

Abstract
Facilitation and inactivation of P/Q-type Ca2+ currents mediated by Ca2+/calmodulin binding to CaV2.1 channels contribute to facilitation and rapid depression of synaptic transmission, respectively. Other calcium sensor proteins displace calmodulin from its binding site and differentially modulate P/Q-type Ca2+ currents, resulting in diverse patterns of short-term synaptic plasticity. Neuronal calcium sensor-1 (NCS-1, frequenin) has been shown to enhance synaptic facilitation, but the underlying mechanism is unclear. We report here that NCS-1 directly interacts with IQ-like motif and calmodulin-binding domain in the C-terminal domain of CaV2.1 channel. NCS-1 reduces Ca2+-dependent inactivation of P/Q-type Ca2+ current through interaction with the IQ-like motif and calmodulin-binding domain without affecting peak current or activation kinetics. Expression of NCS-1 in presynaptic superior cervical ganglion neurons has no effect on synaptic transmission, eliminating effects of this calcium sensor protein on endogenous N-type Ca2+ currents and the endogenous neurotransmitter release machinery. However, in superior cervical ganglion neurons expressing wild-type CaV2.1 channels, co-expression of NCS-1 induces facilitation of synaptic transmission in response to paired pulses and trains of depolarizing stimuli, and this effect is lost in CaV2.1 channels with mutations in the IQ-like motif and calmodulin-binding domain. These results reveal that NCS-1 directly modulates CaV2.1 channels to induce short-term synaptic facilitation and further demonstrate that CaS proteins are crucial in fine-tuning short-term synaptic plasticity.
Keywords: short-term plasticity, neuronal calcium sensor-1, Cav2.1
Introduction
Short-term plasticity on the time scale from milliseconds to seconds causes rapid, bidirectional changes in synaptic strength including facilitation, augmentation, post-tetanic potentiation, and depression (Zucker and Regehr, 2002). Integration of different patterns of short-term plasticity allows synapses to perform filtering and computational functions and, therefore, short-term plasticity is crucial for neural coding and information processing (Abbott and Regehr, 2004). Synaptic facilitation is caused by residual Ca2+ that builds up in the presynaptic terminal during repetitive action potentials and acts on Ca2+ sensors that are distinct from those responsible for fast exocytosis (Blatow et al., 2003; Catterall and Few, 2008; Zucker and Regehr, 2002). However, the effector mechanisms that respond to residual Ca2+ and enhance neurotransmitter release remain elusive.
P/Q-type Ca2+ currents conducted by CaV2.1 channels initiate neurotransmission at most fast conventional synapses (Dunlap et al., 1995; Jun et al., 1999; Llinas et al., 1992; Westenbroek et al., 1995). Facilitation and inactivation of P/Q-type Ca2+ currents are mediated by Ca2+/calmodulin (CaM) binding to the IQ-like motif and calmodulin-binding domain (CBD) in the C-terminal domain of the α12.1 subunit (Catterall and Few, 2008; DeMaria et al., 2001; Lee et al., 2000; Lee et al., 1999; Lee et al., 2003). The steep dependence of neurotransmission on presynaptic Ca2+ entry predicts that this modulation should have a profound impact on short-term plasticity (Dodge and Rahamimoff, 1967). In agreement with this expectation, regulation of CaV2.1 channels by CaM induces facilitation followed by rapid depression of synaptic transmission in transfected superior cervical ganglion (SCG) neurons (Mochida et al., 2008).
CaM is the founding member of a large family of Ca2+ sensor (CaS) proteins that are differentially expressed in the central nervous system and retina (Burgoyne et al., 2004; Burgoyne and Weiss, 2001; Haeseleer et al., 2002). Interaction of Ca2+-binding protein 1 (CaBP1) with CBD induces fast inactivation of CaV2.1 channels, which results in rapid synaptic depression in transfected SCG neurons (Few et al., 2011; Leal et al., 2012; Lee et al., 2002; Mochida et al., 2008). In contrast, visinin-like protein 2 (VILIP-2) binding to CaV2.1 slows the rate of inactivation and enhances short-term synaptic facilitation (Lautermilch et al., 2005; Leal et al., 2012; Nanou et al., 2012).
Neuronal calcium sensor-1 (NCS-1) is the classical example of facilitation of synaptic activity and secretion by CaS proteins (Pongs et al., 1993; Sippy et al., 2003; Tsujimoto et al., 2002; Weiss et al., 2010). Frequenin, the Drosophila homologue of NCS-1, increases synaptic facilitation (Pongs et al., 1993) and modulates Ca2+ entry through functional interaction with the protein encoded by cacophony, the only gene encoding a CaV2 α1 subunit in Drosophila (Dason et al., 2009). In rat hippocampal cell cultures and at the Calyx of Held synapse, NCS-1 induces Ca2+-dependent synaptic facilitation (Sippy et al., 2003; Tsujimoto et al., 2002). In contrast, overexpression of a dominant-negative NCS-1 enhances activity of non-L-type Ca2+ channels in neuroendocrine cells, suggesting that NCS-1 negatively regulates these channels (Weiss et al., 2000; Weiss and Burgoyne, 2001). Direct binding of NCS-1 to the C-terminal domain of CaV2.1 has been shown in biochemical experiments (Lian et al., 2014). However, the effects of NCS-1 interaction with Ca2+ channels in these preparations differ, and the mechanism of NCS-1 regulation of Ca2+ currents and modulation of synaptic plasticity remains uncertain. Here we report that NCS-1 reduces inactivation of CaV2.1 channels through interaction with the IQ-like motif and CBD in the C-terminal domain and thereby enhances short-term synaptic facilitation in SCG neurons. These results reveal the molecular mechanism of NCS-1-induced facilitation and demonstrate that CaS proteins are key players in the diversity of short-term synaptic plasticity.
Experimental methods
Binding assays
Binding assays were performed as previously described (Magupalli et al., 2013; Nanou et al., 2012). NCS-1-MBP or MBP alone was immobilized on amylose beads (New England Biolabs, Beverly, MA) that were extensively washed with PBS buffer. The MBP proteins were incubated with 4 μg of CBD-GST, IQ-GST or GST alone proteins for 2 hrs at 4 °C. The binding buffer contained (in mM): 200 NaCl, 1 MgCl2, 20 Tris-HCl and 0.1% Triton X-100. Ca2+ and EGTA were added as described. After extensive washing, bound proteins were eluted at 97 °C with sample buffer (2×) and separated on a NuPAGE gel (Invitrogen). Immunoblotting was performed with monoclonal anti-GST (Sigma) or anti-MBP (New England Biolabs) antibodies. Bound protein ligands were quantified by immunoblotting of the gel with a secondary antibody against GST or MBP covalently tagged with horseradish peroxidase, and measurement of horseradish peroxidase luminescence in the linear range of the luminometer. Analysis of the blots was done using the National Institutes of Health ImageJ program, and relative binding was normalized to control GST or MBP.
Transfection and voltage clamp recording of tsA-201 cells
Whole-cell voltage clamp recording were acquired from transfected tsA-201 cells (Nanou et al., 2012). Human embryonic kidney tsA-201 cells were grown in DMEM/Ham’s F12 with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 100 units/ml penicillin and streptomycin (Invitrogen) to ~80% confluence at 37 °C and 10% CO2. NCS-1 was subcloned into an eGFP-expressing vector (pEGFP-N1) such that the resulting chimeric protein consisted of NCS-1 followed by eGFP in a single polypeptide. Cells were transfected with cDNA encoding Ca2+ channel subunits α12.1 (2 μg) or α12.1IMAA/ΔCBD (2 μg), β2a (1.5 μg), α2δ (1 μg) and NCS-1-eGFP (0.005 μg) using TransIT-LT1 (Mirus Bio LLC, Madison, WI). Finally, 24h after transfection, the cells were used for electrophysiological recordings. Whole cell voltage clamp recordings were acquired from transfected tsA-201 cells at room temperature. The recordings were done in an extracellular solution containing (in mM) 10 CaCl2 or 10 BaCl2, 150 Tris, 1 MgCl2 (305 mosM) and with an intracellular solution consisting of (in mM) 120 N-methyl-D-glucamine, 60 Hepes, 1 MgCl2, 2 Mg-ATP, and 0.5EGTA (295 mosM). The pH of both solutions was adjusted to pH 7.3 with methanesulfonic acid. Recordings were made with a HEKA EPC 10 patch clamp amplifier using PULSE software (HEKA Elektronik, Lambrecht, Germany) and filtered at 3 kHz. Leak and capacitive transients were subtracted using a P/-4 protocol. Voltage protocols were adjusted to compensate for the more negative voltage dependence of activation in extracellular Ba2+ solution. Data analysis was performed using IGOR (Wave-Metrics, Lake Oswego, OR). Activation curves were fit to determine values for the voltage of half-activation (V1 2) and the slope (k) using the following Boltzmann equation: . All average data represent the mean ± SEM. Statistical significance was determined using Student’s unpaired t test.
Expression of CaV2.1 in cultured SCG neurons
SCG neurons were cultured for 6 weeks as described (Leal et al., 2012; Mochida et al., 2008; Mochida et al., 1996) to allow synapse formation. cDNA encoding α12.1 (2.5 μg) or α12.1IMAA/ΔCBD (2.5 μg), NCS-1-eGFP (0.5 μg) or eGFP were microinjected into the nuclei of SCG neurons through glass micropipettes along with fast green dye as described (Leal et al., 2012; Mochida et al., 2008; Mochida et al., 1996).
Synaptic transmission between SCG neurons
Conventional intracellular recordings were made from two neighboring neurons using microelectrodes filled with 1M KAc (40–60 MΩ). EPSPs were recorded from a non-injected neuron, whereas action potentials were generated in the injected presynpatic neuron expressing CaV2.1 channels and NCS-1 proteins by passing 1–2 nA of current for 5 ms. All endogenous synaptic transmission was blocked by bath application of 3 μM ω-conotoxin GVIA in a modified Kreb’s solution consisting of (in mM) 136 NaCl, 5.9 KCl, 2 CaCl2, 1.2 MgCl2, 11 glucose and 3 Na-Hepes (pH 7.4). Each protocol was repeated 10 times. EPSP amplitudes were averaged and normalized to the first EPSP of each trial and plotted against action potential number. Mean EPSP trains (Fig. 6B) were fit with biexponential functions C + Aexp(−t/τ1) + (1 −A − C)exp(−t/τ2). The absolute values of τ1 describing facilitation were poorly defined by the data and were fixed at reasonable values for fitting. Mean EPSP trains (Figure 5 and Fig. 7) were fit with single exponential functions C + Aexp(−t/τ). Best-fit (solid lines) and 95% confidence estimates for these fits (dotted lines) are provided in the figures.
Figure 6.
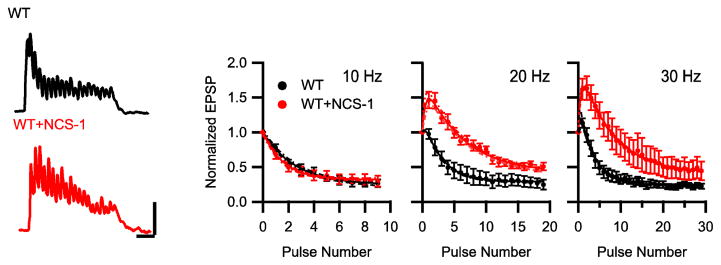
Modulation of short-term plasticity during bursts of neuronal activity by NCS-1. (A) Representative EPSPs evoked by repetitive action potentials at 20 Hz for 1 s in the presynaptic neurons expressing CaV2.1 alone (black) or co-transfected with NCS-1 (red). Data from 10 sweeps repeated every 30 s at each frequency were averaged. (B) Mean normalized EPSP amplitude at 10, 20 and 30 Hz frequency. EPSPs were normalized to the first EPSP of each train and plotted against action potential number for CaV2.1 alone (black) and with NCS-1 (red). Points represent mean ± SEM. n = 6–10
Figure 5.

NCS-1 modulation of synaptic transmission mediated by endogenous N-type Ca2+ current. Mean normalized EPSP amplitude at 30 Hz. EPSPs were normalized to the first EPSP of each train and plotted against action potential number for endogenous CaV2.2 alone (black) and with NCS-1 (red) (n = 6).
Figure 7.
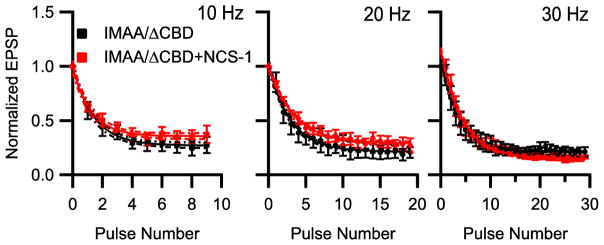
NCS-1 modulation of synaptic transmission mediated by CaV2.1IMAA/ΔCBD. Mean normalized EPSP amplitude at 10, 20 Hz and 30 Hz for neurons injected with CaV2.1IMAA/ΔCBD alone (black) and with NCS-1 (red), n = 6–10.
Results
Interaction between CaV2.1 and NCS-1
Although NCS-1 shares structural and amino acid sequence similarity with other CaS proteins, it was not known prior to this work whether it binds directly to CaV2.1 channels. Therefore, as a first step toward characterizing its potential functional effects on CaV2.1 channels, we used in vitro binding methods to measure its interaction with the CaS protein regulatory site on CaV2.1. To test direct binding of NCS-1 to the IQ-like motif and the CBD in the CaS protein regulatory site, we used MBP-tagged NCS-1 immobilized on amylose beads and GST-tagged IQ-like domain and CBD. In control experiments, GST alone did not bind to either NCS-1-MBP or MBP (Fig. 1A, lanes 1–3). CBD-GST bound specifically to immobilized NCS-1-MBP in the presence of 500 μM Ca2+ (Fig. 1A, lane 4). Binding of CBD-GST to NCS-1-MBD is Ca2+-dependent, as binding was lost in the presence of EGTA (Fig. 1A, lanes 4 and 5). In contrast to CBD-GST, we observed specific binding of the IQ-GST to NCS-1-MBP (Fig. 1A, lanes 6 and 7), but addition of EGTA had no effect (Fig. 1A, lane 8) indicating that binding of IQ-GST to NCS-1-MBP is Ca2+-independent. These results demonstrate differential Ca2+-dependent interactions of NCS-1 with the two components of the CaS protein regulatory site.
Figure 1.

Binding of IQ domain and calmodulin binding domain (CBD) with NCS-1. (A) Binding of GST-tagged IQ domain and CBD domain with MBP-tagged NCS-1 immobilized on amylose resin beads (Bait) in the presence of 200 μM Ca2+, 500 μM Ca2+, or 10 mM EGTA detected by immunoblot with anti-GST antibodies. 1) NCS-1-MBP+GST; 2) MBP+GST; 3) MBP+GST; 4) NCS-MBP+CBD-GST, 500 μM Ca2+; 5) NCS-MBP+CBD-GST, 10 mM EGTA; 6) MBP+IQ-GST; 7) NCS-MBP+IQ-GST; 8) NCS-MBP+IQ-GST; (B) Binding of GST-tagged IQ domain and IM-AA domain with MBP-tagged NCS-1 immobilized on amylose resin beads (Bait) in the presence of 200 μM Ca2+ or 10 mM EGTA detected by immunoblot with anti-GST antibodies. Relative binding to NCS-1 was calculated by determining the ratio between IQ-GST or IMAA-GST and NCS-1-MBP. Bound ligands were quantified by direct photon counting of luminescence from the antibodies bound to epitope tags in the linear range of the luminometer. *p < 0.05, one-way ANOVA, post-hoc Tukey test.
Previous studies have shown that mutation of the IQ-like motif by substitution of two Ala residues for the initial Ile and Met to give the mutant IM-AA reduces binding affinity for CaS proteins (DeMaria et al., 2001; Lee et al., 2003). We used a GST-tagged IM-AA mutant IQ domain to test the effect of this mutation on binding of NCS-1. We observed that alanine substitutions in the IQ-like domain caused a significant decrease in binding (Fig. 1B. Ca2+: IQ + NCS-1, 0.57 ± 0.06 vs. IMAA + NCS-1, 0.24 ± 0.06; EGTA: IQ + NCS-1, 0.56 ± 0.06 vs. IMAA + NCS-1, 0.29 ± 0.08; *p < 0.05). The remaining binding activity was Ca2+ independent as for the wild-type IQ-like motif. These results support the conclusion that NCS-1 binds directly to the CaS protein regulatory site including the IQ-like motif.
Modulation of CaV2.1 channels by NCS-1
We carried out whole-cell voltage clamp studies of tsA-201 cells transfected with CaV2.1 alone or together with NCS-1 during 1-s test pulses. Co-expression of NCS-1 did not alter the peak current amplitude, IV-relationship (Fig. 2A, p = 0.89) or voltage-dependence of half-maximal activation (Fig. 2B, V1/2: 8.28 ± 0.62 mV vs. 8.68 ± 1.01 mV, p = 0.73). The 0 to 63% rise time of the CaV2.1 current was calculated to compare the rate of activation (Fig. 2C). No significant change of the rate of activation was observed (Fig. 2C, 1.62 ± 0.25 ms vs 1.51 ± 0.11 ms, p = 0.78).
Figure 2.

NCS-1 expression does not affect IV-relationship or voltage-dependent activation. (A) Mean IV curve in tsA-201 cells transfected with CaV2.1 alone (black, n = 6) or co-transfected with NCS-1 (red, n = 6). The current was activated by depolarizing steps from −40 mV to 60 mV for 10 ms. (B) Voltage dependence of activation of CaV2.1 channels alone (black; n = 13), or with NCS-1 (red; n = 11) with Ca2+ as permeant ion. (C) The 0 to 63% rise time of CaV2.1 current was calculated in tsA cells transfected with CaV2.1 alone (black, n = 6) or co-transfected with NCS-1 (red, n = 6). (D) Voltage dependence of activation of CaV2.1 channels alone (black; n = 12), or with NCS-1 (red; n = 7) with Ba2+ as permeant ion.
During 1-s test depolarizations, co-expression of NCS-1 with CaV2.1 slowed the rate of inactivation significantly (Fig. 3A). The residual Ca2+ current of NCS-1-expressing cells at the end of a 1-s test pulse was significantly larger than that of cells transfected with CaV2.1 alone (Fig. 3B, 0.28 ± 0.03 vs 0.40 ± 0.04, *p < 0.05). To analyze NCS-1 regulation in response to more physiological stimuli, Ca2+ currents were recorded during trains of repetitive 5 ms depolarizations to 20 mV at 100 Hz. In CaV2.1-transfected cells, the Ca2+ current showed facilitation followed by progressive inactivation, similar to previous studies (Fig. 3C) (Lautermilch et al., 2005; Lee et al., 2000; Nanou et al., 2012). Co-expression of NCS-1 resulted in enhanced facilitation and slowed inactivation of Ca2+ current (Fig. 3C).
Figure 3.

Modulation of CaV2.1 channels by NCS-1. (A) Mean normalized Ca2+ currents in tsA-201 cells transfected with CaV2.1 alone (black, n = 16) or co-transfected with NCS-1 (red, n = 9). *p < 0.05, unpaired t test. (B) Mean ratio of Ca2+ residual current amplitude measured at the end of the 1 s pulse (Ires) over peak current amplitude (Ipeak) from tsA-201 cells transfected with CaV2.1 alone (black) or cotransfected with NCS-1 (red). (C) Mean normalized Ca2+ current during 100 Hz train of 5 ms pulses to +20 mV for CaV2.1 alone (black, n = 15) or co-transfected with NCS-1 (red, n = 11). (D) Mean normalized Ba2+ currents in cells transfected with CaV2.1 alone (black, n = 14) or co-transfected with NCS-1 (red, n = 7). (E) Mean ratio of Ba2+ residual current amplitude measured at the end of the 1 s pulse (Ires) over peak current amplitude (Ipeak) from tsA-201 cells transfected with CaV2.1 alone (black) or cotransfected with NCS-1 (red). (F) Mean normalized Ba2+ current during 100 Hz train for CaV2.1 alone (black, n = 17) or co-transfected with NCS-1 (red, n = 7).
Enhanced facilitation and rapid inactivation of CaV2.1 channels are Ca2+-dependent (Lee et al., 1999). We used Ba2+ substitution for Ca2+ to determine whether the effects of NCS-1 were also Ca2+-dependent. Substitution of Ca2+ with Ba2+ shifted the voltage-dependence activation to more negative membrane potential, but there was no difference in V1/2 between cells transfected with CaV2.1 alone or with NCS-1 (Fig. 2D, V1/2: 2.86 ± 1.28 mV vs. 2.55 ± 1.55 mV; p = 0.88). After replacing Ca2+ with Ba2+ as the charge carrier, the rate of inactivation was similar between cells transfected with CaV2.1 alone or with NCS-1 (Fig. 3D–F), indicating that NCS-1 specifically modulates Ca2+-dependent inactivation. The Ca2+-dependent component of inactivation is caused by association of endogenous CaM with C-terminal domain of CaV2.1 (DeMaria et al., 2001; Lee et al., 1999; Lee et al., 2003). CaM was shown to be endogenously expressed in tsA-201 cells and to bind to the CaS protein regulatory site (Lee et al., 1999). Therefore, our results suggest that NCS-1 binds to the CaS protein regulatory site, displaces CaM, and thereby regulates CaV2.1 channels by reducing Ca2+-dependent inactivation.
Function of the IQ-like motif and CBD in modulation of CaV2.1 channels by NCS-1
To test whether facilitation is mediated by interaction between NCS-1 and CaV2.1, we studied the CaV2.1IMAA/ΔCBD mutant, which is known to abolish the interaction between the CaV2.1 channel and CaS proteins (DeMaria et al., 2001; Lautermilch et al., 2005; Lee et al., 1999; Lee et al., 2003; Nanou et al., 2012). CaV2.1IMAA/ΔCBD was transfected alone or together with NCS-1. Co-expression of NCS-1 with Cav2.1IMAA/ΔCBD had no effect on Ca2+-dependent inactivation during a single depolarization or repetitive depolarizations (Fig. 4A–C) and had no effect on the peak current amplitude (p = 0.86) or voltage-dependence of activation (Fig. 4D V1/2: 8.92 ± 1.3 mV vs. 8.41 ± 1.15 mV; p = 0.78). Overall, these results indicate that association of NCS-1 with the bipartite CaS regulatory site in the C-terminal domain of CaV2.1 is required for NCS-1 modulation of P/Q-type Ca2+ currents.
Figure 4.
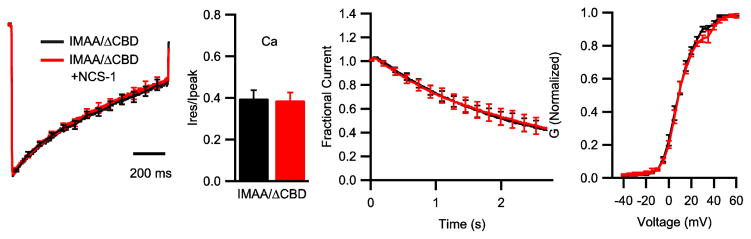
Functions of IQ domain and CBD domain in modulation of CaV2.1 channel by NCS-1. (A) Mean normalized Ca2+ currents in cells transfected with CaV2.1IMAA/ΔCBD (black, n = 11) or co-transfected with NCS-1 (red, n = 9). (B) Mean ratio of Ca2+ residual current amplitude measured at the end of the 1 s pulse (Ires) over peak current amplitude (Ipeak) from tsA-201 cells transfected with CaV2.1IMAA/ΔCBD alone (black) or cotransfected with NCS-1 (red). (C) Mean normalized Ca2+ current during 100 Hz train for CaV2.1IMAA/ΔCBD alone (black, n = 11) or with NCS-1 (red, n = 7). (D) Voltage dependence of activation of CaV2.1IMAA/ΔCBD channels alone (black; n = 6) or with NCS-1 (red; n = 6).
NCS-1 and endogenous neurotransmission in superior cervical ganglion neurons
Cultured SCG neurons transfected with wild-type or mutant calcium channels are an advantageous experimental system for studies of the role of calcium channel regulation in synaptic plasticity (Leal et al., 2012; Magupalli et al., 2013; Mochida et al., 2008; Mochida et al., 2003). Endogenous neurotransmission in cultured SCG neurons is mediated by N-type Ca2+ currents, which can be blocked by ω-conotoxin GVIA (Mochida et al., 2008; Mochida et al., 2003). In response to a train of depolarizing stimuli, endogenous synapses between SCG neurons show rapid depression but no facilitation (Fig. 5). In order to assess the effects of NCS-1 on endogenous neurotransmission in SCG neurons, we transfected NCS-1 by injection of cDNA into the presynaptic neuron (Materials and methods). We found no significant effects of presynaptic expression of NCS-1 on either short-term facilitation or rapid depression of endogenous neurotransmission mediated by N-type Ca2+ currents (Fig. 5). These negative results show that the endogenous N-type calcium channels are not modulated by NCS-1, and they also eliminate effects of NCS-1 on the endogenous exocytosis machinery as important contributors to facilitation in SCG neurons. Therefore, SCG neurons are an appropriate null background for studies of the effects NCS-1 on short-term synaptic plasticity of synaptic transmission mediated by transfected CaV2.1 channels.
NCS-1 modulation of short-term synaptic plasticity mediated by CaV2.1 channels
We transfected CaV2.1 channels without or with NCS-1 in SCG neurons by injection of cDNA in the presynaptic neuron to assure that the expressed proteins have no postsynaptic effects, and we blocked endogenous N-type Ca2+ currents with ω-conotoxin GVIA (see Materials and methods). Since neural information is encoded in bursts of synaptic activity in vivo, we stimulated the synapses at various frequencies to elicit short-term synaptic plasticity (Fig. 6). Repetitive stimulation at 10 Hz produced short-term depression of EPSPs, which was similar in control synapses and NCS-1 synapses (Fig. 5B, EPSP due to stimulus 3: p = 0.48). We observed little or no facilitation followed by rapid depression of synaptic transmission for stimuli at 20 Hz in control synapses (Fig. 6A, B), but synapses expressing NCS-1 showed enhanced facilitation and slower depression at 20 Hz (Fig. 6A, B). More rapid stimulation at 30 Hz induced greater facilitation in control synapses and larger increases in facilitation and in the rate of rapid depression in the presence of NCS-1 (Fig. 6B). Peak synaptic facilitation measured from the EPSPs in response to stimulus 3 was significantly enhanced at 20 Hz and 30 Hz in NCS-1-expressing synapses (CON: 0.78 ± 0.12 vs NCS-1: 1.45 ± 0.11, *p < 0.01 for 20 Hz; 0.95 ± 0.10 vs 1.63 ± 0.17, *p < 0.01 for 30 Hz). These results show that expression of NCS-1 induced enhanced synaptic facilitation during trains of evoked action potentials.
Function of the IQ-like motif and CBD in short-term plasticity
To test whether direct binding of NCS-1 to the CaS protein regulatory site on CaV2.1 is required for modulation of short-term plasticity by NCS-1, we transfected SCG neurons with Cav2.1IMAA/ΔCBD alone or with NCS-1. Co-expression of NCS-1 had no effect on Cav2.1IMAA/ΔCBD-driven synaptic transmission in response to trains of stimulation at 10, 20, and 30 Hz (Fig. 7). Thus, NCS-1 expression did not alter either facilitation or depression in response to trains of stimulation in Cav2.1IMAA/ΔCBD-expressing synapses, indicating that its effects on short-term synaptic plasticity require binding to the CaS regulatory site on CaV2.1 channels.
Modulation of paired-pulse facilitation by NCS-1
Facilitation and rapid depression of synaptic transmission can also be measured in paired-pulse stimulation experiments. To determine whether presynaptic expression of NCS-1 could affect this rapid form of short-term synaptic plasticity, we evoked EPSPs by paired stimuli with different interstimulus intervals as shown in Fig. 8A. Paired pulse ratios (PPRs) for synaptic transmission mediated by CaV2.1 were approximately 1.0 (Fig. 8A, B, CON: PPR30ms, 1.06 ± 0.11, CON: PPR50ms, 1.09 ± 0.09) at the interstimulus intervals we examined using 2 mM [Ca]i, with no indication of paired-pulse facilitation or depression. In contrast, NCS-1-expressing synapses exhibited significant paired-pulse facilitation (PPF) at interstimulus intervals between 30 ms and 50 ms (Fig. 8A–C; NCS-1: PPR30ms, 1.58 ± 0.12, NCS-1: PPR50ms, 1.47 ± 0.13, *p < 0.05). Paired-pulse facilitation induced by NCS-1 was not caused by a change of initial neurotransmitter release probability since there was no difference between amplitudes of the first EPSP between control synapses and NCS-1-expressing synapses (Fig. 8D, left panel, p = 0.56). Moreover, the shapes of EPSPs were identical for control synapses and NCS-1-expressing synapses (Fig. 8A), ruling out changes in the kinetics of the postsynaptic component as the basis of the facilitation. These data show that NCS-1 induces paired-pulse facilitation by changing release probability in an activity-dependent manner.
Figure 8.
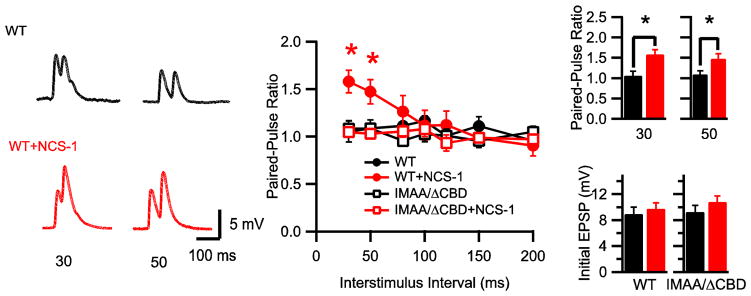
Modulation of paired-pulse plasticity by NCS-1. (A) Representative averages of 10 EPSPs evoked by paired-action potentials with 30-ms or 50-ms interstimulus intervals in presynaptic neurons expressing CaV2.1 alone (black) and in neurons co-injected with NCS-1 (red), recorded in the presence of ω-conotoxin GVIA (3 μM). (B) Paired-pulse ratio (PPR) plotted against interstimulus interval. *p < 0.05, two-way ANOVA, post-hoc Bonferroni test. (C) PPR for 30-and 50-ISI for WT (black), WT+NCS-1 (red) neurons. *p < 0.05. (D) Averaged initial EPSP amplitude (mV) for WT (black, left) and WT+NCS-1 (red, left) and CaV2.1IMAA/ΔCBD (black, right) and CaV2.1IMAA/ΔCBD+NCS-1 (red, right).
In contrast to the results with wild-type CaV2.1 channels, transfection of NCS-1 with Cav2.1IMAA/ΔCBD did not induce facilitation at any interstimulus interval (Fig. 8B). As for wild-type CaV2.1, expression of Cav2.1IMAA/ΔCBD alone or with NCS-1 did not alter the basal neurotransmission compared with synapses expressing CaV2.1 (Fig. 8D, right panel, p = 0.63). These data demonstrate that mutation of the IQ-like motif and deletion of the CBD eliminate the modulation by NCS-1 and therefore suggest that direct binding of NCS-1 to CaV2.1 via the IQ-like motif and CBD is required for NCS-1 modulation of short-term plasticity.
Discussion
Our results show that NCS-1 can modulate P/Q-type Ca2+ currents and short-term synaptic plasticity through direct interaction with the CaS protein regulatory site in the C-terminal domain of presynaptic CaV2.1 channels. These results provide strong evidence that expression of NCS-1 can override ubiquitous CaM-dependent regulation of CaV2.1 channels and allow neurons to change their patterns of short-term synaptic facilitation and depression. Evidently, differential expression of NCS-1 in neurons can contribute to the diversity of presynaptic short-term synaptic plasticity and the resulting broad range of neuronal integration and computation through its modulation of Ca2+-dependent inactivation of presynaptic CaV2.1 channels.
Direct binding between NCS-1 and CaV2.1 channels
We found that NCS-1 interacts with the IQ-like domain of CaV2.1 channels (residues 1848–1964) in a Ca2+-independent manner and binds to the downstream CBD (residues 1959–2035) in a Ca2+-dependent manner. A recent paper published after submission of this work for review also showed that NCS-1 bound directly to the C-terminal domain of CaV2.1 channels; however, it bound to a peptide containing residues 1898–2035 in a Ca2+-dependent manner and did not bind to the CBD (Lian et al., 2014). These two studies are similar in that both show specific, calcium-dependent interactions of NCS-1 with peptides derived from the C-terminal regulatory domain of CaV2.1 channels. However, the details of the binding reaction and its Ca2+-dependent regulation differ between the two sets of results. Differences in the peptide fragments studied and in the ionic conditions used to study the binding interactions may account for these differences in results. Our model for biphasic regulation of CaV2.1 channel function by sequential interactions with the N-and C-terminal lobes of CaS proteins envisions reciprocal Ca2+-dependent conformational changes in the CaS proteins and their interaction site(s) that drive channel regulation (Catterall and Few, 2008; Lee et al., 2003); therefore, it is not surprising that different peptides derived from the interacting sites would have differences in Ca2+-dependent binding properties. Our results presented here connect these binding interactions directly to calcium channel regulation and synaptic plasticity.
NCS-1 binding reduces Ca2+-dependent inactivation of CaV2.1 channels
In transfected tsA-201 cells, we found that Ca2+-dependent inactivation of CaV2.1 channels is specifically reduced by NCS-1. No effect was observed on Ba2+ currents, confirming that the effects of this CaS protein require Ca2+ entry. Previous studies had not resolved the cellular or molecular mechanism through which NCS-1 may regulate the function of CaV2.1 channels. When expressed in dissociated hippocampal neurons, NCS-1 did not alter the non-L-type Ca2+ current, which likely included N-type, P/Q-type, and possibly R-type Ca2+ currents (Sippy et al., 2003). Specific effects of NCS-1 on P/Q-type Ca2+ current may have been obscured by these other Ca2+ currents that are not responsive to NCS-1. Micro-injected NCS-1 protein enhanced facilitation of P/Q-type Ca2+ current at the Calyx of Held synapse via acceleration of voltage-dependent activation (Tsujimoto et al., 2002). This effect could have been mediated indirectly through an intermediary protein, and therefore may be distinct from the modulation of P/Q-type Ca2+ current via direct interaction between NCS-1 and the CaS protein regulatory site on the CaV2.1 channel that we describe here. Alternatively, effects on P/Q-type Ca2+ currents in the Calyx of Held could arise through differential modulation of CaV2.1 channels by CaVβ subunits or other proteins in that synapse.
NCS-1 interaction with CaV2.1 channels induces synaptic facilitation
NCS-1 has been reported to modulate both basal neurotransmission (Olafsson et al., 1995; Wang et al., 2001) and short-term synaptic plasticity (Pongs et al., 1993; Sippy et al., 2003; Tsujimoto et al., 2002) via a variety of molecular mechanisms. Synaptic facilitation depends on elevation of residual Ca2+ during repetitive stimulation (Zucker and Regehr, 2002). One model proposes that facilitation is due to elevation of residual Ca2+ following saturation of local calcium buffers (Zucker and Regehr, 2002). If NCS-1 acts as such a calcium buffer, over-expression of NCS-1 should result in decreased facilitation, which is opposite to our findings. Moreover, in our experiments, it is not likely that NCS-1 enhances facilitation by acting as a fast, saturable buffer (Zucker, 2001), since neither basal neurotransmission nor neurotransmission initiated by N-type Ca2+ currents is affected by NCS-1 expression.
Another model for short-term facilitation is modulation of synaptic vesicle function. Could NCS-1 instead modulate exocytosis machinery? To address this question, we recorded endogenous synaptic transmission driven by CaV2.2 channels in SCG neurons and found that basal neurotransmission was not affected by co-expression of NCS-1 and short-term facilitation remained unchanged. These results indicate that expression of NCS-1 specifically modulates short-term plasticity mediated by CaV2.1 channels and leaves the endogenous exocytosis machinery unaffected. Evidently, NCS-1 primarily acts as a calcium sensor for modulation of CaV2.1 channels and short-term plasticity in the presynaptic nerve terminals of SCG neurons.
Our results show that specific expression of NCS-1 in presynaptic neurons can functionally compete with endogenous CaM for regulation of CaV2.1 channels and synaptic transmission. Endogenous CaS proteins are highly expressed in central neurons, so it is likely that similar regulation of synaptic plasticity and neurotransmission takes place. The interaction between NCS-1 and CaV2.1 channels enhances presynaptic Ca2+ entry by reducing inactivation of P/Q-type Ca2+ current, leading to facilitation of synaptic transmission as predicted by the supralinear dependence of neurotransmission on presynaptic Ca2+ entry (Dodge and Rahamimoff, 1967). The fact that modulation of Ca2+ current and short-term plasticity is largely eliminated when mutant CaV2.1IMAA/ΔCBD channels are expressed demonstrates that direct binding of NCS-1 is required and eliminates effects on other possible presynaptic targets. Our results do not exclude additional mechanisms of NCS-1 regulation of synaptic plasticity in other physiological settings, but regulation of CaV2.1 channels by direct binding to their CaS protein regulatory site is responsible for its effects on short-term synaptic plasticity in transfected SCG neurons.
NCS-1 and diversity of short-term synaptic plasticity
NCS-1, like VILIP-2, CaBP1, and other CaS proteins, has a similar structure to their common ancestor, CaM, with two pairs of EF-hand Ca2+-binding domains separated by a central alpha helix (Burgoyne and Weiss, 2001). NCS-1 is able to bind three Ca2+ ions in the submicromolar range with affinities considerably higher than those of CaM (Mikhaylova et al., 2009). The higher affinity of NCS-1 for Ca2+ likely contributes to its ability to override the regulatory effects of ubiquitously expressed CaM. The highest level of expression of NCS-1 was found in the hippocampus, cerebellum, and retina in rat brain (Braunewell and Gundelfinger, 1999; Paterlini et al., 2000). Therefore, cell-type specific modulation of short-term plasticity by NCS-1 should add to the diversity of the short-term synaptic plasticity in these neurons. Our previous results with CaBP1 and VILIP-2 indicate that these CaS proteins can also override the ubiquitous effects of CaM and modulate CaV2.1 channels and short-term synaptic plasticity in a bi-directional manner, fine-tuning synaptic plasticity by enhancing facilitation or accelerating depression (Leal et al., 2012). Overall, our results suggest that modulation of presynaptic Ca2+ entry due to regulation of facilitation and/or inactivation of P/Q-type Ca2+ current by NCS-1 and other CaS proteins may be a crucial determinant of short-term synaptic plasticity and the encoding properties of neurons through activity-dependent modulation of presynaptic function.
Highlights.
NCS-1 directly interacts with the IQ-like motif and CBD domain of Cav2.1 channels.
NCS-1 reduces Ca2+-dependent inactivation of P/Q-type Ca2+ current.
Presynaptic expression of NCS-1 induces short-term synaptic facilitation.
Presynaptic effects of NCS-1 are lost when the IQ-like motif and CBD are mutated.
Acknowledgments
This work was supported by funds from Research Grant R01 NS22625 from the National Institutes of Health to W. A. C.
Footnotes
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Gundelfinger ED. Intracellular neuronal calcium sensor proteins: a family of EF-hand calcium-binding proteins in search of a function. Cell Tissue Res. 1999;295:1–12. doi: 10.1007/s004410051207. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, O’Callaghan DW, Hasdemir B, Haynes LP, Tepikin AV. Neuronal Ca2+-sensor proteins: multitalented regulators of neuronal function. Trends Neurosci. 2004;27:203–209. doi: 10.1016/j.tins.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Dason JS, Romero-Pozuelo J, Marin L, Iyengar BG, Klose MK, Ferrus A, Atwood HL. Frequenin/NCS-1 and the Ca2+-channel alpha1-subunit co-regulate synaptic transmission and nerve-terminal growth. J Cell Sci. 2009;122:4109–4121. doi: 10.1242/jcs.055095. [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- Few AP, Nanou E, Scheuer T, Catterall WA. Molecular determinants of CaV2.1 channel regulation by calcium-binding protein-1. J Biol Chem. 2011;286:41917–41923. doi: 10.1074/jbc.M111.292417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J Neurosci. 2005;25:7062–7070. doi: 10.1523/JNEUROSCI.0447-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal K, Mochida S, Scheuer T, Catterall WA. Fine-tuning synaptic plasticity by modulation of CaV2.1 channels with Ca2+ sensor proteins. Proc Natl Acad Sci U S A. 2012;109:17069–17074. doi: 10.1073/pnas.1215172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of CaV2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian LY, Pandalaneni SR, Todd PA, Martin VM, Burgoyne RD, Haynes LP. Demonstration of binding of neuronal calcium sensor-1 to the cav2.1 p/q-type calcium channel. Biochemistry. 2014;53:6052–6062. doi: 10.1021/bi500568v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Hillman DE, Cherksey B. Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 1992;15:351–355. doi: 10.1016/0166-2236(92)90053-b. [DOI] [PubMed] [Google Scholar]
- Magupalli VG, Mochida S, Yan J, Jiang X, Westenbroek RE, Nairn AC, Scheuer T, Catterall WA. Ca2+-independent activation of Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain of CaV2.1 calcium channels. J Biol Chem. 2013;288:4637–4648. doi: 10.1074/jbc.M112.369058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova M, Reddy PP, Munsch T, Landgraf P, Suman SK, Smalla KH, Gundelfinger ED, Sharma Y, Kreutz MR. Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc Natl Acad Sci U S A. 2009;106:9093–9098. doi: 10.1073/pnas.0903001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Mochida S, Westenbroek RE, Yokoyama CT, Itoh K, Catterall WA. Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc Natl Acad Sci U S A. 2003;100:2813–2818. doi: 10.1073/pnas.262787299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou E, Martinez GQ, Scheuer T, Catterall WA. Molecular determinants of modulation of CaV2.1 channels by visinin-like protein 2. J Biol Chem. 2012;287:504–513. doi: 10.1074/jbc.M111.292581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olafsson P, Wang T, Lu B. Molecular cloning and functional characterization of the Xenopus Ca2+-binding protein frequenin. Proc Natl Acad Sci U S A. 1995;92:8001–8005. doi: 10.1073/pnas.92.17.8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini M, Revilla V, Grant AL, Wisden W. Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience. 2000;99:205–216. doi: 10.1016/s0306-4522(00)00201-3. [DOI] [PubMed] [Google Scholar]
- Pongs O, Lindemeier J, Zhu XR, Theil T, Engelkamp D, Krah-Jentgens I, Lambrecht HG, Koch KW, Schwemer J, Rivosecchi R, et al. Frequenin--a novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron. 1993;11:15–28. doi: 10.1016/0896-6273(93)90267-u. [DOI] [PubMed] [Google Scholar]
- Sippy T, Cruz-Martin A, Jeromin A, Schweizer FE. Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat Neurosci. 2003;6:1031–1038. doi: 10.1038/nn1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- Wang CY, Yang F, He X, Chow A, Du J, Russell JT, Lu B. Ca2+ binding protein frequenin mediates GDNF-induced potentiation of Ca2+ channels and transmitter release. Neuron. 2001;32:99–112. doi: 10.1016/s0896-6273(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Archer DA, Burgoyne RD. Neuronal Ca2+ sensor-1/frequenin functions in an autocrine pathway regulating Ca2+ channels in bovine adrenal chromaffin cells. J Biol Chem. 2000;275:40082–40087. doi: 10.1074/jbc.M008603200. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD. Voltage-independent inhibition of P/Q-type Ca2+ channels in adrenal chromaffin cells via a neuronal Ca2+ sensor-1-dependent pathway involves Src family tyrosine kinase. J Biol Chem. 2001;276:44804–44811. doi: 10.1074/jbc.M103262200. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Hui H, Burgoyne RD. Neuronal calcium sensor-1 regulation of calcium channels, secretion, and neuronal outgrowth. Cell Mol Neurobiol. 2010;30:1283–1292. doi: 10.1007/s10571-010-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Increased Ca2+ buffering enhances Ca2+-dependent process. J Physiol. 2001;531:583. doi: 10.1111/j.1469-7793.2001.0583h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]