved in MHC-II expression, followed by yeast-2-hybrid-based identification of its putative effector protein ARF7EP. In GST-pulldown experiments Myo1e was found associated with ARF7EP by mass spectrometry, and this association was confirmed in human PBMC by co-immunoprecipitation [39]. Our data, showing a reduced MHC-II surface expression in Myo1e-deficient macrophages and LPS-stimulated DC, thus provide the first evidence from the murine system corroborating a role for Myo1e in regulation of MHC-II cell surface expression. The reduction of MHC-II surface levels in the absence of Myo1e was functionally relevant in that it led to a significant reduction in the capacity of macrophages and DC to elicit antigen-specific proliferation of Ovalbumin-specific CD4+ T cells. This effect is reminiscent of the phenotype observed for siRNA knockdown or overexpression of a dominant-negative version of Myo1c in B lymphocytes by Maravillas-Montero et al. [6], which also leads to impaired antigen-specific CD4+ T-cell stimulation. In addition, the more efficient cleavage of DQ-OVA in Myo1e-deficient DC we observed here may contribute to their reduced T-cell stimulatory capacity through a depletion of the MHC-II compartment of antigenic peptide [40, 41].
The challenge for future experiments will be to determine the mechanisms by which Myo1e controls macrophage spreading, chemokine secretion and MHC-II antigen presentation. In addition to the analysis of the cell biology of Myo1e, the regulation of its localization and interaction with binding partners by site-specific phosphorylation need to be studied in detail. The involvement of Myo1e in several key functional aspects of macrophage biology (such as cell spreading and motility, chemokine secretion and antigen presentation), shown here in vitro, suggests that this motor protein may also play an important role in innate immune cell recruitment and function during inflammatory and infectious disease processes in vivo.
Materials and methods
Mice
C57BL/6 and Myo1e −/− mice [14] were maintained at the Department of Laboratory Animal Resources, SUNY Upstate Medical University (Syracuse, NY). All protocols for animal studies and cell collection using these mice were approved by the SUNY Upstate Medical University IACUC. Bone marrow of C57BL/6 and Myo1e −/− mice was controlled deep-frozen in FCS containing 10% DMSO, shipped on dry ice from USA to Germany and stored in liquid nitrogen until usage. C57BL/6 and OT-II mice were maintained at the Franz Penzoldt Center of the Medical Faculty at the University Erlangen-Nuremberg.
Reagents
LPS (Escherichia coli 0111:B4) was purchased from Sigma-Aldrich. Murine IFN-γ was delivered from PeproTech. Antibodies to ERK1/2, phosphorylated (p)-ERK1/2, p38, p-p38, were obtained from Cell Signaling Technology and to Grb2 from BD Biosciences. Myo1e antibody was obtained from Proteintech. MHC-II (I-A/I-E) FACS antibody conjugated to eFluor®450 (clone M5/114.15.2) was purchased from eBioscience. Antibodies against CD4/FITC (clone GK1.5), CD11b/allophycocyanin (clone M1/70) and CCL2/PE (clone 2H5) were delivered from BioLegend.
Macrophage and DC differentiation
Mouse bone marrow (BM) stored in liquid nitrogen was thawed in a water bath at 37°C and immediately transferred into pre-warmed Dulbecco’s modified Eagle’s medium (DMEM). BM was washed once in 30 ml DMEM to eliminate DMSO, required for deep-freezing. Macrophage differentiation occurred in hydrophobic Teflon bags as described by Wiese M. et al [18]. To obtain DC, BM cells were cultured on Petri dishes (Nunc) for 8 days in cRPMI 1640 containing 10% X63-cell conditioned medium as a source for GM-CSF. After 3 and 6 days 10 ml of cRPMI + 10% GM-CSF was added to the cell culture. Differentiated cells were stimulated with 100 ng/ml LPS or 50 ng/ml IFN-γ for respective experiments.
siRNA
siRNA oligonucleotides directed against Actr3 (ID 74117), Fmn1 (ID 14260), Fhod1 (ID 234686), Coro1b (ID 23789), Mtss1 (ID 211401), Lsp1 (NM_019391), Eps8 (ID 13860), Myo1f (ID 17916) and non-silencing siRNA (nsRNA) were purchased from Dharmacon (Thermo Fisher Scientific) using Dharmacon’s pre-validated siRNA database. Each specific siRNA set composed of a pool of four specifically directed siRNAs. Lyophilized siRNAs samples were resuspend in 1-fold siRNA buffer from Dharmacon to a final concentration of 20 μM, heated for 1 min to 90°C and incubate at 37°C for 60 min on a thermo shaker. Dissolved samples were stored in aliquots at −80° C. Delivery of siRNA into primary MØ was done by electroporation as described [18]. In brief, 20 μl of the 20 μM siRNA stock were transferred into 4-mm cuvette (Peqlab, Germany) and filled up to a volume 50 μl with Opti-MEM (Invitrogen). 50 μl of the cell suspension (containing 2×106 macrophages) were added, incubated 5 min on ice and subsequently pulsed in a GenePulser Xcell (Bio-Rad, München, Germany). Pulse conditions were 400 V, 150 μF, and 100 Ω. Electroporated cells were transferred into required well plates and incubated at 37°C. 1–2 h upon electroporation pure RPMI 1640 was aspirated and exchanged by addition of cRPMI and incubated 48 h at 37° C, 5% CO2 prior LPS to stimulation.
qRT-PCR
KD efficiency was measured by qRT-PCR. At least 2×105 electroporated cells were transferred into 24-well cell culture plate (Nunc) 48 h upon stimulation. RNA was extracted via RNeasy Mini Kit (Qiagen) and cDNA synthesis was subsequently performed by high-capacity cDNA kit (Applied Biosystems). Samples were treated as described in the manufacturer’s instructions. The qRT-PCR was done by using a 7900HT Fast Real-Time PCR System from Applied Biosystem. Expression levels of the housekeeping gene Hprt as well as Actr3, Fmn1, Fhod1, Coro1b, Mtss1, Lsp1, Eps8, Myo1f and Myo1e were analyzed using primer/probe combinations selected from the Roche Universal Probe Library. Fold changes were calculated with the ΔΔCT method using calibrators as indicated.
Cell spreading
48 h or 24 h prior to LPS stimulation [100 ng/ml] 2,5×104 electroporated or KO cells, respectively, were transferred to 8-well Permanox chamber slides (Nalge Nunc International) respectively. Cells were fixed at indicated time points in pre-warmed 2% PFA/PBS for 20 min, followed by CD11b/allophycocyanin surface staining and subsequently samples were covered in DAPI containing mounting medium (Dianova). Fluorescence images were obtained using a Zeiss AxioVert 200M (Germany) widefield microscope. Contact areas of activated macrophages were automatically determined as described [15].
Western Blot
To determine protein content or MAPK activation, 106 cells/well were cultured in 1 ml cDMEM in 12 well plates and lysed at indicated time points after LPS stimulation. 20 μl of lysate were loaded on 10% PAA gel containing SDS. After blotting, the membrane was blocked in TBS buffer containing 3% BSA and 0.01% Tween20, and proteins were detected with the respective antibodies.
ELISA
Supernatants of LPS-stimulated cells were collected from the respective conditions and cytokine and chemokine concentration was measured by ELISA kits from R&D. CCL2 ELISA kit was obtained from BioLegend. Samples were treated as described in the manufacturer’s instructions.
FACS analysis
FACS staining was performed according to standard protocols with antibodies against MHC-II (I-A/I-E) and CD4. Detailed informations listed in “Reagents”. Data were recorded on a FACSCanto II (BD Bioscience) and analyzed using FACSDiva Software V6.12 and FlowJo 7.6.5 software. Staining and washing procedures were performed directly in 96-well plates used to stimulate the macrophages with LPS or IFNγ. Cells were fixed with 1% PFA in PBS overnight before a final washing step and taking up in PBS for flowcytometric analysis. The vast majority of macrophages detached from the plate, making any special further detachment treatment unnecessary. For detection of intracellular CCL2 protein, macrophages were stimulated with LPS in the presence of Brefeldin A (5 μg/ml), fixed with 2% PFA for 20 minutes, washed and permeabilized with saponin before staining with anti-CCL2-PE antibody.
OT-II stimulation assay
Differentiated bone-marrow MØ or DC were cultured for 24 h in the presence or absence of ovalbumin (Sigma-Aldrich). The APCs were washed at least 5 times with pure RPMI before co-cultivation with purified CD4+ cells from OT-II mice. Numbers of APC were titrated as indicated while constant numbers of 2–3×105 CD4+ T cells were added to the suspension. CD4+ T cells were purified from spleen and lymph nodes of OT-II mice by magnetic cell sorting (Miltenyi Biotec) and their purity was determined by FACS to be >93%. To analyze proliferation capacity, cells were pulsed after 72 h of culture with 3H-thymidine (1 μCi/well) (PerkinElmer, Rodgau, Germany) for 20 h. Cells were harvested onto glassfiber filtermats using an ICH-110 harvester (Inotech, Dottikon, Switzerland) and filters were counted in a 1450 microplate (Wallac, Turku, Finland).
CFSE dilution assay
APCs and CD4+ T cells were prepared as described in “OT-II stimulation assay”. In advance to co-cultivation the initial CD4+ T cell population was stained with CFSE (#422701, BioLegend) according to the standard protocol. After 72 h T-cell division was determined by FACS analysis.
DQ-OVA Assay
4×105 bone-marrow derived macrophages or DC per well were incubated in 24-well plates for the indicated time points in the presence of 20 μg/ml DQ-OVA (Invitrogen). To stop DQ-OVA processing, cells were washed twice with PBS and fixed in 2% PFA/PBS for at least 30 minutes. To determine antigen processing, the increased fluorescence of cleaved DQ-OVA was analyzed by FACS. To prevent DQ-OVA endocytosis and processing, control cells were incubated at 4° C under constant ovalbumin presence.
Statistical analysis
If not described otherwise in the Figure legends, results were expressed as mean ± SEM. Graphs were generated with Graph Pad Prism and statistical significance was determined with Student t test for unpaired conditions (ns=not significant; * p <0.05; * * p <0.001, * * * p <0.0001).
Acknowledgments
Supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 643, TP A10 to R.L.) and by the NIH award 1R01DK083345 to M.K.. The sponsor was not involved in study design, data collection, data interpretation, or the decision to publish the study.
The authors thank Elisabeth Zinser and Gerhard Krönke for provision of OT-II mice and support with the T-cell stimulation assay. Furthermore, the authors appreciate the skilled technical support of Barbara Bodendorfer and Sharon Chase.
Abbreviations
- APC
antigen-presenting cell
- DC
dendritic cell
- KD
knockdown
Footnotes
Conflict of interest
The authors declare no commercial or financial conflict of interest.
References
- 1.Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 4.Krendel M, Mooseker MS. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda) 2005;20:239–251. doi: 10.1152/physiol.00014.2005. [DOI] [PubMed] [Google Scholar]
- 5.Santos-Argumedo L, Maravillas-Montero JL, Lopez-Ortega O. Class I myosins in B-cell physiology: functions in spreading, immune synapses, motility, and vesicular traffic. Immunol Rev. 2013;256:190–202. doi: 10.1111/imr.12105. [DOI] [PubMed] [Google Scholar]
- 6.Maravillas-Montero JL, Gillespie PG, Patino-Lopez G, Shaw S, Santos-Argumedo L. Myosin 1c participates in B cell cytoskeleton rearrangements, is recruited to the immunologic synapse, and contributes to antigen presentation. J Immunol. 2011;187:3053–3063. doi: 10.4049/jimmunol.1004018. [DOI] [PubMed] [Google Scholar]
- 7.Maravillas-Montero JL, Lopez-Ortega O, Patino-Lopez G, Santos-Argumedo L. Myosin 1g regulates cytoskeleton plasticity, cell migration, exocytosis, and endocytosis in B lymphocytes. Eur J Immunol. 2013 doi: 10.1002/eji.201343873. [DOI] [PubMed] [Google Scholar]
- 8.Dart AE, Tollis S, Bright MD, Frankel G, Endres RG. The motor protein myosin 1G functions in FcgammaR-mediated phagocytosis. J Cell Sci. 2012;125:6020–6029. doi: 10.1242/jcs.109561. [DOI] [PubMed] [Google Scholar]
- 9.Kim SV, Mehal WZ, Dong X, Heinrich V, Pypaert M, Mellman I, Dembo M, et al. Modulation of cell adhesion and motility in the immune system by Myo1f. Science. 2006;314:136–139. doi: 10.1126/science.1131920. [DOI] [PubMed] [Google Scholar]
- 10.Krendel M, Osterweil EK, Mooseker MS. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett. 2007;581:644–650. doi: 10.1016/j.febslet.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J, Grassart A, Drubin DG. Myosin 1E coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol Biol Cell. 2012;23:2891–2904. doi: 10.1091/mbc.E11-04-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chase SE, Encina CV, Stolzenburg LR, Tatum AH, Holzman LB, Krendel M. Podocyte-specific knockout of myosin 1e disrupts glomerular filtration. Am J Physiol Renal Physiol. 2012;303:F1099–1106. doi: 10.1152/ajprenal.00251.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krendel M, Kim SV, Willinger T, Wang T, Kashgarian M, Flavell RA, Mooseker MS. Disruption of Myosin 1e promotes podocyte injury. J Am Soc Nephrol. 2009;20:86–94. doi: 10.1681/ASN.2007111172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenzel J, Held C, Palmisano R, Teufel S, David JP, Wittenberg T, Lang R. Measurement of TLR-Induced Macrophage Spreading by Automated Image Analysis: Differential Role of Myd88 and MAPK in Early and Late Responses. Front Physiol. 2011;2:71. doi: 10.3389/fphys.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 17.Weintz G, Olsen JV, Fruhauf K, Niedzielska M, Amit I, Jantsch J, Mages J, et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol Syst Biol. 2010;6:371. doi: 10.1038/msb.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiese M, Castiglione K, Hensel M, Schleicher U, Bogdan C, Jantsch J. Small interfering RNA (siRNA) delivery into murine bone marrow-derived macrophages by electroporation. J Immunol Methods. 2010;353:102–110. doi: 10.1016/j.jim.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Boczkowska M, Rebowski G, Petoukhov MV, Hayes DB, Svergun DI, Dominguez R. X-ray scattering study of activated Arp2/3 complex with bound actin-WCA. Structure. 2008;16:695–704. doi: 10.1016/j.str.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dettenhofer M, Zhou F, Leder P. Formin 1-isoform IV deficient cells exhibit defects in cell spreading and focal adhesion formation. PLoS ONE. 2008;3:e2497. doi: 10.1371/journal.pone.0002497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schonichen A, Mannherz HG, Behrmann E, Mazur AJ, Kuhn S, Silvan U, Schoenenberger CA, et al. FHOD1 is a combined actin filament capping and bundling factor that selectively associates with actin arcs and stress fibers. J Cell Sci. 2013;126:1891–1901. doi: 10.1242/jcs.126706. [DOI] [PubMed] [Google Scholar]
- 22.Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–929. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandhi M, Achard V, Blanchoin L, Goode BL. Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol Cell. 2009;34:364–374. doi: 10.1016/j.molcel.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McConnell RE, Tyska MJ. Leveraging the membrane - cytoskeleton interface with myosin-1. Trends Cell Biol. 2010;20:418–426. doi: 10.1016/j.tcb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung G, Hammer JA., 3rd The actin binding site in the tail domain of Dictyostelium myosin IC (myoC) resides within the glycine- and proline-rich sequence (tail homology region 2) FEBS Lett. 1994;342:197–202. doi: 10.1016/0014-5793(94)80500-8. [DOI] [PubMed] [Google Scholar]
- 26.Rump A, Scholz T, Thiel C, Hartmann FK, Uta P, Hinrichs MH, Taft MH, et al. Myosin-1C associates with microtubules and stabilizes the mitotic spindle during cell division. J Cell Sci. 2011;124:2521–2528. doi: 10.1242/jcs.084335. [DOI] [PubMed] [Google Scholar]
- 27.Menegazzi R, Busetto S, Dri P, Cramer R, Patriarca P. Chloride ion efflux regulates adherence, spreading, and respiratory burst of neutrophils stimulated by tumor necrosis factor-alpha (TNF) on biologic surfaces. J Cell Biol. 1996;135:511–522. doi: 10.1083/jcb.135.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt A, Caron E, Hall A. Lipopolysaccharide-induced activation of beta2-integrin function in macrophages requires Irak kinase activity, p38 mitogen- activated protein kinase, and the Rap1 GTPase. Mol Cell Biol. 2001;21:438–448. doi: 10.1128/MCB.21.2.438-448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev. 2007;219:143–156. doi: 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 30.Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol Rev. 2013;256:222–239. doi: 10.1111/imr.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu SL, May JR, Helgeson LA, Nolen BJ. Insertions within the actin core of actin-related protein 3 (Arp3) modulate branching nucleation by Arp2/3 complex. J Biol Chem. 2013;288:487–497. doi: 10.1074/jbc.M112.406744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta P, Gauthier NC, Cheng-Han Y, Zuanning Y, Pontes B, Ohmstede M, Martin R, et al. Myosin 1E localizes to actin polymerization sites in lamellipodia, affecting actin dynamics and adhesion formation. Biol Open. 2013;2:1288–1299. doi: 10.1242/bio.20135827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cervero P, Himmel M, Kruger M, Linder S. Proteomic analysis of podosome fractions from macrophages reveals similarities to spreading initiation centres. Eur J Cell Biol. 2012;91:908–922. doi: 10.1016/j.ejcb.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Schiller HB, Friedel CC, Boulegue C, Fassler R. Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 2011;12:259–266. doi: 10.1038/embor.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diakonova M, Bokoch G, Swanson JA. Dynamics of cytoskeletal proteins during Fcgamma receptor-mediated phagocytosis in macrophages. Mol Biol Cell. 2002;13:402–411. doi: 10.1091/mbc.01-05-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bi J, Chase SE, Pellenz CD, Kurihara H, Fanning AS, Krendel M. Myosin 1e is a component of the glomerular slit diaphragm complex that regulates actin reorganization during cell-cell contact formation in podocytes. Am J Physiol Renal Physiol. 2013;305:F532–544. doi: 10.1152/ajprenal.00223.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schietroma C, Yu HY, Wagner MC, Umbach JA, Bement WM, Gundersen CB. A role for myosin 1e in cortical granule exocytosis in Xenopus oocytes. J Biol Chem. 2007;282:29504–29513. doi: 10.1074/jbc.M705825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel DY, Heijnen PD, Breur M, de Vries HE, Tool AT, Amor S, Dijkstra CD. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J Neuroinflammation. 2014;11:23. doi: 10.1186/1742-2094-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paul P, van den Hoorn T, Jongsma ML, Bakker MJ, Hengeveld R, Janssen L, Cresswell P, et al. A Genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell. 2011;145:268–283. doi: 10.1016/j.cell.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 40.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307:1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 41.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]