

Abstract
We report a new methodology for the synthesis of polymer-drug conjugates from “compound”—all in one—prodrug monomers that consist of a cyclic polymerizable group that is appended to a drug through a cleavable linker. We show that organocatalyzed ring-opening polymerization can polymerize these monomers into well-defined polymer prodrugs that are designed to self-assemble into nanoparticles and release drug in response to a physiologically relevant stimulus. This method is compatible with structurally diverse drugs and allows different drugs to be copolymerized with quantitative conversion of the monomers. The drug loading can be controlled by adjusting the monomer(s) to initiator feed ratio and drug release can be encoded into the polymer by the choice of linker. Initiating these monomers from a polyethylene glycol macroinitiator yields amphiphilic diblock copolymers that spontaneously self-assemble into micelles with a long plasma circulation, which is useful for systemic therapy.
Keywords: Polymerizable prodrug, Ring-opening polymerization, Polymer-drug conjugate, Nanoparticle, Cancer therapy
Most small-molecule drugs utilized in the clinic have poor bioavailability and suboptimal pharmacokinetics because of their hydrophobicity and low molecular weight. Polymeric drug delivery systems can improve the efficacy of these drugs by increasing their water solubility, prolonging their circulation time, increasing the amount of drug deposited in the target tissue, and decreasing their exposure to normal tissues.[1] Conjugation of hydrophobic drugs to hydrophilic polymers can address these problems,[2] and is typically carried out by separate synthesis of the polymer, drug and linker, and sequential conjugation of the three entities to create the polymer-drug conjugate. This conventional strategy requires multiple reaction steps with limited yield, and has limited control of the site and degree of drug loading. New methods are hence needed to synthesize polymer-drug conjugates that have the following attributes: (1) are compatible with a structurally diverse set of drugs; (2) enable more than one drug to be conjugated to the same polymer with tunable control of the loading of the two drugs; (3) proceed with high yield and enable easy purification of the conjugate; and (4) enable release of the drug by a biologically relevant trigger.
Motivated by this rationale, we report herein a new method to synthesize polymer-drug conjugates by living ring-opening polymerization (ROP) of prodrugs (Scheme 1). This scheme inverts the conventional approach of conjugating a drug to a polymer post-synthesis, and instead directly incorporates the drug during synthesis of the polymer. These polymer prodrugs are composed of a “compound” monomer that consists of three covalently linked moieties: (1) a cyclic group that can undergo ROP to yield a biodegradable main chain, that is attached to (2) a cleavable linker, and which is attached to (3) a drug of interest. Living ROP of the compound monomer leads to the synthesis of a polymer with a biodegradable main chain with pendant drug molecules that are linked to the main chain via a cleavable linker. We show that this method is suitable for structurally diverse drugs. More one drug can be copolymerized by this methodology and the drug loading and release can be readily controlled by adjusting the monomer to initiator feed ratio and by the design of linkers that are cleaved by relevant in vivo triggers, such as the reductive environment of the cell cytosol. We further show that by the appropriate choice of an initiator—in this case poly(ethylene glycol) methyl ether (mPEG)—the second drug-loaded segment can be directly grown from the mPEG macroinitiator, leading to the formation of an amphiphilic diblock copolymer that spontaneously self-assembles into PEGylated—stealth—micelles with a size and pharmacokinetics that are suitable for systemic therapy of solid tumors.
Scheme 1.
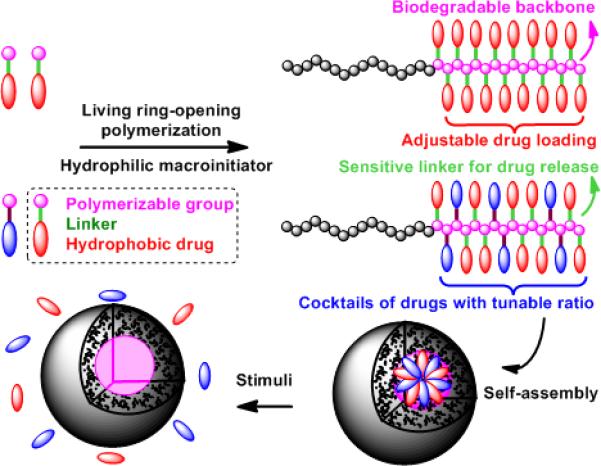
Schematic illustration of the design and synthesis of biodegradable polymer prodrugs by living ROP of “compound”—all in one—prodrug monomers, as well as their self-assembly into nanoscale micelles.
Polymerization of prodrugs was previously reported by conventional condensation polymerization.[3] Recently, living radical and ring-opening metathesis polymerization of prodrugs have been explored to prepare polymer therapeutics,[4] however, polymer-drug conjugates synthesized by these methods are non-biodegradable, which limits their clinical application. We chose organocatalytic ROP[5] for synthesis of polymer prodrugs because it is a powerful method for the synthesis of aliphatic polyesters,[6] polycarbonates,[7] polypeptides,[8] and polyphospho-esters,[9] that are biodegradable. As the starting point for the synthesis of the compound monomer, we chose a commercially available functional ester intermediate, pentafluorophenyl 5-methyl-2-oxo-1, 3-dioxane-5-carboxylate (Carb-C6F5). Our initial choice of anticancer drug was chlorambucil (CL) because the clinical application of CL is limited by its toxic side effects such as nausea, myelotoxicity, and neurotoxicity.[11] The prodrug 2 (CarbCL) consisting of a polymerizable cyclic carbonate linked to an ethylene glycol linker and CL was synthesized by reaction between hydroxyl functionalized 1 and Carb-C6F5 in tetrahydrofuran using CsF as catalyst (Scheme 2a). Details of the synthesis and characterization of 1 and 2 are described in the Supporting Information.
Scheme 2.
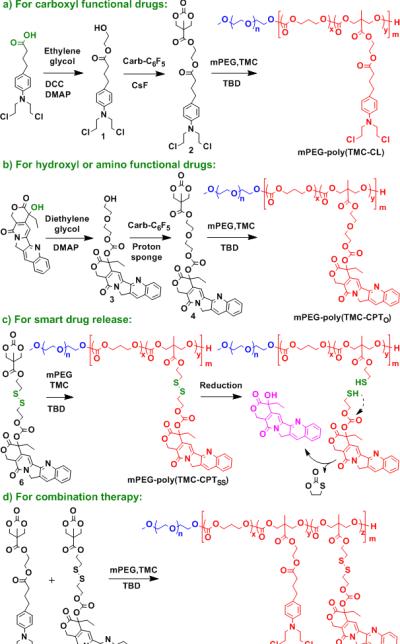
Detailed synthetic routes of polymerizable prodrugs and their polymers.
We investigated ROP of CarbCL using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as the organocatalyst and mPEG as macroinitiator. We chose mPEG as the macroinitiator because the resulting diblock copolymer, consisting of mPEG and the polymer prodrug, is amphiphilic and is likely, we hypothesized, to self-assemble into long circulating nanoparticles by virtue of PEGs’ stealth-like properties. Trimethylene carbonate (TMC), a commercial available cyclic carbonate monomer, was used as a co-monomer to tune the degree of drug loading.
We investigated the copolymerization of CarbCL and TMC in chloroform at room temperature (RT) with different monomer/initiator feed ratios (Table 1). As shown in Figure 1a, the ROP of CarbCL and TMC exhibited a linear evolution of Mn with increasing monomer conversion that is characteristic of a living polymerization. Gel permeation chromatography (GPC) showed monomodal and symmetric elution peaks for mPEG-poly(TMC-CL)s that exhibited a clear shift to a higher molecular weight with reaction time (Figure 1a, inset). In comparison with mPEG, GPC elution curves of mPEG-poly(TMC-CL)s showed no visible residual mPEG peak, indicating high initiation efficacy of ROP. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectra of these polymers showed the absence of a peak at ~5 kDa, suggesting almost complete consumption of the macroinitiator (Figure S9). Furthermore, almost quantitative conversion of the monomers was achieved after ~17 h reaction according to the proton nuclear magnetic resonance (1H NMR) spectra (Figure S8a and Table S1). Interestingly, complete consumption of CarbCL was observed within 10 min indicating its higher reactivity than TMC. Similar results have been reported for copolymerization of TMC with other cyclic carbonate monomers.[12] The quantitative conversion of the comonomers also facilitated the purification of mPEG-poly(TMC-CL) with a high yield of ~85% by precipitation from chloroform into diethyl ether. 1H NMR spectrum revealed that >99% of the polymer chains have hydroxyl end-groups, verifying the homogeneity of the polymer (Figure S8c). In addition, the degree of polymerization (DP) of poly(TMC-CL) could be conveniently adjusted by tuning the monomer/initiator feed ratio (Table 1, entry 1-4). As given in Figure 1b, the drug loading of mPEG-poly(TMC-CL) could be tuned from 10 wt.% to ~30 wt.% by increasing the CarbCL/mPEG molar feed ratio from 5.0 to 15. Taken together, these results confirm that organocatalyzed ROP of prodrugs enables the facile synthesis of polymer prodrugs with quantitative monomer conversion and polymerization initiation efficiency, and with an adjustable degree of drug loading.
Table 1.
Summary of all polymer prodrugs.[a]
| Entry | Molar feed ratio M1:M2:M3:M4:I | DP M1:M2:M3:M4 | Mn g mol−1 | Mw/Mn | Dh nm | PDI |
|---|---|---|---|---|---|---|
| 1 | 45 : 5 : 0 : 0 :1 | 49 : 4 : 0 : 0 | 12100 | 1.17 | 35 | 0.06 |
| 2 | 43 : 8 : 0 : 0 : 1 | 48 : 7 : 0 : 0 | 13400 | 1.19 | 40 | 0.06 |
| 3 | 40 : 10 : 0 : 0 : 1 | 46 : 13 : 0 : 0 | 16300 | 1.23 | 43 | 0.04 |
| 4 | 35 : 15 : 0 : 0 : 1 | 41 : 16 : 0 : 0 | 17400 | 1.29 | 49 | 0.07 |
| 5 | 40 : 0 : 10 : 0 : 1 | 45 : 0 : 9 : 0 | 15800 | 1.19 | 37 | 0.04 |
| 6 | 37 : 0 : 3 : 0 : 1 | 43 : 0 : 2 : 0 | 10600 | 1.27 | 31 | 0.01 |
| 7 | 40 : 0 : 0 : 5 : 1 | 44 : 0 : 0 : 4 | 12400 | 1.19 | 30 | 0.04 |
| 8 | 43 : 5 : 0 : 3 : 1 | 47 : 5 : 0 : 2 | 14000 | 1.20 | 33 | 0.05 |
M1, M2, M3, M4, and I are TMC, CarbCL, Carb-O-CPT, Carb-SS-CPT, and mPEG-5k, respectively. The DP was calculated by 1H NMR. The Mn and Mw/Mn were obtained from GPC. The Dh and PDI of the self-assembled micelles were determined by DLS.
Figure 1.

a, Linearly increasing Mn by 1H NMR as a function of monomer conversion for ROP of CarbCL and TMC using mPEG (Mn of 5 kDa) as macroinitiator. The inset shows representative GPC curves after 10 min (red), 1 h (green), and 4 h (pink) reaction. b, Plot of drug loading vs. molar feed ratio of CarbCL/mPEG. c, Plot of the I339.2/I334.9 ratio from pyrene excitation spectra as a function of the concentration of the polymer prodrugs on a log scale (log C). d, Representative cryo-TEM image of mPEG-poly(TMC46-CL13) micelles.
The amphiphilic nature of these polymer prodrugs also drives their self-assembly in aqueous media. The critical micellization concentration (CMC) was characterized by using pyrene as a probe.[13] As shown in Figure 1c, the CMCs of mPEG-poly(TMC- CL) slightly decreased from 2.5 to 1.1 μg mL−1 with an increase in the polycarbonate content from 58 wt.% to 71 wt.%. These relatively low CMC values indicate that the mPEG-poly(TMC-CL) micelles are quite stable in water. Dynamic light scattering (DLS) showed that the size of the micelles was tunable by control of the molecular weight of the hydrophobic polycarbonate segment. The average hydrodynamic diameter (Dh) of mPEG-poly(TMCCL) micelles increased from 35 to ~50 nm as the CarbCL to mPEG ratio increased from 5.0 to 15 (Table 1, entry 1-4). Transmission electron microscopy (TEM) images further showed that these amphiphilic polymer prodrugs self-assembled into spherical micelles with a size that agreed well with DLS results (Figure 1d, Figure S14 and Table S2).
We next investigated the generality of this methodology by asking the question whether it could be used with other hydrophobic drugs, for example, bearing hydroxyl, amine, or other functional groups? To answer this question, we chose camptothecin (CPT), a hydroxyl functionalized anticancer drug for synthesis of a polymer prodrug. CPT is a highly potent, naturally occurring alkaloid with a wide spectrum of antitumor activity through inhibition of topoisomerase I and HIF-1α.[14] However, its systemic delivery is problematic because of its low aqueous solubility. As shown in Scheme 2b, CPT was first activated by triphosgene in the presence of 4-(dimethylamino)pyridine (DMAP) and then reacted with excess diethylene glycol to produce intermediate 3. Finally, prodrug 4 (Carb-O-CPT) consisting of a polymerizable cyclic carbonate, a diethylene glycol linker, and CPT was successfully synthesized by reaction between 3 and Carb-C6F5 in dimethyl sulfoxide using 1,8-bis(dimethylamino)naphthalene (proton sponge) as the catalyst. Copolymerization of Carb-O-CPT and TMC was performed in chloroform at RT using TBD and mPEG as the catalyst and macroinitiator, respectively. The conversion of Carb-O-CPT was nearly complete, as indicated by 1H NMR (Table S1, entry 5, 6). Polymer prodrugs of mPEG-poly(TMCCPTO) (N.B.: the subscript o indicates a chemically stable ether linker) with a CPT loading from 6.6 wt.% to 21 wt.% were obtained by increasing the Carb-O-CPT/mPEG molar feed ratio from 3.0 to 10 (Table 1, entry 5, 6).
Having synthesized two polymer prodrugs with tunable loading, we next turned our attention to devising a suitable release mechanism of the drug from the polymer. To address this challenge, a reduction-responsive prodrug 6 (Carb-SS-CPT) was designed and synthesized, as shown in Scheme 2c, motivated by previous studies on reduction sensitive polymer nanoparticles.[15] The synthetic route of 6 is similar to that of Carb-O-CPT except that 2,2’-dithiodiethanol was used as a linker. As expected, mPEG-poly(TMC-CPTSS) (N.B. the subscript SS indicates a reduction cleavable disulfide linker) was synthesized by ROP of Carb-SS-CPT and TMC, using TBD and mPEG as catalyst and macroinitiator, respectively (Table 1, entry 7). The drug release kinetics of mPEG-poly(TMC-CPTSS) under physiological environment were investigated with or without treatment with glutathione (GSH). As shown in Figure 2a, rapid drug release was observed for mPEG-poly(TMC-CPTSS) micelles in the presence of 10 mM GSH. The cumulative released CPT reached ~75% after 24 hours incubation, indicating the reduction-responsiveness of mPEG-poly(TMCCPTSS). Liquid chromatography–mass spectrometry analysis of the released products from mPEG-poly(TMC-CPTSS) showed that the disulfide bond was cleaved by GSH (Figure S15), which induced the breakdown of the neighboring carbonate bond to generate free CPT.[16] In contrast, only ~15% released drug was observed for mPEG-poly(TMC-CPTSS) micelles when incubated without GSH. The nonresponsive control, mPEG-poly(TMCCPTO) micelles also displayed slow drug release even upon addition of 10 mM GSH. Furthermore, very limited drug leakage was observed by incubating the micelles in serum (Figure S16b). These release profiles showed that mPEG-poly(TMC-CPTSS) micelles have good stability under normal physiological conditions, but exhibit rapid drug release in a reductive environment.
Figure 2.

a, Cumulative drug release from mPEG-poly(TMC-CPTSS) micelles in PBS at 37 °C with (■) or without (●) 10 mM GSH. Nonresponsive mPEG-poly(TMC-CPTO) in the presence of GSH was used as a control (▲). b, Cell viability treated with mPEG-poly(TMC-CL) (● C26 cells; ▼ 4T1 cells) or free CL (■ C26 cells; ▲ 4T1 cells). c, cell viability treated with mPEG-poly(TMC CPTSS) (● C26 cells; ◄ 4T1 cells), mPEG-poly(TMC-PCPTO) (▲ C26 cells; ► 4T1 cells), or free CPT (■ C26 cells; ▼ 4T1 cells). d, 4T1 cell viability treated with mPEG-poly(TMC-CL-CPTSS) (●) or free CPT (■). The cells were incubated for 72 hours and the % cell viability is normalized against the blank cells in the same experiment. e, Plot of carbonate bond content in mPEG-poly(TMC)46 vs. incubation time in lipase solution at 37 °C. f, Plasma drug concentration of mPEG-poly(TMC-CPTSS) (●) and free CPT (■) as a function of time post-i.v. injection in mice.
To complete our exploration of its flexibility, we next examined whether this synthetic methodology was amenable to the copolymerization of two different drug containing monomers, as many cancers are treated with a cocktail of different drugs.[17] We hence copolymerized CarbCL, Carb-SS-CPT and TMC using TBD and mPEG as the catalyst and macroinitiator, respectively (Scheme 2d). A diblock copolymer of mPEG with a random block of poly(TMC-CL-CPTSS) was easily synthesized with quantitative conversion of the monomers (Table S1, entry 8). The mPEG-poly(TMC-CL-CPTSS) diblock copolymer contained a CL/CPT molar ratio of 2.5, close to the CarbCL/Carb-SS-CPT molar feed ratio (Table 1, entry 8). This suggests that the total drug loading and ratio of the two drugs of mPEG-poly(TMC-CLCPTSS) can be controlled by adjusting the molar feed ratio of the two drug containing co-monomers.
We then evaluated the anticancer effects of these polymer prodrugs in vitro by a cell viability assay in murine C26 colon and 4T1 breast cancer cell lines; these cell lines were chosen because they have been reported to be sensitive to CL and CPT.[18] It was found that both polymer prodrugs exhibited dose-dependent inhibition of C26 and 4T1 cells. The doses of mPEG-poly(TMC-CL) required for 50% cytotoxicity (IC50) against C26 and 4T1 cells were 39 and 1.2×102 μM respectively, which were ~2 fold higher than those for free CL (Figure 2b). These results are encouraging because the calssical CL prodrugs often show significantly lower in vitro cytotoxicity than free CL.[11] The IC50 of mPEG-poly(TMC-CPTSS) for C26 and 4T1 cells were 0.32 and 1.4 μM, respectively, which were much lower than the IC50 of mPEG-poly(TMC-CPTO)–a polymer prodrug wherein the drug is attached to the polymer through a stable ether linker–in the same cell lines (Figure 2c). The enhanced cytotoxicity of mPEG-poly(TMC-CPTSS) compared to mPEG-poly(TMC-CPTO) is likely due to its reduction-sensitive linker, which facilitates CPT release in cells (Figure S17). We note that extracellular release of camptothecin might occur in cell culture and in vivo because of the presence of thiols secreted by cells that would lead to cleavage of this non-sterically hindered disulfide bond between the drug and polymer.[19] Surprisingly, the IC50 value of mPEG-poly(TMC-CPTSS) against 4T1 cells was even lower than that of free CPT. The higher cytotoxicity of CPT in the reducible polymer conjugate than free drug also translated to a more potent combination polymer prodrug, as the IC50 of mPEG-poly(TMC-CL-CPTSS ) for 4T1 cells was ~0.15 μM, which is 14-fold lower than a cocktail of the free drugs administered at the same drug ratio (Figure 2d). Together, these in vitro studies clearly demonstrate that these polymer prodrugs have similar— and in one case—greater cytotoxicity than the free drug(s), and that this effect can be modulated by the design of the linker.
To confirm that the base polymer has no intrinsic cytotoxicity, we cultured C26 and 4T1 cells with mPEG-poly(TMC)46, a diblock copolymer devoid of drug, and observed that the polymer exhibited no cytotoxicity even at a high concentration of up to 1.0 mg mL−1 (Figure S19). We also examined the in vitro degradation of the base polymer in the presence of lipase that is known to degrade PTMC and other aliphatic polycarbonates.[20] The results showed that ~60% carbonate bonds in mPEG-poly(TMC)46 were degraded after 6 days of incubation with lipase (Figure 2e), demonstrating the biodegradability of the polycarbonate.
Finally, the in vivo plasma concentration of mPEG-poly(TMCCPTSS) was measured as a function of time after i.v. injection into mice. The data were fitted to a two-compartment pharmacokinetic model, yielding a plasma AUC of 6.1×102 μMh (Figure 2f). The AUC for mice treated with the same dose of free CPT was only 71 μMh. This ~9-fold increase in plasma AUC suggests that these polymer prodrug micelles have a long plasma circulation by virtue of PEGs’ stealth-like properties and are likely to preferentially accumulate in solid tumors, as compared to free drug.
In summary, we report a new methodology for the synthesis of well-defined polymer prodrugs that are designed to self-assemble into nanoparticles and release drug in response to a physiologically relevant stimulus from compound monomers that consist of a cyclic polymerizable group that is appended to a drug through a cleavable linker. Initiating ROP of these prodrug monomers from a PEG macroinitiator yields amphiphilic diblock copolymers that spontaneously self-assemble into spherical nanoscale micelles in which the drug(s) are sequestered in the core of the micelle with a PEG corona that imparts a long plasma circulation to the micelles that is ideal for systemic therapy of solid tumors. These results set the stage for a thorough evaluation of their in vivo efficacy, studies that are currently ongoing.
Supplementary Material
Acknowledgements
We would like to thank Michelle Gignac for the TEM, Yingrong Xu for the MALDI-TOF MS and Xiaomeng Wan for the SEC measurements. This work was supported by funding from the NIH (R01-EB 000188 and R01-DK092665) to A.C.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat. Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; b Lukyanov AN, Torchilin VP. Adv. Drug Delivery Rev. 2004;56:1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]; c Kataoka K, Harada A, Nagasaki Y. Adv. Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]; d Duncan R. Nat. Rev. Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]; e Lee CC, MacKay JA, Frechet JM, Szoka FC. Nat. Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]; f Davis ME, Chen Z, Shin DM. Nat. Rev. Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 2.a Ulbrich K, Šubr V. Adv. Drug Delivery Rev. 2004;56:1023–1050. doi: 10.1016/j.addr.2003.10.040. [DOI] [PubMed] [Google Scholar]; b Johnson JA, Lu YY, Burts AO, Xia Y, Durrell AC, Tirrell DA, Grubbs RH. Macromolecules. 2010;43:10326–10335. doi: 10.1021/ma1021506. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Proc. Natl. Acad. Sci. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]; d del Rosario LS, Demirdirek B, Harmon A, Orban D, Uhrich KE. Macromol. Biosci. 2010;10:415–423. doi: 10.1002/mabi.200900335. [DOI] [PubMed] [Google Scholar]
- 3.a Rosario-Meléndez R, Harris CL, Delgada-Rivera R, Yu L, Uhrich KE. J. Controlled Release. 2012;162:538–544. doi: 10.1016/j.jconrel.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rosario-Meléndez R, Yu W, Uhrich KE. Biomacromolecules. 2013;14:3542–3548. doi: 10.1021/bm400889a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ouimet MA, Griffin J, Carbone-Howell AL, Wu WH, Stebbins ND, Di R, Uhrich KE. Biomacromolecules. 2013;14:854–861. doi: 10.1021/bm3018998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Hu XL, Hu JM, Tian J, Ge ZS, Zhang GY, Luo KF, Liu SY. J. Am. Chem. Soc. 2013;135:17617–17629. doi: 10.1021/ja409686x. [DOI] [PubMed] [Google Scholar]; b Liao LY, Liu J, Dreaden EC, Morton SW, Shopsowitz KE, Hammond PT, Johnson JA. J. Am. Chem. Soc. 2014;136:5896–5899. doi: 10.1021/ja502011g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Kiesewetter MK, Shin EJ, Hedrick JL, Waymouth RM. Macromolecules. 2010;43:2093–2107. [Google Scholar]; b Nederberg F, Zhang Y, Tan JPK, Xu KJ, Wang HY, Yang C, Gao SJ, Guo XD, Fukushima K, Li LJ, Hedrick JL, Yang Y-Y. Nat. Chem. 2011;3:409–414. doi: 10.1038/nchem.1012. [DOI] [PubMed] [Google Scholar]
- 6.a Albertsson AC, Varma IK. Adv. Polym. Sci. 2002;157:1–40. [Google Scholar]; b Jérôme C, Lecomte P. Adv. Drug Delivery Rev. 2008;60:1056–1076. doi: 10.1016/j.addr.2008.02.008. [DOI] [PubMed] [Google Scholar]; c Gerhardt WW, Noga DE, Hardcastle KI, García AJ, Collard DM, Weck M. Biomacromolecules. 2006;7:1735–1742. doi: 10.1021/bm060024j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Rokicki G. Prog. Polym. Sci. 2000;25:259–342. [Google Scholar]; b Feng J, Zhuo RX, Zhang XZ. Prog. Polym. Sci. 2012;37:211–236. [Google Scholar]; c Zhu KJ, Hendren RW, Jensen K, Pitt CG. Macromolecules. 1991;24:1736–1740. [Google Scholar]
- 8.a Cheng JJ, Deming TJ. Top. Curr. Chem. 2012;310:1–26. doi: 10.1007/128_2011_173. [DOI] [PubMed] [Google Scholar]; b Huang J, Heise A. Chem. Soc. Rev. 2013;42:7373–7390. doi: 10.1039/c3cs60063g. [DOI] [PubMed] [Google Scholar]
- 9.a Wang YC, Yuan YY, Du JZ, Yang XZ, Wang J. Macromol. Biosci. 2009;9:1154–1164. doi: 10.1002/mabi.200900253. [DOI] [PubMed] [Google Scholar]; b Zhang SY, Zou J, Zhang FW, Elsabahy M, Felder SE, Zhu JH, Pochan DJ, Wooley KL. J. Am. Chem. Soc. 2012;134:18467–18474. doi: 10.1021/ja309037m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Sanders DP, Fukushima K, Coady DJ, Nelson A, Fujiwara M, Yasumoto M, Hedrick JL. J. Am. Chem. Soc. 2010;132:14724–14726. doi: 10.1021/ja105332k. [DOI] [PubMed] [Google Scholar]; b Engler AC, Chan JMW, Coady DJ, O'Brien JM, Sardon H, Nelson A, Sanders DP, Yang YY, Hedrick JL. Macromolecules. 2013;46:1283–1290. [Google Scholar]
- 11.a Dorr RT, Fritz WL. Cancer Chemotherapy Handbook. Elsevier Science; New York: 1982. p. 486. [Google Scholar]; b Beyer U, Roth T, Schumacher P, Maier G, Unold A, Frahm AW, Fiebig HH, Unger C, Kratz F. J. Med. Chem. 1998;41:2701–2708. doi: 10.1021/jm9704661. [DOI] [PubMed] [Google Scholar]
- 12.Pratt RC, Nederberg F, Waymouth RM, Hedrick JL. Chem. Commun. 2008:114–116. doi: 10.1039/b713925j. [DOI] [PubMed] [Google Scholar]
- 13.Wilhelm M, Zhao CL, Wang YC, Xu RL, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991;24:1033–1040. [Google Scholar]
- 14.a Hsiang YH, Hertzberg R, Hecht S, Liu LF. J. Biol. Chem. 1985;260:4873–4878. [PubMed] [Google Scholar]; b Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- 15.a Hao JH, Kwissa M, Pulendran B, Murthy N. Int. J. Nanomedicine. 2006;1:97–103. doi: 10.2147/nano.2006.1.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cerritelli S, Velluto D, Hubbell J. Biomacromolecules. 2007;8:1966–1972. doi: 10.1021/bm070085x. [DOI] [PubMed] [Google Scholar]
- 16.Lee MH, Yang ZG, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS. Chem. Rev. 2013;113:5071–5109. doi: 10.1021/cr300358b. [DOI] [PubMed] [Google Scholar]
- 17.Smalley RV, Carpenter J, Bartolucci A, Vogel C, Krauss S. Cancer. 1977;40:625–632. doi: 10.1002/1097-0142(197708)40:2<625::aid-cncr2820400206>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 18.a Goff RD, Thorson JS. J. Med. Chem. 2010;53:8129–8139. doi: 10.1021/jm101024j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Davis ME. Adv. Drug Delivery Rev. 2009;61:1189–1192. doi: 10.1016/j.addr.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Brülisauer L, Valentino G, Morinaga S, Cam K, Bukrinski JT, Gauthier MA, Leroux J-C. Angew. Chem. Int. Ed. 2014;53:8392–8396. doi: 10.1002/anie.201404238. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126:8532–8536. [Google Scholar]
- 20.a Tsutsumi C, Nakagawa K, Shirahama H, Yasuda H. Macromol. Biosci. 2002;2:223–232. [Google Scholar]; b Zhang Z, Kuijer R, Bulstra SK, Grijpma DW, Feijen J. Biomaterials. 2006;27:1741–1748. doi: 10.1016/j.biomaterials.2005.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.